

Summary
MLL/SET methyltransferases catalyze methylation of histone 3 lysine 4 and play critical roles in development and cancer. We assessed MLL/SET proteins and found that SETD1A is required for survival of acute myeloid leukemia (AML) cells. Mutagenesis studies and CRISPR-Cas9 domain screening, showed the enzymatic SET domain is not necessary for AML cell survival but that a newly identified region, termed the FLOS (Functional Location on SETD1A) domain, is indispensable. FLOS disruption suppresses DNA damage response genes and induces p53-dependent apoptosis. The FLOS domain acts as a Cyclin K-binding site that is required for chromosomal recruitment of Cyclin K, and for DNA repair-associated gene expression in S phase. These data identify a connection between the chromatin regulator SETD1A and the DNA damage response that is independent of histone methylation, and suggests that targeting SETD1A and Cyclin K complexes may represent a therapeutic opportunity for AML and potentially other cancers.
In-brief
Independent of its enzymatic activity, H3K4 methyltransferase SETD1A promotes leukemic cell survival by regulating DNA damage response.
Introduction
Histone 3 lysine 4 methylation (H3K4me) is a post-translational modification that is associated with transcriptional regulation in multiple organisms from yeast to humans (Barski et al., 2007; Briggs et al., 2001; Ng et al., 2003; Shilatifard, 2008). The enzymes that catalyze this modification are important for the maintenance of developmental gene expression in multicellular organisms and are frequently mutated in human cancer (Bledau et al., 2014; Denissov et al., 2014; Ernst et al., 2004; Kandoth et al., 2013). H3K4 histone methyltransferases (HMTs) are defined by an enzymatic SET domain (Gu et al., 1992; Jones and Gelbart, 1993; Tschiersch et al., 1994). Yeast have a single H3K4 HMT, SET1, that is present in a multiprotein (COMPASS) complex responsible for all H3K4 methylation (Schneider et al., 2005). Mammals have 6 homologs of yeast SET1 that possess H3K4 HMT activity, including MLL1-4 (KMT2A-D) and SETD1A/B (KMT2F/G). Each of these enzymes is found in a multiprotein complex with overlapping subunits. While each of these complexes have been shown to possess H3K4 HMT activity, the extent to which their cellular functions are largely a result of SET-domain associated enzymatic activity is unclear.
One of the most well characterized mammalian SET1 homologs is MLL1. MLL1 is found in fusion proteins generated by chromosomal translocation of the MLL1 gene to one of more than 60 different partner genes (Krivtsov and Armstrong, 2007). Pediatric acute leukemia cases with MLL1-rearrangement (MLL-r) have a poor prognosis (Balgobind et al., 2009). Recent studies have shown that wild-type MLL proteins play a role in MLL-r leukemias but the mechanistic details and whether these roles are dependent on H3K4 HMT activity is just beginning to be explored (Milne et al., 2010; Santos et al., 2014; Thiel et al., 2010; Wong et al., 2015). Wild-type MLL1 is also indispensable for several subtypes of AML beyond MLL-r AML including MN1-expressing leukemia, NPM1-mutated leukemia and NUP98-rearranged leukemia (Kuhn et al., 2016; Riedel et al., 2016; Xu et al., 2016). However, the catalytic SET domain of the MLL1 protein is dispensable for leukemia growth suggesting a non-catalytic function of wild-type MLL1 and redundancies such that other SET1 homologs might contribute H3K4 HMT activity (Kuhn et al., 2016; Mishra et al., 2014). Since the enzymatic domains of the SET1 vertebrate homologs are highly conserved during evolution, these homologs might have both redundant and unique functions in regulating H3K4 methylation and leukemia cell proliferation. Distinguishing the dependencies on enzymatic and non-enzymatic functions in leukemia will shape small molecule drug development focused on targeting chromatin associated, epigenetic mechanisms.
Notable functional differences between closely-related SET1 homologs have been characterized in leukemia. For instance, although MLL3 and MLL4 are homologs of Drosophila Trithorax-related (Trr), MLL4 has a tumor-promoting role in MLL-r leukemia (Santos et al., 2014), while MLL3 is a tumor suppressor in −7/del(7q) AML (Chen et al., 2014). Differences such as these highlight the importance of defining unique functions of SET1 homologs. Homolog specific-binding proteins are likely one of the factors that influence the biologic activity of the different MLL complexes. For instance, MEN1 (also known as Menin) is a specific binding partner of MLL1/MLL2, and it is essential for chromatin binding and leukemogenic activity of MLL1 fusion proteins (Hughes et al., 2004; Yokoyama et al., 2004). Importantly, drugs targeting SET1 homolog binding partners have been identified as potential therapeutic tools against MLL-r/non-MLL-r leukemia and other solid cancers (Borkin et al., 2015; Cao et al., 2014; Funato et al., 2014; Grembecka et al., 2012; Kuhn et al., 2016). Thus, to identify novel therapeutic drug targets in cancer, we need to better understand the unique protein-protein interactions and the functional roles of H3K4 HMT complexes in normal cells and in cancer cells.
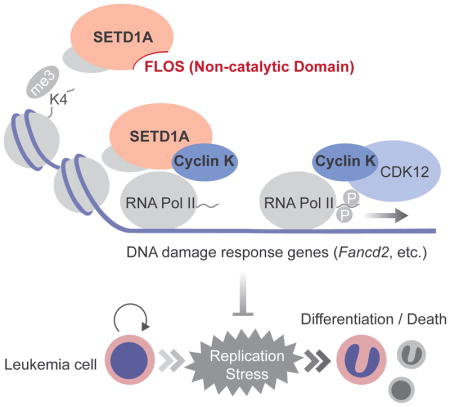
Here we report the essential roles of SETD1A in cell proliferation and define a novel protein-interacting domain in SETD1A. We show that the domain is required for the regulation of DNA damage response genes and that this novel domain binds Cyclin K. Our data also demonstrate that SETD1A/Cyclin K control DNA damage response gene expression during the S-phase of the cell cycle. These results reveal a non-redundant and non-enzymatic role of SETD1A, and suggest a potential therapeutic opportunity for leukemia and potentially other tumors.
Results
SETD1A/COMPASS complex components are required for growth of MLLAF9 leukemia cells
To identify MLL/SET/COMAPSS complex subunits important for leukemia cell growth, we performed a shRNA-based screen with murine MLL-AF9 leukemia cells. We prepared 42 individual-shRNA vectors against MLL family members and COMPASS subunits, and then performed a growth competition assay (Figure 1A). Toxicity from Setd1a, Cxxc1, Hcfc1 and Wdr5 shRNAs suggested that the SETD1A/COMPASS complex might have critical roles in MLL-AF9 leukemia cells (Figure 1A). Efficacy and specificity of shRNAs were confirmed by rescue experiments with an exogenous SETD1A construct (Figure S1A–C). To evaluate the effect of Setd1a knockdown in greater detail, we generated murine MLL-AF9 leukemia single cell clones which highly express a doxycycline inducible Setd1a shRNA. After doxycycline administration, two Setd1a shRNAs showed significant suppression of Setd1a expression (Figure 1B), and inhibition of leukemia cell proliferation (Figure 1C). Setd1a shRNAs induced apoptosis, cell cycle arrest, and neutrophilic differentiation (Figures 1D and 1E, S1D). To examine and compare the functional role of Setd1a and Setd1b on H3K4 methylation, we also performed an inducible knockdown of either Setd1a or Setd1b, or both genes in murine MLL-AF9 leukemia cells (Figures 1F and S1E–S1I). Even in this setting, Setd1a knockdown predominantly induced growth arrest and cell differentiation (Figures S1H–I). Surprisingly, Setd1a knockdown did not show reduction of global H3K4me3 (Figure 1F). H3K4me3 ChIP-seq also showed no obvious change in H3K4me3 after knockdown of Setd1a (Figure S1J). To confirm these effects on other leukemia cells, we suppressed SETD1A in the MOLM-13 human leukemia cell line (Figures S1K). Consistent with murine cells, SETD1A shRNA expression in MOLM-13 cells induced growth arrest and differentiation (Figures S1L–M). To examine a role for SETD1A in leukemia progression in vivo, we performed a Setd1a knockdown experiment in a MLL-AF9 murine leukemia model. At 3 weeks post-transplantation, Setd1a shRNAs significantly suppressed the frequency of leukemia cells in the PB (Figure 1G). In addition, Setd1a shRNA constructs significantly prolonged the survival of recipient mice (Figures 1H). Consistent with data from the PB, Setd1a knockdown strongly suppressed the growth of leukemia cells in bone marrow (Figure S1N). In mice that did get sick in the Setd1a knockdown group, we did not observe GFP++ cells in their bone marrow (BM) (Figure S1O) demonstrating loss of Setd1a shRNA expression. When we sorted GFP++ cells expressing Setd1a shRNAs from the bone marrow and spleen at earlier timepoints, these cells showed comparable H3K4me3 levels (Figure S1P), lower Setd1a expression (Figure S1Q), higher percentage of apoptotic cells (Figure 1I), and decreased proliferation (Figure S1R). The morphology and Gr-1 expression of the GFP++ cells also indicated induction of differentiation by Setd1a suppression in vivo (Figure 1J and S1S). These data show that Setd1a has indispensable roles in leukemia cell proliferation and leukemia progression.
Figure 1. SET/COMPASS complex is required for leukemia cell survival and H3K4me3 modification.

A. The indicated MLL/SET/COMPASS complex subunit shRNAs with a GFP reporter were expressed in mouse MLL-AF9 leukemia cells. This experiment was repeated 2 times with 3 biological replicates in each experiment.
B. Setd1a expression was analyzed in mouse MLL-AF9 leukemia cells that were transfected with doxycycline (dox) -inducible Renilla luciferase (shRen) control or Setd1a shRNAs (shSetd1a-1 and shSetd1a-2). Cells were analyzed at 4 days post-dox. Representative data from one out of 3 independent experiments with 3 biological replicates are shown.
C. Numbers of dox-inducible Ren or Setd1a shRNA-expressing cells were counted every 3 days post-dox. This experiment was repeated 2 times with 3 biological replicates in each experiment.
D. Annexin V+DAPI− population in dox-inducible Ren or Setd1a shRNA-expressing cells was assessed at 4 days post-dox. This experiment was repeated 2 times with 3 biological replicates in each experiment.
E. Dox-inducible Ren or Setd1a shRNA-expressing cells were stained with May-Grünwald/Giemsa at 4 days post-dox. Representative images from 3 independent experiments are shown. Bottom graph indicates the percentage of blasts (red), monocytes/macrophages (grey) and neutrophils (black).
F. Western blot analysis of Ren (Ren/Ren), Setd1b (Ren/Setd1b), Setd1a (Setd1a/Ren), and Setd1a/b (Setd1a/Setd1b) shRNA-expressing cells was performed at 4 days post-dox. Quantified intensity is normalized by GAPDH signal and fold changes are shown as a relative value to Control. Representative images from 2 independent experiments are shown.
G. Percentage of GFP+ shRNA-expressing leukemia cells in PB was analyzed at 3 weeks post-transplant. Representative data from one out of 2 independent experiments are shown.
H. Survival of recipient mice harboring dox-inducible shRNA-expressing leukemia cells was plotted. Representative data from one out of 2 independent experiments are shown (n = 5/group).
I. Annexin V+DAPI− population in Ren or Setd1a shRNA-expressing (GFP++, See also Figure S1N) leukemia cells in BM and spleen was analyzed at 3 weeks post-transplant. Representative data from one out of 2 independent experiments are shown (n = 4/group).
J. Ren or Setd1a shRNA-expressing leukemia cells in BM were sorted and stained with May-Grünwald/Giemsa at 3 weeks post-transplant. Representative images of 3 biological replicates are shown.
An internal region of SETD1A, but not the SET domain, is indispensable for leukemia growth
To evaluate the functional dependency on SETD1A, we performed rescue experiments with exogenous SETD1A mutant constructs (Figures 2A, 2B and S2A). As expected, wild-type SETD1A expression restored the growth of RFP+ Setd1a shRNA expressing cells (Figure S2B). Although the RRM domain deletion mutant failed to fully rescue cell growth at 12 days post-doxycycline, deletion of NSET and SET domains completely rescued the growth of Setd1a knockdown cells (Figures S2C). These results show that the H3K4 HMT activity of SETD1A is dispensable for MLL-AF9 leukemia growth.
Figure 2. Internal region on SETD1A encodes a critical functional domain for leukemia cell survival.

A. The human SETD1A deletion mutant constructs are shown as schematic illustrations.
B. Western blot analysis was performed using SETD1A deletion mutant-expressing 293T cells. These constructs were used for the following rescue experiments. Representative images from 2 independent experiments are shown.
C. Human SETD1A deletion mutant-expressing mouse MLL-AF9 leukemia cells were transfected with dox-inducible mouse Setd1a 3′UTR targeting shRNAs. This experiment was performed with 2 independent shRNAs and repeated 2 times with 3 biological replicates in each experiment.
D. Evolutionarily conserved motifs were identified inside of the functional region on SETD1A and named as FLOS1-4 (upper right panel). SETD1A mutant-expressing mouse MLL-AF9 leukemia cells were transfected with mouse Setd1a shRNAs. This experiment was performed with 2 independent shRNAs and repeated 2 times with 3 biological replicates in each experiment.
E. Empty (GFP), SETD1A wild-type (WT) and FLOS1/alanine mutant (F1-5A)–expressing cells were transfected with mouse Setd1a 3′UTR shRNAs, and the annexin V+DAPI− populations were analyzed at 4 days post-dox. This experiment was performed with 2 independent shRNAs and repeated 2 times with 3 biological replicates in each experiment.
F. Domain-focused CRISPR strategy was performed with Setd1a or Setd1b sgRNAs in inducible Cas9 (iCas9)-expressing MLL-AF9 leukemia cells or NIH3T3 cells. The relative location of each sgRNA to the SETD1A or SETD1B protein is indicated along the x-axis. Empty vector or Rpa3 sgRNAs were used as negative or positive controls, respectively. Representative data from one out of 3 independent experiments are shown.
G. Domain-focused CRISPR strategy was performed with Setd1a sgRNAs in human leukemia and sarcoma cell lines. Empty vector or RPA3 sgRNAs were used as negative or positive controls, respectively. This experiment was repeated 2 times with 3 biological replicates in each experiment.
The RRM domain of SETD1A is located at the N-terminus, and the NSET and SET domains are located at the C-terminus. Any role for the large internal region of SETD1A is unknown. We constructed eight additional SETD1A mutants, lacking parts of its internal region, and performed rescue experiments (Figures 2A and 2B). The SETD1A constructs that encode proteins lacking the entire internal region, as well as three independent deletion mutants (Δ387–586, Δ587–786, and Δ787–1026) showed strong functional defects (Figure 2C). These three mutant proteins were still mainly localized in the nucleus (Figure S2D). To identify potential functional domains spaning these regions, we compared the amino acid sequences of SETD1A and SETD1B across vertebrate species (Figures 2D and S2E). We found four highly conserved regions within 387–786 aa in SETD1A (Figure S2E). Here, we named these regions FLOS1-4 (Functional location on SETD1A: F1-4) and constructed alanine-substituted SETD1A mutants. Alanine-substituted mutants of FLOS1 and FLOS2 ablated SETD1A function (Figure 2D). We constructed seven independent mutants spanning the aa region 587–786 in SETD1A. However we found that every construct rescued leukemia cell growth after Setd1a knockdown (Figure 2D). These data suggest that the most critical regions of aa 387–1026 are FLOS1 and FLOS2. Furthermore, FLOS1 mutated SETD1A was unable to suppress apoptosis after Setd1a knockdown (Figure 2E). We have thus identified a novel functional region of SETD1A.
The FLOS domain supports leukemia cell growth
In order to further validate the FLOS domain as critical for leukemia cell survival, we targeted the FLOS and SET domain of endogenous SETD1A protein in leukemia cells using a domain-focused CRISPR-mediated mutagenesis approach (Shi et al., 2015). Only FLOS domain targeted MLL-AF9 leukemia cells were significantly depleted. Cells with other Setd1a targeted sgRNAs, as well as Setd1b targeting sgRNAs, were not depleted (Figure 2F and S2F). In order to assess whether targeting these specific regions of SETD1A suppressed growth of all proliferating cells, we also analyzed the roles of these domains in NIH3T3 fibroblast cells. Remarkably, the FLOS domain targeting sgRNAs did not affect the ratio of GFP-positive cells in growth competition assays (Figure 2F). In contrast, an Rpa3 sgRNA, which targets an essential gene, showed strong toxicity even in NIH3T3 cells (Figure 2F). We performed both a SURVEYOR assay and deep-sequencing analysis of mutation abundance following doxycycline treatment (Figures S2G–J). As expected, both MLL-AF9 cells and NIH3T3 cells showed high mutation frequencies, and the mutations from non-functional region targeted cells (NF-1/2) were stably detected in culture over time (Figures S2H–J). Although sgRNA-expressing NIH3T3 cells still harbored the FLOS1 mutated allele 12 days after doxycycline treatment, MLL-AF9 leukemia cells did not show the FLOS1 mutated allele 9 days after doxycycline treatment (Figures S2G, S2I–J). FLOS1/2 targeting sgRNAs were also tolerated in normal hematopoietic stem/progenitor cells, which formed colonies in vitro (Figure S2K). We also designed sgRNA constructs for the human SETD1A FLOS1 domain and expressed them in two MLL-r leukemia (MOLM-13, MV4-11), two non-MLL-r leukemia (U937, K562), and three sarcoma cell lines (A673, HSSYII, RH30) (Figure 2G). Other than RH30, the cell lines were sensitive to sgRNAs which target the FLOS1 domain, but not the SET domain. These studies indicate that the FLOS domain of endogenous SETD1A is more critical for cancer cells than other cell types such as fibroblasts.
To confirm the endogenous SETD1A function in leukemia, we generated MLL-AF9 transformed Setd1afl/fl;CreERT2 cells (Figure 3A). After tamoxifen treatment, Setd1afl/fl cells showed significant loss of colony-forming ability in culture (Figure 3B and S3A). Wild-type or SET domain deletion mutants completely rescued colony formation after Setd1a deletion, but neither FLOS1/2 mutants of SETD1A nor wild-type SETD1B rescued colony forming activity (Figure 3B). These results suggest that the FLOS domain may be responsible for the unique function of SETD1A in leukemia. To evaluate this, we performed domain swap experiments using expression constructs encoding a chimeric protein with SETD1B RRM/NSET/SET domains and SETD1A FLOS domain (Figure 3C and S3B). SETD1B variants showed comparable expression level in Setd1afl/fl;CreERT2 leukemia cells (Figures 3D). The SETD1 chimera encoding the 387–586 aa from SETD1A (B-FS) strongly supports cell survival and colony formation. The SETD1 chimera encoding the 387–1026 aa from SETD1A (B-FL) completely compensate the function of SETD1A (Figures 3E–G and S3C–D). SETD1 chimera-expressing Setd1afl/fl;CreERT2 leukemia cells showed complete deletion of Setd1a alleles (Figure 3H). These results indicate that the FLOS1 domain supports a non-redundant and non-enzymatic function of SETD1A in leukemia cell growth.
Figure 3. SETD1A FLOS domain provides a non-redundant role in leukemia cell proliferation.

A. Relative expression level of Setd1a in MLL-AF9 leukemia cells was quantified by qRT-PCR at 4 days post-tamoxifen treatment. This experiment was repeated 3 times with 3 biological replicates in each experiment.
B. MLL-AF9 leukemia cells were transfected with SETD1A mutant constructs. The cells were treated with tamoxifen, then assayed for colony-forming potential. Representative data from one out of 2 independent experiments with 3 biological replicates are shown.
C. The chimeric constructs of human SETD1A and human SETD1B are shown as schematic illustrations.
D. Setd1afl/fl;CreER cells were transfected with SETD1 chimeric constructs and relative exogenous gene expression level was quantified by Taqman probes which can detect either human SETD1A or SETD1B. This experiment was repeated 2 times with 3 biological replicates in each experiment.
E. MLL-AF9 leukemia cells were transfected with SETD1 chimeric constructs and monitored for cell number increase within 3 days from day 4 post-tamoxifen treatment. This experiment was repeated 3 times with 3 biological replicates in each experiment.
F. Annexin V+DAPI− population in Setd1afl/fl;CreER cells expressing SETD1 chimeric constructs was analyzed at day 5 post-tamoxifen treatment. This experiment was repeated 3 times with 3 biological replicates in each experiment.
G. Representative morphology of colonies in Setd1afl/fl;CreER cells expressing SETD1 chimeric constructs are shown.
H. Single cell-derived colonies of Setd1afl/fl;CreER cells expressing SETD1 chimeric constructs were isolated after tamoxifen-treatment followed by colony-forming assay. Setd1a alleles of 5 clones are genotyped by PCR.
Disruption of SETD1A suppresses the DNA damage response and induces p53-dependent apoptosis in AML cells
We performed RNA-seq analysis using Setd1a+/+ and Setd1aΔ/Δ MLL-AF9 leukemia cells (Figure 4A). Through gene ontology (GO) analysis and gene set enrichment analysis (GSEA), we found a significant decrease of DNA repair and Fanconi pathway-associated genes and a significant increase of p53 targets in Setd1aΔ/Δ MLL-AF9 leukemia cells (Figures 4B and S4A–B). Genes critical for the DNA damage response including Fancd2 and Mlh1 were downregulated in Setd1aΔ/Δ cells, and these findings were confirmed by western blotting (Figures S4C) (Ceccaldi et al., 2016). Fancd2 expression was downregulated 24 hours after Setd1a inactivation and preceded apoptosis induction (Figure 4C and S4D) supporting a direct effect of Setd1a suppression on Fancd2 expression. Complete knockout of Setd1a also did not affect the abundance of H3K4me3 and H3K27Ac marks on the Fancd2 or Mlh1 locus (Figure 4D, S4E–F and Table S2). Expression of wild-type SETD1A rescued Fancd2 expression whereas FLOS domain mutants did not rescue expression (Figure 4E). SETD1A/B chimeras containing the SETD1A FLOS1 domain also rescued Fancd2 expression (Figure 4F). Consistent with a role in the DNA damage response, Setd1aΔ/Δ MLL-AF9 leukemia cells showed higher frequencies of chromosomal gaps/breaks (Figure 4G, Table S1). The SETD1A FLOS domain is therefore required for maintenance of genes that are important for the DNA damage response.
Figure 4. SETD1A FLOS domain regulates DNA damage response.

A. A volcano plot is used to depict RNA-seq data. Three independent samples on Setd1a+/+ and Setd1aΔ/Δ leukemia cells were analyzed. Significantly downregulated DNA repair genes classified by GO analysis (see also Figure S4A) are shown as red dots.
B. Gene set enrichment analysis was performed on RNA-seq data from MLL-AF9 leukemia cells. The scatter plot shows normalized enrichment score (NES) and false discovery rate (FDR) q-values comparing MSigDB curated gene set enrichment (left). Gene sets upregulated or downregulated in Setd1aΔ/Δ leukemia cells have positive or negative NES values, respectively. Representative enrichment plot from a DNA repair gene set is shown (right).
C. qRT-PCR was performed to analyze Fancd2 expression in Setd1afl/fl;CreER MLL-AF9 leukemia cells from 24 to 120 hours post-tamoxifen. This experiment was repeated 3 times with 3 biological replicates in each experiment.
D. Histograms depict H3K4me3, H3K27Ac and mRNA peaks on the Fancd2 locus in Setd1a knockout leukemia cells.
E. MLL-AF9 leukemia cells were transfected with SETD1A mutant cDNA and qRT-PCR analysis of Fancd2 was performed at 4 days post-tamoxifen. Representative data from one out of 2 independent experiments with 3 biological replicates are shown.
F. MLL-AF9 leukemia cells were transfected with SETD1A/B mutant cDNA and qRT-PCR analysis of Fancd2 was performed at 4 days post-tamoxifen. Representative data from one out of 2 independent experiments with 3 biological replicates are shown.
G. Images are shown of representative karyotypes in control and Setd1a-deficient leukemia cells. Red arrows indicate breaks/gaps on the chromosome. See Table 1.
H. MLL-AF9 leukemia cells were treated with tamoxifen and p53 protein expression was analyzed by western blot analysis at 4 days post-tamoxifen.
I. p53 sgRNAs with a tRFP657 reporter and Setd1a FLOS1 sgRNAs with a GFP reporter were sequentially transfected into iCas9-expressing MLL-AF9 leukemia cells. Representative data from one out of 2 independent experiments with 3 biological replicates are shown.
J. Setd1a FLOS1 sgRNAs with a GFP reporter were transduced into p53−/− derived iCas9-expressing MLL-AF9 leukemia cells. Representative data from one out of 2 independent experiments with 3 biological replicates are shown.
K. MLL-AF9 leukemia cells were transfected with p53 shRNA and treated with tamoxifen for 24 hours. Relative expression level of Fancd2 was quantified by qRT-PCR at 4 days post-tamoxifen. Representative data from one out of 2 independent experiments with 3 biological replicates are shown.
Since Fancd2 null HSCs can be rescued by p53 suppression (Ceccaldi et al., 2012), we wanted to determine if Setd1a inactivation induced p53-dependent cell death. First, we found that p53 accumulated after Setd1a deletion (Figure 4H). To assess a potential functional role for p53 in Setd1a-induced apoptosis, we performed serial disruption of p53 (Figure S4G) and then the SETD1A FLOS1 domain by CRISPR. Remarkably, leukemia cells that harbored p53 sgRNAs were resistant to the affects of two independent Setd1a sgRNAs targeting the FLOS1 domain whereas control cells harboring sgRNAs targeting the Rosa locus were sensitive to the Setd1a sgRNAs (Figures 4I and S4H). p53-dependency was also confirmed with MLL-AF9/Cas9-expressing cells derived from p53−/− BM LSK (Lineage− Sca-1+ c-KIT+) cells (Figure 4J). p53 knockdown did not affect the silencing of Fancd2 in Setd1a-deficient cells (Figure 4K). These results demonstrate that SETD1A is required to regulate the expression of genes important for the DNA damage response, and to protect leukemia cell chromosomal integrity thus preventing p53-dependent apoptosis.
The FLOS domain of SETD1A binds Cyclin K
Having identified this critical domain in SETD1A, we set out to identify proteins that bound to this region. We expressed Flag-tagged SETD1A in 293T cells and performed co-immuprecipitation (Co-IP) followed by mass spectrometry (Figure 5A). We identified 68 potential SETD1A binding proteins including several known SETD1A binding partners. GO analysis found that the proteins involved in the DNA damage response were enriched in the SETD1A binding proteins (Figures 5A and S5A). Of particular interest was Cyclin K, which is known to control transcription of multiple genes involved in the DNA damage response much like we have found for SETD1A (Blazek et al., 2011). To evaluate the Cyclin K binding region in SETD1A, we performed Co-IP experiments in 293T cells using the SETD1A mutants in Figure 2A. Wild-type SETD1A was co-isolated with endogenous Cyclin K, but SETD1A lacking aa 387–586 completely lost Cyclin K-binding (Figure 5B). Remarkably, a fragment of SETD1A from aa 387–586 could be co-isolated with Cyclin K and this association was disrupted by deletion or alanine substitution mutants of the FLOS1 domain (Figure 5C). Mutation of FLOS1, but not FLOS2, in full-length SETD1A was sufficient to deplete Cyclin K binding (Figures 5D). FLOS1/2 mutation did not decrease the binding with COMPASS subunits, other known SETD1A complex subunits, or TCERG1, a RNAP2 regulator, which are all detected in mass spectrometry (Figure 5D). Interestingly, strong Cyclin K association was only observed with SETD1A, but not SETD1B, indicating that the unique interaction between SETD1A and Cyclin K might provide SETD1A specific functions in leukemia cells (Figure 5E). Deletion of the N- or C-terminal portions surrounding FLOS1 also disrupted the binding of Cyclin K, which suggests that the structures surrounding the FLOS1 core sequence also support the interaction between SETD1A and Cyclin K (Figure S5B). Cyclin K is observed at the H3K4me3 positive TSS and Setd1a disruption reduced the signal for Cyclin K but not H3K4me3 (Figure 5F and S5C). To identify the SETD1A binding domain in Cyclin K, we performed Co-IP experiments with domain deletion mutants of Cyclin K, and we identified the cyclin box domains, which are required for the regulation of CDK12/13 kinase activity, as the binding region for SETD1A (Figure 5G). These results show that FLOS1 is critical for association with Cyclin K, and that the SETD1A-Cyclin K pathway is indispensable for leukemia cell growth.
Figure 5. SETD1A FLOS domain binds Cyclin K.

A. SETD1A-binding proteins were identified by Co-IP MS. Proteins were visualized by silver stain (left) and excised from the gel. 68 proteins were identified as SETD1A-binding proteins (right box outlined in grey) and 11 proteins were classified as DNA damage response-associated proteins in GO analysis (red circle). The ranks of signal intensity are shown in brackets.
B. 293T cells were transfected with SETD1A deletion mutant constructs and the protein extracts were used for the Co-IP experiment.
C. 293T cells were transfected with internal region specific fragments of SETD1A constructs. FLAG-tagged fragments illustrated on the left were used for Co-IP.
D. 293T cells were transfected with SETD1A FLOS1 or FLOS2 alanine mutant constructs.
E. 293T cells were transfected with SETD1A and SETD1B constructs. CXXC1, a common binding partner of SETD1A/B, was detected as a positive control.
F. Average ChIP-seq signal of H3K4me3 (upper) and CCNK (lower) at H3K4me3 positive TSS from Setd1aΔ/Δ and Setd1a+/+ leukemia cells.
G. 293T cells were transfected with Cyclin K mutants. FLAG-tagged Cyclin K mutants illustrated on the left were used for Co-IP.
SETD1A-Cyclin K/CDK12 axis is indispensable for AML cell
Having identified the interaction between SETD1A and Cyclin K, we wanted to assess the functional role of Cyclin K in leukemia. We first performed a CRISPR domain scan using 188 sgRNAs spanning the Cyclin K coding region and discovered an indispensable role for the cyclin box domains in leukemia cell proliferation (Figure 6A). Next, we mutated Cyclin K using two sgRNAs that we identified in the CRISPR domain scan, and its CDK partners, CDK12/13, using 4 sgRNAs each (Figure 6B). The sgRNAs targeting kinase domains of CDK12 showed strong negative selection in leukemia cells similar to Cyclin K, whereas the guides targeting CDK13 were not selected against. These results support the idea that the Cyclin K/CDK12 pathway is an effector of SETD1A function in leukemia. RNA-seq analysis of cells expressing sgRNAs that target Cyclin K showed that 190 genes overlap with the downregulated gene set in Setd1a knockout cells (Figure 6C). DNA damage response genes, including Fancd2, Atr and Mlh1, were commonly downregulated (Figures 6D, 6E and S6A–D). Disruption of Cdk12 also caused downregulation of Fancd2, further supporting the concept that SETD1A regulates the function of the Cyclin K/CDK12 complex (Figure 6F). The doxycyline inducible Cyclin K shRNAs led to acute reduction of Fancd2 mRNA levels in leukemia cells (Figure S6E–F). Similar to those of FLOS domain mutations, the effects of Cyclin K sgRNAs were partially rescued by p53 depletion (Figure S6G). Both SETD1A and Cyclin K interact with CDK12, which suggested the functional importance of a SETD1A/Cyclin K/CDK12 pathway (Figure 6G). The Cyclin K/CDK12 complex has been shown to regulate RNAP2 CTD phosphorylation (Blazek et al., 2011), but SETD1A disruption did not show strong effects on the global level of RNAP2 CTD phosphorylation, nor on Cyclin K protein levels in MLL-r leukemia cells (Figure 6H). These studies show that the Cyclin K/CDK12 pathway is required for leukemia cell growth similar to SETD1A, but that SETD1A does not affect global RNAP2 CTD phosphorylation.
Figure 6. SETD1A regulates the Cyclin K/CDK12 axis in MLL-r leukemia.

A. A domain focused CRISPR strategy was used to analyze the functional regions of the Ccnk gene in mouse MLL-AF9 leukemia cells. Representative data from one out of 3 independent experiments are shown.
B. Cdk12/13 kinase domain–targeting sgRNAs with a RFP reporter were used for competitive growth assay in iCas9-expressing MLL-AF9 leukemia cells. Representative data from one out of 2 independent experiments with 3 biological replicates are shown.
C. RNA-seq analysis was performed with iCas9-expressing MLL-AF9 leukemia cells harboring Ccnk sgRNAs. Four independent Ccnk sgRNAs were used. The overlap in downregulated genes between Ccnk sgRNA-expressing cells and Setd1a knockout cells are shown.
D. Gene ontology analysis of 190 commonly downregulated genes is shown in (C). The bar for DNA damage stimulus is shown in red.
E–F. qRT-PCR analysis of Fancd2 in Ccnk sgRNA (E) or Cdk12 sgRNA (F) –expressing leukemia cells at 4 days post-dox. Representative data from one out of 2 independent experiments with 3 biological replicates are shown.
G. 293T cells were transfected with FLAG-tagged Cyclin K or FLAG-tagged SETD1A and CDK12 expression vectors. FLAG-tag antibody was used for the Co-IP experiment. Representative images from 2 independent experiments are shown.
H. Western blot analysis was performed to check the global level of Cyclin K and RNAP2 CTD (Ser2 or Ser5) phosphorylation in SETD1A-deficient MLL-AF9 leukemia cells. Representative images from 2 independent experiments are shown.
SETD1A regulates FANCD2 expression in a cell cycle-dependent manner
Given that SETD1A and Cyclin K are both required for appropriate expression of DNA damage response genes, but that global RNAP2 phosphorylation is not affected by SETD1A suppression, we wondered if the SETD1A function may be locus specific and therefore control Cyclin K/CDK12 function at select loci. We assessed this possibility in NIH3T3 cells. While NIH3T3 cells showed significantly less sensitivity to Setd1a sgRNAs in competitive growth assays (Figure 2F), they still showed downregulation of FANCD2 and MLH1 expression, after Setd1a knockdown (Figure S7A). To examine the locus-dependent effects of the SETD1A-RNAP2 axis, we performed ChIP-seq analysis for SETD1A and RNAP2 in control and Setd1a knockdown NIH3T3 cells (Figures 7A–B). SETD1A protein was detected on the promoter and TSS regions, and it decreased in cells where Setd1a was suppressed by shRNA knockdown (Figure 7A–B). In contrast, RNAP2 peaks were increased on the SETD1A positive TSS in SETD1A knockdown cells (Figure 7A–B). Increasing of RNAP2 at TSS suggests promoter-proximal pausing after SETD1A knockdown. To monitor the Cyclin K and RNAP2 CTD-Ser2 phosphorylation, we performed ChIP-qPCR at the Fancd2 locus, and confirmed the binding of SETD1A on the Fancd2 TSS and observed significant decrease of Cyclin K and RNAP2 CTD-Ser2 phosphorylation (Figure 7C and S7B–C). These results show that SETD1A is crucial for the recruitment of Cyclin K, which promotes CDK12 occupancy/activation, RNAP2 phosphorylation and gene expression.
Figure 7. SETD1A-Cyclin K regulates FANCD2 transcription in G1/S-phase.

A. Density of ChIP-seq reads for SETD1A (red) and RNAP2 (blue) of SETD1A positive TSS (n=18396) from Ren shRNA or Setd1a shRNA–expressing NIH3T3 cells are shown. Regions are ranked according to log2 fold change of SETD1A signal intensity between Ren and Setd1a shRNA.
B. Average ChIP-seq signal of SETD1A (upper) and RNAP2 (lower) of SETD1A positive TSS from Ren shRNA or Setd1a shRNA-expressing NIH3T3 cells are shown.
C. ChIP-qPCR was performed for SETD1A on the Fancd2 locus in Ren shRNA (blue) or Setd1a shRNA (red) –expressing NIH3T3 cells at 4 days post-dox. Rabbit IgG was used as negative control in ChIP (dot lines). Representative data from 3 independent experiments with 3 biological replicates are shown.
D. Cell cycle stage was visualized by flow cytometry in a FUCCI positive 293T single cell clone (left panel). DNA contents in each fraction are shown in the right panel.
E. 293T-FUCCI cells were fractionated by cell sorting. Western blot analysis was performed using the indicated antibodies. Representative data from 3 independent experiments are shown.
F. FANCD2 transcriptional level during the cell cycle was monitored by qRT-PCR. This experiment was repeated 3 times with 3 biological replicates in each experiment.
G. Scramble shRNA (Scr, blue) and SETD1A shRNA (red) –expressing 293T-FUCCI cells were analyzed at 4 days post-transfection. Expressions of SETD1A (left) and FANCD2 (right) are shown. See also Figure S7E.
H. FANCD2 transcription in Scr shRNA (blue) and CCNK shRNA (red) –expressing 293T-FUCCI cells were analyzed at 4 days post-transfection. See also Figure S7F.
I. FANCD2 transcription in DMSO (blue) and 100 nM THZ531 (red) –treated 293T-FUCCI cells were analyzed at 3 days post-treatment. Representative data from one out of 2 independent experiments with 3 biological replicates are shown.
Previous studies have shown that molecules such as FANCD2, a SETD1A-Cyclin K target gene, are necessary in S phase of the cell cycle (Ceccaldi et al., 2016). We hypothesized that the SETD1A-Cyclin K complex might regulate gene expression at the G1-S transition. We employed a system to monitor FANCD2 transcription during the cell cycle in a manner that did not require molecules that induced DNA damage or cellular toxicity. We labeled the 293T cell line with FUCCI (Fluorescent, Ubiquitination-based cell cycle indicator) and enriched the cells in each cell cycle stage by cell sorting (Figure 7D) (Sakaue-Sawano et al., 2008). Interestingly, RNA as well as protein levels for FANCD2 were increased in the S phase-enriched fraction (C+G+) (Figures 7E–F). Cyclin K-SETD1A interaction did not change through the G1-S transition (Figure S7D), but either SETD1A or Cyclin K knockdown significantly suppressed the cell cycle-dependent induction of FANCD2 expression (Figures 7G–H and S7E–F). In addition, pharmacological inhibition of CDK12/13 with THZ531, a covalent inhibitor (Zhang et al., 2016), also suppressed the transcriptional activation of FANCD2 in S phase (Figure 7I).
SETD1A/Cyclin K/CDK12 pathway regulates cell growth of human leukemia cells
To evaluate the effect of CyclinK/CDK12 suppression in human leukemia, we treated human leukemia cell lines as well as MLL-AF9 leukemia cells derived from human CD34+ cord blood with THZ531. THZ531 inhibited growth of MLL-r/non-MLL-r leukemia cell lines, and induced apoptosis in the MOLM-13 cell line (Figure S7G–S7H). We also confirmed the importance of the SETD1A-CCNK axis in MLL-AF9 transduced human cord blood cells through shRNA-based suppression of either SETD1A or CCNK (Figure S7I). These results demonstrate that the SETD1A/Cyclin K/CDK12 pathway regulates cell-cycle dependent expression of DNA damage repair genes and that inhibition of these mechanisms leads to significant inhibition of growth of human leukemia cells.
Discussion
There is increasing interest in the function of the MLL family of H3K4 HMTs as they have been shown to play diverse roles in organismal and cancer development (Rao and Dou, 2015). Furthermore, accumulating evidence suggests that multiple members of this family may be therapeutic targets in cancer (Cao et al., 2014; Chen et al., 2014; Kuhn et al., 2016; Santos et al., 2014; Xu et al., 2016). The cellular roles of MLL family members is presumed to be due to H3K4 methylation, but in only a few studies has the HMT activity been demonstrated to be of importance. Indeed, recent studies indicate that the SET domain of MLL1 is dispensable for leukemias that require MLL1 for continued proliferation (Kuhn et al., 2016; Mishra et al., 2014). The data presented here show that leukemia cells tolerate inactivation of the SET domain of SETD1A even though they are absolutely dependent on SETD1A for continued proliferation and survival. Given that SETD1A and SETD1B are thought be responsible for much of the cellular H3K4 methylation, this suggests functional redundancy for H3K4 methylation between these highly homologous proteins. However, it is clear that there is a critical non-redundant function for SETD1A in leukemia cells which prompted further study. These data support the concept that MLL family members play critical roles in cell biology that are independent of H3K4 methylation and cautions against necessarily focusing on the SET domain as critical for a given phenotype.
Through detailed mutagenesis and CRISPR-Cas9 domain scanning, we found a previously unidentified domain in SETD1A that is critical for leukemia proliferation and survival. We call this the functional location on SETD1A (FLOS) domain. The FLOS domain is critical for SETD1A-dependent expression of genes involved in the DNA damage response and interacts with Cyclin K. This is in keeping with the recent demonstration that the Cyclin K/CDK12 complex is a regulator of DNA damage response genes (Blazek et al., 2011; Greifenberg et al., 2016). It also shows the functional importance of the SETD1A/Cyclin K interaction in leukemia cells. Importantly, SETD1A depletion reduces Cyclin K occupancy on the Fancd2 gene locus, without affecting the global Cyclin K protein levels, thus demonstrating that the SETD1A FLOS domain is essential for locus-specific recruitment of Cyclin K. These data are important not only because they highlight a relationship between SETD1A, Cyclin K and the DNA damage response but also because recent efforts toward the drug development of CDK12/13 inhibitors suggest this pathway may be an important target for cancer drug development (Zhang et al., 2016).
We show that SETD1A is required for appropriate expression of DNA damage response genes. Since Fancd2 was the most sensitive gene to SETD1A disruption, we assessed Fancd2 dependence in leukemia and found an essential role in the maintenance of MLL-AF9 leukemia cells. Activation of p53 is observed upon Fancd2 deficiency and p53 knockout restores the Fancd2−/− BM LSK cell population and functions in vivo (Ceccaldi et al., 2012; Kim et al., 2003). Consistent with these results, cellular differentiation and cell death induced by targeting the FLOS domain was rescued by p53 knockout. Beyond Fancd2, Atr expression was also dependent on the SETD1A. Atr disruption also induces myeloid differentiation and loss of genome integrity in MLL-r leukemia cells much like SETD1A inactivation (Ruzankina et al., 2007; Santos et al., 2014). Our results strongly support the idea that SETD1A coordinately regulates DNA damage response pathways that are critical for leukemia proliferation via its FLOS domain.
Cyclin K was originally identified as a cyclin that could rescue the lethality of G1 cyclin-deficient yeast, but the cell-cycle dependent functions of Cyclin K have not been deeply characterized (Edwards et al., 1998). Here we found the highest levels of Cyclin K protein in S phase using a non-invasive cell cycle indicator, FUCCI (Sakaue-Sawano et al., 2008). Moreover, our data demonstrate that SETD1A is functionally important for S phase dependent FANCD2 expression. Our findings provide new insights into how epigenetic regulators functionally modulate gene transcription in a cell-cycle dependent manner. Importantly, FLOS-domain specific targeting was toxic in leukemia cells, but not fibroblast cells suggesting that there may be an enhanced dependency in leukemia cells.
Taken together, our work shows that the SETD1A FLOS domain regulates expression of genes involved in the DNA damage response via interaction with Cyclin K. Furthemore, MLL family HMTs can work as regulators of gene expression that are independent of their enzymatic functions. This newly defined function for SETD1A could be a novel therapeutic target for cancer drug development and therefore detailed structural analysis of the FLOS domain and its interaction with Cyclin K will be illuminating and potentially important for future drug development. Finally, SETD1A is a multifunctional modulator of gene expression with mechanisms that are overlapping and non-overlapping with SETD1B. This is likely true for other MLL family members. This is an attribute that should be considered as drugs are developed to target this family of proteins.
STAR Methods
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-CXXC1 | Abcam | Cat#ab56035 |
| Rabbit polyclonal anti-H3K4me1 | Abcam | Cat#ab8895 |
| Rabbit polyclonal anti-H3K4me2 | Millipore | Cat#07-030 |
| Rabbit polyclonal anti-H3K4me3 | Abcam | Cat#ab8580 |
| Rabbit polyclonal anti-H3K27Ac | Abcam | Cat#ab4729 |
| Rabbit polyclonal anti-Histone H3 | Abcam | Cat#ab1791 |
| Rabbit monoclonal anti-GAPDH | Cell Signaling | Cat#2118S |
| Rabbit monoclonal anti-WDR5 | Abcam | Cat#ab178410 |
| Rabbit monoclonal anti-ASH2L | Cell Signaling | Cat#5019S |
| Rabbit polyclonal anti-RbBP5 | Bethyl Laboratories | Cat#A300-109A |
| Rabbit polyclonal anti-WDR82 | Abgent | Cat#AP4812a-ev |
| Rabbit polyclonal anti-CA150(TCERG1) | Bethyl Laboratories | Cat#A300-360A |
| Rabbit monoclonal anti-FANCD2 | Abcam | Cat#ab108928 |
| Rabbit monoclonal anti-MLH1 | Abcam | Cat#ab92312 |
| Mouse monoclonal anti-p53 | Abcam | Cat#ab28 |
| Rabbit polyclonal anti-FLAG | GenScript | Cat#A00170-40 |
| Mouse monoclonal anti-FLAG M2 Affinity Gel | Sigma | Cat#A2220 |
| Rabbit polyclonal anti-Cyclin K | Bethyl Laboratories | Cat#A301-939A |
| Rabbit polyclonal anti-SETD1A | Abcam | Cat#ab70378 |
| Rabbit polyclonal anti-RNA polymerase II CTD repeat phospho S2 | Abcam | Cat#ab5095 |
| Rabbit polyclonal anti-RNA polymerase II CTD repeat phosphor S5 | Abcam | Cat#ab5131 |
| Mouse monoclonal anti-RNA polymerase II | diagenode | Cat#C15100055 |
| Rabbit polyclonal anti-Lamin B1 | Abcam | Cat#ab16048 |
| Chemicals | ||
| Nutlin-3 | Sigma | Cat#N6287 |
| THZ531, CDK12/CDK13 inhibitor | Laboratory of Nathaneal Gray | N/A |
| Deposited Data | ||
| Raw and analyzed data of RNA-seq and ChIP-seq | This paper | GEO: GSE108615 |
| Raw data of Mass spectrometry analysis | This paper | MassIVE: MSV000080012 |
| Experimental Models: Cell Lines | ||
| Human: 293T cells | ATCC | CRL-3216 |
| Human: Plat-E cells | Cell Biolabs | RV-101 |
| Human: MV4-11 cells | ATCC | CRL-9591 |
| Human: U937 cells | ATCC | CRL-1593.2 |
| Human: K562 cells | ATCC | CCL-243 |
| Human: KG-1 cells | ATCC | CCL-246 |
| Human: HL-60 cells | ATCC | CCL-240 |
| Human: THP-1 cells | ATCC | TIB-202 |
| Human: MOLM-13 cells | DSMZ | ACC-554 |
| Human: NB-4 cells | DSMZ | ACC-207 |
| Human: ML-2 cells | DSMZ | ACC-15 |
| Human: Jurkat cells | DSMZ | ACC-282 |
| Human: A673 cells | ATCC | CRL-1598 |
| Human: SJCRH30 (RH30) cells | ATCC | CRL-2061 |
| Human: HSSYII cells | Laboratory of Stefan Fröhling | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Setd1atm1a(EUCOMM)Wtsi | EMMA | EM: 06857 |
| Mouse: Act-Flpe; B6.Cg-Tg(ACTFLPe)9205Dym/J | The Jackson Laboratory | JAX: 005703 |
| Mouse: Rosa26-Cas9 knock-in; Gt(ROSA)26Sortm1.1(CAG-cas9*,-EGFP)Fezh/J | The Jackson Laboratory | JAX: 024858 |
| Mouse: B6.129S2-Trp53tm1Tyj/J | The Jackson Laboratory | JAX: 002101 |
| Oligonucleotides | ||
| shRNA (see Table S3) | This paper | N/A |
| sgRNA (see Table S3) | This paper | N/A |
| Genotyping DNA Primers (see Table S4) | This paper | N/A |
| TaqMan Probes (see Table S4) | Life Technologies | Cat#4331182, Cat#4351372 |
| Recombinant DNA | ||
| Plasmid: TRIN miR-E | Laboratory of Scott Lowe | N/A |
| Plasmid: RT3GEPIR miR-E | MSKCC RNAi Core | N/A |
| Plasmid: pMSCV-MLL-AF9-ires-Neo | Laboratory of Scott Armstrong | N/A |
| Plasmid: pMSCV-MLL-AF9-ires-GFP | Laboratory of Scott Armstrong | N/A |
| Plasmid: pMSCV-rtTA-pgk-hygro | Laboratory of Scott Armstrong | N/A |
| Plasmid: pLKO5.sgRNA.EFS.GFP | (Heckl et al., 2014) | Addgene #57822 |
| Plasmid: pLKO5.sgRNA.EFS.tRFP657 | (Heckl et al., 2014) | Addgene #57824 |
| Plasmid: pCW-Cas9 | (Wang et al., 2014) | Addgene #50661 |
| Plasmid: pMD2.G | Laboratory of Didier Trono | Addgene #12259 |
| Plasmid: psPAX2 | Laboratory of Didier Trono | Addgene #12260 |
| Plasmid: ES-FUCCI | (Sladitschek and Neveu, 2015) | Addgene #62451 |
| Plasmid: pLKO.1 puro | (Stewart et al., 2003) | Addgene #8453 |
| Plasmid: pCMV6-hSETD1A(NM_014712)-Myc-DDK | Origene | Cat#RC214996 |
| Plasmid: pCVM6-hSETD1B(NM_015048)-Myc-DDK | Origene | Cat#RC227091 |
| Plasmid: pcDNA3.1-CCNK(NM_001099402)-(K)DYK | GenScript | Cat#OHu30814D |
| Plasmid: pCl-Neo-His-Pitaire(CDK12)-Myc | (Ko et al., 2001) | Addgene #26066 |
| Software and Algorithms | ||
| Prism 7 | GraphPad softare | N/A |
| CRISPResso ver 0.8.4 | (Pinello et al., 2016) | https://github.com/lucapinello/CRISPResso |
| EaSeq | (Lerdrup et al., 2016) | http://easeq.net |
| Analysis of NGS data | Basepair software | https://www.basepairtech.com |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Scott A. Armstrong (Scott_Armstrong@dfci.harvard.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Setd1atm1a(EUCOMM)Wtsi mice (EMMA) were crossed with Act-Flpe mice (The Jackson Laboratory, 005703) to generate a floxed allele. Rosa26-Cas9 knock-in mice (024858) and p53 knockout mice (002101) were obtained from The Jackson Laboratory. C57BL/6 female mice (CD45.2, Charles River) were used as transplantation recipients. Mice used in this study have been maintained in the Research Animal Resource Center of MSKCC and the DFCI Animal Resource Center, following a vertebrate animal protocol approved by MSKCC’s and DFCI’s Institutional Animal Care and Use Committee (IACUC), respectively. Both female and male mice were used in our leukemia studies and no obvious sex-dependent differences were observed.
Human Cord Blood Samples
Normal cord blood units, designated for research use, were obtained from the New York Blood Center (NYBC, New York, NY) under IRB approval from the National Cord Blood Program (NCBP). Both male and female samples were randomly assigned and used in this study.
Cell Lines and Cell Culture
293T, Plat-E, A673 and HSSYII cell lines were maintained in DMEM medium containing 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. MV4-11, K562, KG-1, HL-60 cell lines were maintained in IMDM medium containing 10% FBS and 1% penicillin-streptomycin. U937, THP-1, MOLM-13, NB-4, ML-2, Jurkat and RH30 cell lines were maintained in RPMI1640 medium containing 10% FBS and 1% penicillin-streptomycin. Cells were maintained in a humidified incubator at 37 °C, 5% CO2.
METHOD DETAILS
Murine and human MLL-AF9 leukemia model
BM cells were obtained from femoral and tibial bones, and stained with Lineage-Biotin (559971, BD Biosciences), Sca-1-PE (122508, BioLegend), c-KIT-Alexa Flour 647 (105818, BioLegend) antibodies and Streptoavidin-FITC (554060, BD Biosciences), then LSK cells were isolated with FACS Aria (BD Biosciences). LSK cells were cultured in RPMI medium 1640 supplemented with 10% FBS, 20 ng/ml rmSCF, 10 ng/ml rmIL3, 10 ng/ml rmIL6. After 24 h of cultivation, spin infection was performed by adding retrovirus carrying MLL-AF9-ires-Neo or MLL-AF9-ires-GFP in the media containing 8 μg/ml polybrene to LSK cells and centrifuging at 1,400 × g for 90 min. MLL-AF9 expressing LSK cells were serially re-plated with methylcellulose medium (STEMCELL Technologies, MethoCult M3234) containing the above cytokines for 3 weeks. To obtain the fully transformed leukemia cells, immortalized cells were transplanted into lethally irradiated (9.5 Gy) syngeneic recipient mice along with 5 × 105 bone marrow cells from wild-type mice. Human cord blood CD34+ cells were enriched by microbeads (Miltenyi Biotec) and MLL-AF9 transduced cells were transplanted to NOD/SCID mice. AML cells were harvested from bone marrow or spleen and cultured in IMDM medium supplemented with 20% FBS, beta-mercaptoethanol, 20 ng/ml rhIL6/rhTPO/rhSCF/rhGM-CSF, 10 ng/ml IL3.
shRNA
miR-30 shRNA retroviral vector library and TRIN miR-E retroviral vector were obtained from Scott Lowe’s lab (Zuber et al., 2011). RT3GEPIR miR-E retroviral vector was obtained from the RNAi Core in MSKCC (Fellmann et al., 2013). Plat-E retroviral packaging cells were transiently transfected with retroviral vector plasmids using X-tremegene 9 (Roche), and culture supernatants containing retroviruses were collected 48 h after transfection. For the doxycycline-inducible TRIN miR-E shRNA system, we transduced MSCV-rtTA-pgk-hygro transgene to MLL-AF9-ires-GFP expressing MLL-AF9 leukemia cells with retrovirus. In miR-30 shRNA experiments, infected cells were analyzed by flow cytometer every 3 days from 2 days post-infection. In TRIN miR-E shRNA experiments, cells were selected with 1mg/ml G418 for 2 days from 24 h post-infection to enrich the infected cells. The cells were treated with doxycycline, and analyzed at 2 days post-doxycycline to obtain the percentage of shRNA expressing cells. Doxycycline treated cells were analyzed again after 24 h, and analyzed every 3 days until 9 days or 12 days post-doxycycline. For in vitro studies, 1 μg/ml doxycycline was treated for every experiment. RT3GEPIR miR-E vector infected MLL-AF9 leukemia cells were selected with 2.5 μg/ml puromycin, and transplanted to sublethally irradiated (6.0 Gy) syngenic recipient mice. After 1-week post-transplant, we administrated doxycycline to the recipient mice by both food-intake (625 mg/kg diet, Harlan) and drinking water (2 mg/ml doxycycline with 5% sucrose). For human gene knockdown, 293T cells were transiently transfected with lentiviral vector plasmid pLKO1-puro with both pPAX2 and pMD2 packaging plasmids, and culture supernatants containing lentiviruses were collected 48 h after transfection. We transduced lentiviral vector by spin infection. shRNA expressing cells were selected with 2.5 μg/ml puromycin for 24 h and maintained in media containing 0.5 μg/ml puromycin.
Isolation of Leukocytes
PB cells were obtained from the facial vein. BM cells were obtained from femoral and tibial bones by aspiration. Splenocytes were isolated by pressing the tissues through a cell strainer (BD). Erythrocytes were removed with PB Pharm Lyse Lysing Buffer (BD).
Apoptosis, Cell cycle and Cell Differentiation
Apoptosis was evaluated with Annexin V Apoptosis Detection Kit APC (eBioscience). Cell cycle stage was analyzed with either Click-iT EdU Imaging Kit (Thermo Fisher Scientific) or BrdU Flow Kit (BD). Cell differentiation was monitored by microscopy. Cells were centrifuged onto glass slides and stained with the Jorvet Dip Quick stain kit (Fisher).
Sequence Alignment
Amino acid sequence of Homo sapiens SETD1A (NP_055527)/SETD1B (NP_055863), Mus musculus SETD1A (NP_821172)/SETD1B (NP_001035488), Mesocricetus auratus SETD1A (XP_005064405), Xenopus tropicalis SETD1A (XP_004919748), Alligator mississippiensis SETD1A (XP_006262890), Danio rerio SETD1A (XP_001920852), Poecilia reticulate SETD1A (XP_008414548) were compared with Vector NTI (Invitrogen).
CRISPR
pLKO5.sgRNA.EFS.GFP (Addgene #57822) and pLKO5.sgRNA.EFS.tRFP657 (Addgene #57824) constructs were a gift from Benjamin Ebert (Heckl et al., 2014). pCW-Cas9 lentiviral vector (Addgene #50661) was a gift from Eric Lander & David Sabatini (Wang et al., 2014). We designed 3–4 individual guide RNA sequences for each domain by CRISPR Design Tool (http://crispr.mit.edu/). 293T cells were transiently transfected with lentiviral vector plasmids with both pPAX2 and pMD2 packaging plasmids, and culture supernatants containing retroviruses were collected 48 h after transfection. First, we infected the doxycycline-inducible Cas9 vector to MLL-AF9-ires-GFP expressing MLL-AF9 leukemia cells, and established the parental clone which can induce high expression level of Cas9 by doxycycline. Next, we infected sgRNA vector, and then performed the experiments with these cells from 2 days post-infection. GFP+ cell frequencies were measured by BD FACSCanto II or LSRFortessa (BD Biosciences). To evaluate the genome editing frequency, we performed SURVEYOR assays with Suveyor mutation detection kit (IDT). For deep-sequencing analysis of CRISPR-targeting sites, genomic DNA was amplified using the primers on Table S4. Amplified PCR products were purified by PCR purification kit (Qiagen). Libraries were prepared and sequenced on Mi-Seq instrument (illumina). The reference amplicon, annotated coding sequence, and the paired end reads were then used with CRISPResso (version 0.8.4; http://github.com/lucapinello/CRISPResso), with a quality filter of 10 and sequence trimming via Trimmomatic (http://www.usadellab.org/cms/index.php?page=trimmomatic). Cas9-expressing normal hematopoietic stem/progenitor cells were isolated by cell sorting of BM LSK cells from Rosa26-Cas9 knockin mice, and LSK cells were cultured in StemSpan SFEM (STEMCELL Technologies) containing 100 ng/ml rmSCF/rmFLT3/rmTPO. After 24 hours of cultivation, we infected sgRNA vector by spin infection. sgRNA-expressing LSK cells were isolated by cell sorter and were plated with methylcellulose medium (STEMCELL Technologies, MethoCult GF M3434) for 10 days. For the CRISPR domain scan of Ccnk gene, 188 sgRNAs with a RFP reporter were used for competitive growth assay. We purified sgRNA plasmid vectors with 96-well Miniprep Kit (Millipore) and performed a competitive growth assay in 96-well plates.
cDNA mutagenesis and rescue experiment
Myc- and DDK-tagged human SETD1A (NM_014712) and SETD1B (NM_015048) cDNA were obtained from Origene and DDK-tagged human CCNK (NM_001099402) cDNA was obtained from GenScript. Site-directed mutagenesis was performed by inverse PCR, with 5′-phosphorylated primers and CloneAmp HiFi PCR premix (Clontech). Each sequence of mutated cDNA was verified by Sanger sequencing, and protein expression was confirmed by western blotting. For the cDNA rescue experiment, SETD1A or SETD1B cDNA was cloned into pMSCV-ires-GFP vector, and retroviruses were generated in Plat-E cells. cDNA expression vectors were transduced into rtTA-expressing MLL-AF9 leukemia cells or Setd1aflox MLL-AF9 leukemia cells by retrovirus, and GFP positive cells were sorted by FACSAria.
Immunoprecipitation
293T cells were cultured in 6-well plates, and transfected with Xtremegene 9 transfection reagent. Cells were harvested at 72 hrs post-transfection, and lysed with 1x cell lysis buffer (Cell signaling) containing proteinase inhibitors (Biotool). 400 μg of protein lysate was loaded on pre-washed FLAG M2 affinity gel, and incubated overnight. After three TBS washes, we eluted the FLAG-tagged protein with 40 μl of TBS containing 3x FLAG peptides (ApexBio). Binding proteins were analyzed by western blotting or mass spectrometry. For FUCCI-expressing 293T cells, cells were sorted and lysed with 1x cell lysis buffer. Protein lysate was mixed with Rabbit IgG or CCNK antibody, and incubated overnight. The lysate was loaded on pre-washed Protein A/G Dynabeads, and incubated for 2 hours at 4°C, then eluted by 2x SDS sample buffer. Proteins were analyzed by western blotting.
Western blotting
Cells were lysed in 1x cell lysis buffer (Cell Signaling Technology) containing protease inhibitor cocktail (Bimake). Lysis was completed via ultrasonication and proteins were quantified with BCA Protein Assay Kit (Thermo Fisher Scientific). Lysate was mixed with 4xLDS sample buffer (Invitrogen) and beta-mercaptoethanol, and proteins were denatured by boiling. Denatured proteins were separated on a 4–12% Bis-Tris gel (ThermoFisher) in NuPAGE MOPS SDS running buffer (Thermo Fisher Scientific) and transferred to a PVDF membrane (Millipore) in NuPAGE Transfer Buffer (Thermo Fisher Scientific) with XCell II Blot Module (Thermo Fisher Scientific). Blots were blocked with 5% skim milk in TBST for 30 min, and incubated with primary antibodies (which are listed in the Key Resource Table) at 4 °C overnight. Immunocomplexes were labeled by HRP-conjugated anti-mouse IgG or anti-rabbit IgG and visualized using Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific). The signals were deteceted with ImageQuant LAS 4000 (GE Healthcare) and signal intensities were quantified using Image J software.
ChIP and RNA analyses
For ChIP-seq, Setd1aΔ/Δ and Setd1a+/+ leukemia cells were analyzed at day 3 post-tamoxifen. shRNA-expressing NIH3T3 cells were analyzed at day 4 post-doxycycline. Cells were fixed with 2mM DSG for 30 min followed by 1% formaldehyde for 10 min and treated with 0.125M Glycine for 5 min. Fixed cells were washed twice with cold PBS. Washed cells were suspended in ChIP Lysis Buffer and shredded using Bioruptor (Diagenode) or E220 Focused-ultrasonicator (Covaris). Appropriate amounts of antibodies were added into the chromatin and incubated overnight at 4 °C. The immune complex was collected with protein A/G agarose or magnetic beads and washed sequentially in the low salt wash buffer (20mM Tris pH8, 150mM NaCl, 0.1% SDS, 1% Triton-X100, 2mM EDTA), the high salt wash buffer (20mM Tris pH8, 500mM NaCl, 0.1% SDS, 1% Triton-X100, 2mM EDTA), the LiCl wash buffer (10mM Tris pH8, 250mM LiCl, 1% NP-40, 1% Sodium Dexyxholate, 1mM EDTA) and TE. Chromatin was eluted with elution buffer (1% SDS, 0.1 M NaHCO3), and then reverse cross-linked with 0.2M NaCl at 65 °C for 4 hours. DNA was purified with a PCR purification kit (Qiagen) or by PC8 extraction and isopropanol precipitation. For RNA-seq, Setd1aΔ/Δ and Setd1a+/+ leukemia cells were analyzed at day 4 post-tamoxifen. Ccnk sgRNA-expressing cells were analyzed at day 4 post-doxycycline. For RNA, total RNA was purified with RNeasy Mini kit (Qiagen). cDNA was synthesized with Tetro cDNA synthesis kit (Bioline). cDNA fragments were quantified by a TaqMan Gene expression assay (Applied Biosystems). ChIP DNA or cDNA fragments were quantified by SYBR Green Real-time PCR or a TaqMan Gene expression assay (Applied Biosystems) with a ViiA 7 Real time PCR system. Libraries were prepared by using a ThruPLEX DNA-seq Kit (Rubicon Genomics). The DNA library was validated using TapeStation (Agilent Technologies) and was quantified using a Qubit 2.0 Fluorometer (Thermo Fisher Scientific). Libraries were pooled and sequenced on Illumina HiSeq2000 or Illumina NextSeq500. Data were analyzed by workflows on Basepair (https://www.basepairtech.com). Heatmaps of ChIP-seq data were generated by using EaSeq software (http://easeq.net).
FUCCI
ES-FUCCI (Citrine-Geminin(1-110)/mCherry-hCdt1(30-120)) was a gift from Pierre Neveu (Addgene plasmid #62451) (Sladitschek and Neveu, 2015). We transduced the vector into the 293T cell line and established single cell-clones that stably express the reporters. Cells in each cell cycle stage were sorted by FACSAria.
Mass spectrometry analysis
Unless otherwise specified, all chemicals were from Sigma-Aldrich at the highest available purity. Proteins were separated with gradient Bis-Tris Protein Gel (Thermo Fisher Scientific) and stained with Pierce Silver Stain for Mass Spectrometry kit (Thermo Fisher Scientific). Lanes containing PAGE-resolved proteins were excised from the gel and divided into eight pieces, that were de-stained using 30 mM potassium hexa-cyanoferrate (III)/100 mM sodium thiosulfate, washed with 25 mM ammonium bicarbonate pH8.4/25% (v/v) Optima LC/MS acetonitrile (Thermo Fisher Scientific, Fair Lawn, NJ) and de-hydrated using a vacuum centrifuge. Proteins were reduced (10 mM DTT/100 mM ammonium bicarbonate pH8.4, 56°C for 60 minutes) and alkylated (55 mM iodoacetamide/100 mM ammonium bicarbonate pH8.4, 25°C for 30 minutes, dark), and excess iodoacetamide was removed by three cycles of dehydration (100 μl C2H3N) and rehydration (100 μl of 100 mM ammonium bicarbonate pH8.4) prior to final dehydration in a vacuum centrifuge. Gel slabs were re-hydrated using 50 mM ammonium bicarbonate pH8.4 containing 0.04 μg sequencing grade modified porcine trypsin (Promega, Madison, WI) per reaction and proteolysis was allowed to proceed for 16 hours at 37°C. Proteolysis inhibition and tryptic peptide elution from the polyacrylamide slabs was achieved by two consecutive 30 minute incubations in 5% formic acid/70% acetonitrile (v/v) under continuous shaking. Eluates from each sample were pooled, lyophilized and stored at −80°C until analysis.
Peptide pellets were resuspended in 0.1% formic acid (99+%, Thermo Fisher Scientific, Rockford, IL)/3% acetonitrile and 5% of the solution was analyzed by LC/MS. The LC system consisted in a vented trap-elute setup (Ekspert nanoLC 425, Eksigent, Redwood city, CA) coupled to the Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific, San Jose, CA) via an nano electro-spray DPV-565 PicoView ion source (New Objective, Woburn, MA). The trap column was fabricated capping a 5 cm × 150 μm internal diameter silica capillary (Polymicro Technologies, Phoenix, AZ) with a 2 mm silicate frit, and pressure loaded with Poros R2-C18 10 μm particles (Life Technologies, Norwalk, CT). The analytical column consisted of a 25 cm × 75 μm internal diameter column with integrated electrospray emitter (New Objective, Woburn, MA), and was packed with ReproSil-Pur C18-AQ 1.9 μm particles (Dr. Maisch, Ammerbuch-Entringen, Germany). Samples were loaded on the trap column at 1 μl/minute with analytical column excluded from the flow path. The analytical column was then put in-line with the trap, and peptides were resolved over 120 minutes using a 5–40% gradient acetonitrile/0.1% formic acid (buffer B) gradient in water/0.1% formic acid (buffer A) at 300 nl/minute. Precursor ion scans were recorded from 400–2000 m/z in the Orbitrap (120,000 resolution at m/z 200) with automatic gain control target set 105 ions and maximum injection time of 50 ms. We used data-dependent mass spectral acquisition with monoisotopic precursor selection, charge ion selection (2–7), dynamic exclusion (60 sec, 5 ppm tolerance), HCD fragmentation (normalized collision energy 32, isolation window 1 Th) using the top speed algorithm with a duty cycle of 2 sec. Product ion spectra were recorded in the linear ion trap (“normal” scan rate, automatic gain control= 104 ions, maximum injection time = 150 ms). Raw files are submitted to the MassIVE repository with accession number MSV000080012.
Raw files were submitted to MaxQuant 1.4.1.2 to be searched against the human UniProt database (version 04/22-14, containing isoforms) with FDR<0.01 at both peptide and protein level. Mass tolerance was set at 5 ppm for orbitrap spectra and 0.5 Da for linear trap spectra. C-carbamidomethylation was set as fixed modification, while phosphorylation of S/T/Y and M-oxidation were allowed as variable. The software was set to align chromatographic features (Matching time window= 1 min, Alignment time window =20 min). Identified proteins mapping in the common contaminants database cRAP were removed from the output. GO term analysis was performed with GOTermFinder (http://go.princeton.edu/cgi-bin/GOTermFinder).
THZ531, CDK12/13 covalent inhibitor
The half-maximal inhibitory of THZ531 in 293T-FUCCI cells was determined (IC50 = 60 nM). 100 nM THZ531 was treated for 72 hours and cells from each cell cycle stage were collected by cell sorter.
QUANTIFICATION AND STATISTICAL ANALYSIS
Error bars in all of the data represent a standard deviation. Number of replicates and number of repeated experiments are reported in the Figure legends. For statistical comparison, we performed a Student’s t test or one-way ANOVA followed by Tukey’s test. Data with statistical significance (*P < 0.05, **P < 0.01) are shown in figures. Statistical analyses were performed using Prism 7 software (GraphPad).
DATA AND SOFTWARE AVAILABILITY
The accession number for the RNA-seq and ChIP-seq data reported in this paper is NCBI GEO: GSE108615. The accession number for the mass spectrometry analysis data reported in this paper is MassIVE: MSV000080012. The accession number for the raw data reported in this paper is Mendeley Data: https://doi:10.17632/hhjhsrgkz7.1.
Supplementary Material
The summary of the karyotyping analysis in Setd1a-knockout MLL-AF9 leukemia cells is shown.
RNA downregulated genes in Setd1a knockout cells were classified with the promoter/TSS H3K4me3 level.
The list of shRNAs and sgRNAs used in this study is shown.
The list of DNA primers and Taqman probes used in this study is shown.
Highlights.
H3K4 methyltransferase SETD1A has non-redundant functions with SETD1B.
Non-enzymatic domain of SETD1A is indispensable for leukemia cell growth.
SETD1A FLOS domain regulates expression of genes involved in DNA damage response.
SETD1A FLOS domain interacts with Cyclin K to regulate gene expression.
Acknowledgments
We would like to thank Jessica Brady and Zhaohui Feng for administrative help with this project. We are grateful to Hideki Terai and Kunihiro Hinohara for techenial advice. We are also grateful to Nathaneal Gray for providing THZ531. HT was supported by a grant from Kanae foundation for the promotion of medical science, and Program for Advancing Strategic International Networks to Accelerate the Circulation of Talented Researchers, JAPAN. SL was supported by NCI grants CA190261. AK was supported by NCI grants R01 CA204396 and P30 CA008748. SAA was supported by the Leukemia and Lymphoma Society and NCI grants CA66996, CA140575.
Footnotes
Declaration of interests
SAA consults for Epizyme Inc, Imago Biosciences, C4 Therapeutics.
Author Contributions
Conceptualization, H.T., S.A.A.; Investigation, H.T., P.C., Z.F.; Resources, C.H., C.C., C.D.D., S.L., A.K.; Formal Analysis, H.T., R.K.; Writing-Original Draft, H.T.; Writing-Review & Editing, C.D.D., S.A.A.; Supervision, S.A.A.; Funding Acquisition, S.A.A.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Balgobind BV, Raimondi SC, Harbott J, Zimmermann M, Alonzo TA, Auvrignon A, Beverloo HB, Chang M, Creutzig U, Dworzak MN, et al. Novel prognostic subgroups in childhood 11q23/MLL-rearranged acute myeloid leukemia: results of an international retrospective study. Blood. 2009;114:2489–2496. doi: 10.1182/blood-2009-04-215152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Blazek D, Kohoutek J, Bartholomeeusen K, Johansen E, Hulinkova P, Luo Z, Cimermancic P, Ule J, Peterlin BM. The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes & development. 2011;25:2158–2172. doi: 10.1101/gad.16962311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bledau AS, Schmidt K, Neumann K, Hill U, Ciotta G, Gupta A, Torres DC, Fu J, Kranz A, Stewart AF, et al. The H3K4 methyltransferase Setd1a is first required at the epiblast stage, whereas Setd1b becomes essential after gastrulation. Development (Cambridge, England) 2014;141:1022–1035. doi: 10.1242/dev.098152. [DOI] [PubMed] [Google Scholar]
- Borkin D, He S, Miao H, Kempinska K, Pollock J, Chase J, Purohit T, Malik B, Zhao T, Wang J, et al. Pharmacologic Inhibition of the Menin-MLL Interaction Blocks Progression of MLL Leukemia In Vivo. Cancer cell. 2015;27:589–602. doi: 10.1016/j.ccell.2015.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs SD, Bryk M, Strahl BD, Cheung WL, Davie JK, Dent SY, Winston F, Allis CD. Histone H3 lysine 4 methylation is mediated by Set1 and required for cell growth and rDNA silencing in Saccharomyces cerevisiae. Genes & development. 2001;15:3286–3295. doi: 10.1101/gad.940201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao F, Townsend EC, Karatas H, Xu J, Li L, Lee S, Liu L, Chen Y, Ouillette P, Zhu J, et al. Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Molecular cell. 2014;53:247–261. doi: 10.1016/j.molcel.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R, Parmar K, Mouly E, Delord M, Kim JM, Regairaz M, Pla M, Vasquez N, Zhang QS, Pondarre C, et al. Bone marrow failure in Fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell stem cell. 2012;11:36–49. doi: 10.1016/j.stem.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R, Sarangi P, D’Andrea AD. The Fanconi anaemia pathway: new players and new functions. Nature reviews Molecular cell biology. 2016;17:337–349. doi: 10.1038/nrm.2016.48. [DOI] [PubMed] [Google Scholar]
- Chen C, Liu Y, Rappaport AR, Kitzing T, Schultz N, Zhao Z, Shroff AS, Dickins RA, Vakoc CR, Bradner JE, et al. MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer cell. 2014;25:652–665. doi: 10.1016/j.ccr.2014.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denissov S, Hofemeister H, Marks H, Kranz A, Ciotta G, Singh S, Anastassiadis K, Stunnenberg HG, Stewart AF. Mll2 is required for H3K4 trimethylation on bivalent promoters in embryonic stem cells, whereas Mll1 is redundant. Development (Cambridge, England) 2014;141:526–537. doi: 10.1242/dev.102681. [DOI] [PubMed] [Google Scholar]
- Edwards MC, Wong C, Elledge SJ. Human cyclin K, a novel RNA polymerase II-associated cyclin possessing both carboxy-terminal domain kinase and Cdk-activating kinase activity. Molecular and cellular biology. 1998;18:4291–4300. doi: 10.1128/mcb.18.7.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst P, Fisher JK, Avery W, Wade S, Foy D, Korsmeyer SJ. Definitive hematopoiesis requires the mixed-lineage leukemia gene. Developmental cell. 2004;6:437–443. doi: 10.1016/s1534-5807(04)00061-9. [DOI] [PubMed] [Google Scholar]
- Fellmann C, Hoffmann T, Sridhar V, Hopfgartner B, Muhar M, Roth M, Lai DY, Barbosa IA, Kwon JS, Guan Y, et al. An optimized microRNA backbone for effective single-copy RNAi. Cell reports. 2013;5:1704–1713. doi: 10.1016/j.celrep.2013.11.020. [DOI] [PubMed] [Google Scholar]
- Funato K, Major T, Lewis PW, Allis CD, Tabar V. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science. 2014;346:1529–1533. doi: 10.1126/science.1253799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greifenberg AK, Honig D, Pilarova K, Duster R, Bartholomeeusen K, Bosken CA, Anand K, Blazek D, Geyer M. Structural and Functional Analysis of the Cdk13/Cyclin K Complex. Cell reports. 2016;14:320–331. doi: 10.1016/j.celrep.2015.12.025. [DOI] [PubMed] [Google Scholar]
- Grembecka J, He S, Shi A, Purohit T, Muntean AG, Sorenson RJ, Showalter HD, Murai MJ, Belcher AM, Hartley T, et al. Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nature chemical biology. 2012;8:277–284. doi: 10.1038/nchembio.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Nakamura T, Alder H, Prasad R, Canaani O, Cimino G, Croce CM, Canaani E. The t(4;11) chromosome translocation of human acute leukemias fuses the ALL-1 gene, related to Drosophila trithorax, to the AF-4 gene. Cell. 1992;71:701–708. doi: 10.1016/0092-8674(92)90603-a. [DOI] [PubMed] [Google Scholar]
- Heckl D, Kowalczyk MS, Yudovich D, Belizaire R, Puram RV, McConkey ME, Thielke A, Aster JC, Regev A, Ebert BL. Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nature biotechnology. 2014;32:941–946. doi: 10.1038/nbt.2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes CM, Rozenblatt-Rosen O, Milne TA, Copeland TD, Levine SS, Lee JC, Hayes DN, Shanmugam KS, Bhattacharjee A, Biondi CA, et al. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Molecular cell. 2004;13:587–597. doi: 10.1016/s1097-2765(04)00081-4. [DOI] [PubMed] [Google Scholar]
- Jones RS, Gelbart WM. The Drosophila Polycomb-group gene Enhancer of zeste contains a region with sequence similarity to trithorax. Molecular and cellular biology. 1993;13:6357–6366. doi: 10.1128/mcb.13.10.6357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Lee JE, Cho EJ, Liu JO, Youn HD. Menin, a tumor suppressor, represses JunD-mediated transcriptional activity by association with an mSin3A-histone deacetylase complex. Cancer Res. 2003;63:6135–6139. [PubMed] [Google Scholar]
- Ko TK, Kelly E, Pines J. CrkRS: a novel conserved Cdc2-related protein kinase that colocalises with SC35 speckles. Journal of cell science. 2001;114:2591–2603. doi: 10.1242/jcs.114.14.2591. [DOI] [PubMed] [Google Scholar]
- Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nature reviews Cancer. 2007;7:823–833. doi: 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- Kuhn MW, Song E, Feng Z, Sinha A, Chen CW, Deshpande AJ, Cusan M, Farnoud N, Mupo A, Grove C, et al. Targeting Chromatin Regulators Inhibits Leukemogenic Gene Expression in NPM1 Mutant Leukemia. Cancer discovery. 2016;6:1166–1181. doi: 10.1158/2159-8290.CD-16-0237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerdrup M, Johansen JV, Agrawal-Singh S, Hansen K. An interactive environment for agile analysis and visualization of ChIP-sequencing data. Nature structural & molecular biology. 2016;23:349–357. doi: 10.1038/nsmb.3180. [DOI] [PubMed] [Google Scholar]
- Milne TA, Kim J, Wang GG, Stadler SC, Basrur V, Whitcomb SJ, Wang Z, Ruthenburg AJ, Elenitoba-Johnson KS, Roeder RG, et al. Multiple interactions recruit MLL1 and MLL1 fusion proteins to the HOXA9 locus in leukemogenesis. Molecular cell. 2010;38:853–863. doi: 10.1016/j.molcel.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra BP, Zaffuto KM, Artinger EL, Org T, Mikkola HK, Cheng C, Djabali M, Ernst P. The histone methyltransferase activity of MLL1 is dispensable for hematopoiesis and leukemogenesis. Cell reports. 2014;7:1239–1247. doi: 10.1016/j.celrep.2014.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng HH, Robert F, Young RA, Struhl K. Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Molecular cell. 2003;11:709–719. doi: 10.1016/s1097-2765(03)00092-3. [DOI] [PubMed] [Google Scholar]
- Pinello L, Canver MC, Hoban MD, Orkin SH, Kohn DB, Bauer DE, Yuan GC. Analyzing CRISPR genome-editing experiments with CRISPResso. Nature biotechnology. 2016;34:695–697. doi: 10.1038/nbt.3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao RC, Dou Y. Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nature reviews Cancer. 2015;15:334–346. doi: 10.1038/nrc3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel SS, Haladyna JN, Bezzant M, Stevens B, Pollyea DA, Sinha AU, Armstrong SA, Wei Q, Pollock RM, Daigle SR, et al. MLL1 and DOT1L cooperate with meningioma-1 to induce acute myeloid leukemia. The Journal of clinical investigation. 2016;126:1438–1450. doi: 10.1172/JCI80825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzankina Y, Pinzon-Guzman C, Asare A, Ong T, Pontano L, Cotsarelis G, Zediak VP, Velez M, Bhandoola A, Brown EJ. Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell stem cell. 2007;1:113–126. doi: 10.1016/j.stem.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 2008;132:487–498. doi: 10.1016/j.cell.2007.12.033. [DOI] [PubMed] [Google Scholar]
- Santos MA, Faryabi RB, Ergen AV, Day AM, Malhowski A, Canela A, Onozawa M, Lee JE, Callen E, Gutierrez-Martinez P, et al. DNA-damage-induced differentiation of leukaemic cells as an anti-cancer barrier. Nature. 2014;514:107–111. doi: 10.1038/nature13483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider J, Wood A, Lee JS, Schuster R, Dueker J, Maguire C, Swanson SK, Florens L, Washburn MP, Shilatifard A. Molecular regulation of histone H3 trimethylation by COMPASS and the regulation of gene expression. Molecular cell. 2005;19:849–856. doi: 10.1016/j.molcel.2005.07.024. [DOI] [PubMed] [Google Scholar]
- Shi J, Wang E, Milazzo JP, Wang Z, Kinney JB, Vakoc CR. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nature biotechnology. 2015;33:661–667. doi: 10.1038/nbt.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilatifard A. Molecular implementation and physiological roles for histone H3 lysine 4 (H3K4) methylation. Curr Opin Cell Biol. 2008;20:341–348. doi: 10.1016/j.ceb.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sladitschek HL, Neveu PA. MXS-Chaining: A Highly Efficient Cloning Platform for Imaging and Flow Cytometry Approaches in Mammalian Systems. PloS one. 2015;10:e0124958. doi: 10.1371/journal.pone.0124958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PA, et al. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA. 2003;9:493–501. doi: 10.1261/rna.2192803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel AT, Blessington P, Zou T, Feather D, Wu X, Yan J, Zhang H, Liu Z, Ernst P, Koretzky GA, et al. MLL-AF9-induced leukemogenesis requires coexpression of the wild-type Mll allele. Cancer cell. 2010;17:148–159. doi: 10.1016/j.ccr.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschiersch B, Hofmann A, Krauss V, Dorn R, Korge G, Reuter G. The protein encoded by the Drosophila position-effect variegation suppressor gene Su(var)3–9 combines domains of antagonistic regulators of homeotic gene complexes. The EMBO journal. 1994;13:3822–3831. doi: 10.1002/j.1460-2075.1994.tb06693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SH, Goode DL, Iwasaki M, Wei MC, Kuo HP, Zhu L, Schneidawind D, Duque-Afonso J, Weng Z, Cleary ML. The H3K4-Methyl Epigenome Regulates Leukemia Stem Cell Oncogenic Potential. Cancer cell. 2015;28:198–209. doi: 10.1016/j.ccell.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Valerio DG, Eisold ME, Sinha A, Koche RP, Hu W, Chen CW, Chu SH, Brien GL, Park CY, et al. NUP98 Fusion Proteins Interact with the NSL and MLL1 Complexes to Drive Leukemogenesis. Cancer cell. 2016;30:863–878. doi: 10.1016/j.ccell.2016.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama A, Wang Z, Wysocka J, Sanyal M, Aufiero DJ, Kitabayashi I, Herr W, Cleary ML. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Molecular and cellular biology. 2004;24:5639–5649. doi: 10.1128/MCB.24.13.5639-5649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Kwiatkowski N, Olson CM, Dixon-Clarke SE, Abraham BJ, Greifenberg AK, Ficarro SB, Elkins JM, Liang Y, Hannett NM, et al. Covalent targeting of remote cysteine residues to develop CDK12 and CDK13 inhibitors. Nature chemical biology. 2016;12:876–884. doi: 10.1038/nchembio.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The summary of the karyotyping analysis in Setd1a-knockout MLL-AF9 leukemia cells is shown.
RNA downregulated genes in Setd1a knockout cells were classified with the promoter/TSS H3K4me3 level.
The list of shRNAs and sgRNAs used in this study is shown.
The list of DNA primers and Taqman probes used in this study is shown.