

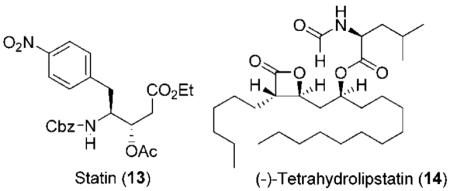
Abstract
An asymmetric synthesis of anti-aldol segments via a nonaldol route is described. The strategy involves a highly diastereoselective synthesis of functionalized tetrahydrofuran derivatives from optically active 4-phenylbutyrolactone. Treatment of the tetrahydrofuran derivatives with a Lewis acid and acetic anhydride provided the corresponding ring-opened styrene derivatives. Oxidative cleavage of the styrene derivatives provided access to the anti-aldol segments. The utility of this methodology was demonstrated by the synthesis of statine derivatives and pancreatic lipase inhibitor, (—)-tetrahydrolipstatin.
Introduction
The anti-α-alkyl-β-hydroxycarbonyl motifs are prevalent in a wide variety of natural and unnatural bioactive organic molecules.1 An asymmetric aldol addition of a suitable carbonyl derivative to an aldehyde stands out as the most straightforward approach to their synthesis. Consequently, a number of asymmetric methodologies have been developed over the years. Particularly, a number of chiral auxiliary-controlled highly diastereoselective anti-aldol additions have been reported.2 However, issues related to limited reaction scope, functional group compatibility, ready availability of reagents and auxiliaries, and operation complexity continue to attract much interest for alternative approaches. One notable nonaldol process has been developed by Jung and co-workers.3 Recently, in the context of the synthesis of cryptophycin 52, optically active phenylbutyrolactone 1 was efficiently converted to functionalized tetrahydrofuran derivative 2a (Figure 1).4 Both new stereogenic centers in 2a were created based upon the chirality of 4-phenylbutyrolactone, which in turn was prepared on a multigram scale by using Corey–Bakshi–Shibata (CBS) reduction as the key step.5 Opening of the tetrahydrofuran ring via an acyloxonium ion intermediate afforded functionalized β-hydroxy ester 3a, which was converted to cryptophycin 52 (4). The overall process is practical and may provide convenient access to a variety of γ-substituted ester derivatives. Of particular interest, this functionalized hexenoate derivative can serve as a useful anti-aldol surrogate since olefin cleavage would provide the anti-aldol product. Generation of such anti-aldol products would not be straightforward with the use of a direct aldol reaction. Encouraged by the good diastereoselectivity and overall efficiency of the route, we have now investigated the generality of this “nonaldol anti-aldol” strategy. We have applied this nonaldol strategy to the synthesis of statine derivatives which are widely utilized in the design and synthesis of aspartyl protease inhibitors. Also, we have demonstrated the utility of this method in the synthesis of (—)-tetrahydrolipstatin, a potent inhibitor of pancreatic lipase.
Figure 1.
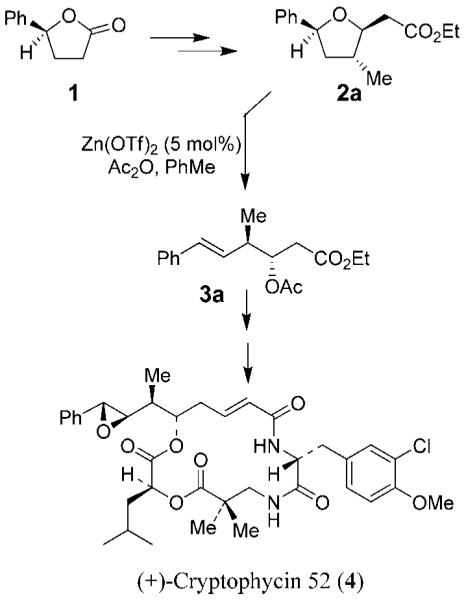
Nonaldol route to key anti-aldol precursor 3.
Results and Discussion
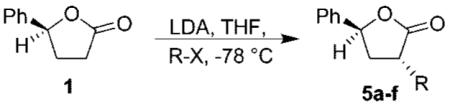
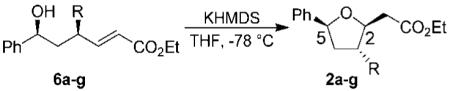
On the basis of our previous work, we decided to expand the scope and utility of functionalized tetrahydrofurans in synthesis.4 In particular, we planned to study the opening of the tetrahydrofuran ring to unravel the cyclic stereochemistry into an acyclic form. As shown in Scheme 1, our synthesis utilized known lactone 1, which was obtained in multigram quantities by employing CBS reduction as the key step.5 Alkylation of lactone 1 with LDA, and a variety of electrophiles, provided substituted lactones 5a–f in good diastereoselectivity (1H NMR analysis) and excellent yields as shown in Table 1. Reduction of lactones 5a–f and 1 by using DIBAL-H afforded the corresponding lactols. Wittig olefination of the resulting lactols gave rise to α,β-unsaturated esters 6a–g with high E-selectivity as shown by 1H NMR analysis. Treatment of the α,β-unsaturated esters 6a–g with KHMDS in THF at −78 °C furnished tetrahydrofuran derivatives 2a–g. As shown in Table 2, these tetrahydrofuran derivatives were formed in excellent yield and diastereoselectivity.
Scheme 1.
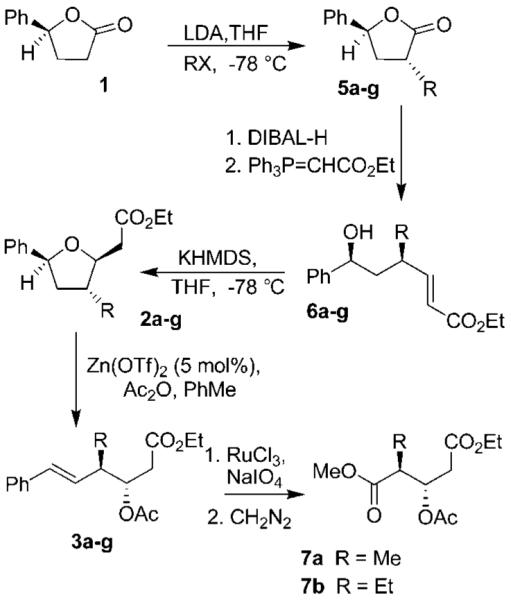
Synthesis of anti-Aldol Segments
Table 1. The Alkylation of Lactone 1 with Various Electrophiles.
| |||||
|---|---|---|---|---|---|
| entry | R | X | product | % yield | dra |
| 1 | −CH3 | I | 5a | 75 | 16:1 |
| 2 | −CH2CH3 | I | 5b | 67 | 17:1 |
| 3 | −CH2CN | Br | 5c | 76 | 17:1 |
| 4 | −CH2C6H5 | Br | 5d | 63 | 99:1 |
| 5 | −CH2-C6H5-pNO2 | Br | 5e | 71 | 99:1 |
| 6 | −CH2CH2CHCH2 | Br | 5f | 32 | 20:1 |
The dr was determined by 1H NMR of the crude reaction mixture.
Table 2. Synthesis of Substituted Tetrahydrofurans.
| ||||
|---|---|---|---|---|
| entry | R | product | % yield | dra |
| 1 | −CH3 | 2a | 70 | 11:1 |
| 2 | −CH2CH3 | 2b | 91 | 60:1 |
| 3 | −CH2CN | 2c | 82 | 11:1 |
| 4 | −CH2-C6H5 | 2d | 91 | 49:1 |
| 5 | −CH2-C6H5-pNO2 | 2e | 70 | 14:1 |
| 6 | −CH2CH2CHCH2 | 2f | 86 | 100:1 |
| 7 | −H | 2g | 55 | 1:1 |
The dr was determined by 1H NMR of the crude reaction mixture.
Having made the requisite tetrahydrofurans 2a–g, we then explored the key ring-opening reaction. Previously, we have carried out a ring-opening reaction of the methyl-substituted derivative 2a by using a catalytic amount (6 mol %) of ZnCl2 in the presence of acetic anhydride.4,6 However, these conditions were not optimal for tetrahydrofuran derivatives containing electron-rich substituents at C-3. After surveying a number of Lewis acids, we found that a catalytic amount of Zn(OTf)2 provided the best results. Thus, exposure of furans 2a–g to Zn(OTf)2 (5 mol %) and acetic anhydride in refluxing toluene gave rise to the anti-aldol precursors 3a–g as shown in Table 3.
Table 3. Synthesis of Substituted β-Acetoxy Ester.

| |||
|---|---|---|---|
| entry | R | product | % yield |
| 1 | −CH3 | 3a | 76 |
| 2 | −CH2CH3 | 3b | 60 |
| 3 | −CH2CN | 3c | 53 |
| 4 | −CH2-C6H5a | 3d | |
| 5 | −CH2-C6H5-pNO2 | 3e | 90 |
| 6 | −CH2CH2CHCH2 | 3f | 86 |
| 7 | −H | 3g | 78 |
See Scheme 2 for benzyl example.
Initially, we explored the ring-opening reaction of unsubstituted tetrahydrofuran 2g (R = H), which proceeded smoothly in the presence of a catalytic amount (5 mol %) of Zn(OTf)2 and excess acetic anhydride to furnish the styrene 3g in 78% yield. The reactions with methyl- and ethyl-substituted derivatives 2a and 2b also gave the respective styrene derivatives 3a and 3b in good yield (Table 3, entries 1 and 2). The ring-opening reaction of nitrile-substituted derivative 2c failed to furnish any appreciable amount of the corresponding styrene derivative, probably due to the strong coordination of the nitrile group with Zn(OTf)2. When the reaction was carried out in the presence of 3 equiv of Zn(OTf)2, the desired styrene derivative 3c was obtained in 53% yield. We then explored the ring-opening reaction with benzyl- and allyl-substituted tetrahydrofurans. The ring-opening reaction of the allyl-substituted tetrahydrofuran provided a complex mixture of products due to cation–olefin cyclization. The ring-opening reaction with homoallyl derivative 2f (Table 3, entry 6) proceeded smoothly to afford styrene derivative 3f in 86% yield. Our attempts to open 2d with Zn(OTf)2 under a variety of reaction conditions provided a complex mixture of opened ring products as well as substituted tetraline derivatives. As shown in Scheme 2, exposure of 2d to 5 mol % of Zn(OTf)2 resulted in a 1:4 mixture of styrene derivative 3d with a diastereomeric mixture (3:1) of tetraline derivatives 11a and 11b. This mixture was separated by HPLC and the identity of products 11a and 11b was confirmed by extensive NMR studies.
Scheme 2.

Lewis Acid Catalyzed Ring-Opening of Benzyl Derivative 2d
The rationale for the ring-opening reaction and the formation of tetraline derivatives 11a and 11b is shown in Scheme 2. Activation of acetic anhydride by Zn(OTf)2 leads to the formation of acyloxonium ion 8. The ring-opening by cleavage of the C–O bond at C-5 would give rise to a more stable benzylic carbonium ion 9. Cleavage of the C–O bond at C-2, on the other hand, would provide a less stable carbonium ion. Also, elimination of an α-proton of the ester followed by cleavage of the C–O bond may lead to the formation of an α,β-unsaturated ester derivative. However, no product corresponding to the cleavage of the C–O bond at C-2 was isolated. The formation of products 3d, 11a, and 11b can be rationalized through the formation of benzylic carbonium ion 9. The loss of a proton from 9 (path a) presumably resulted in the styrene derivative 3d. The tetraline derivatives 11a and 11b were formed due to a competing intramolecular trapping of the benzylic carbonium ion intermediate 9 as shown in path b. The observed ratio of tetralines 11a and 11b can be rationalized on the basis of proposed transition states TS-11a and TS-11b shown in Figure 2. The transition state TS-11a is preferred over TS-11b because of the developing 1,3-diaxial interaction in TS-11b.
Figure 2.
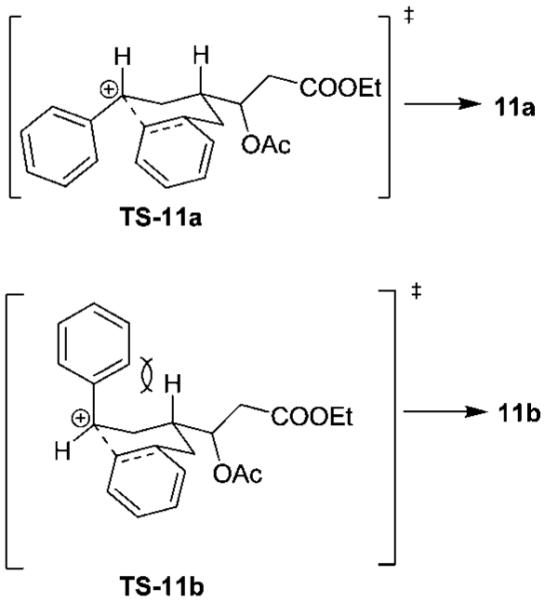
Proposed transition state for the intramolecular trapping of the benzylic carbonium ion.
Upon the basis of this insight, we speculated that an electron-withdrawing group on the aromatic ring would slow down the competing intramolecular trapping of the benzylic carbonium ion and would favor the formation of the desired styrene opening product through pathway a in Scheme 2. Indeed, Lewis acid-catalyzed reaction with p-NO2 benzyl derivative 2e furnished styrene derivative 3e in 90% yield.
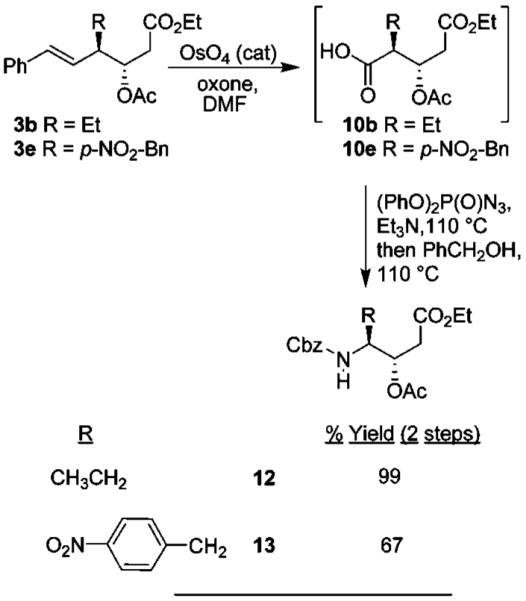
Various styrene derivatives resulting from the Lewis acid-catalyzed ring opening are suitable precursors of the anti-aldol segments. As shown in Scheme 1, oxidative cleavage of styrenes 3a and 3b with RuCl3/NaIO4 gave rise to the corresponding acids which were subsequently converted to the methyl esters 7a and 7b by treatment with diazomethane. However, better yields for the cleavage of styrenes were obtained by using OsO4 and oxone in DMF.7 Accordingly, styrenes 3b and 3e were converted to their respective acids 10b and 10e and then subjected to Curtius rearrangement.8 The resulting acids without further purification were exposed to 2.2 equiv of diphenyl phosphorazidate and 2.2 equiv of triethylamine in toluene at reflux. Benzyl alcohol was added and the resulting mixture was heated at reflux for 12 h to provide Cbz derivatives 12 and 13 in 99% and 67% yields, respectively, over two steps (Scheme 4). These statine derivatives have been extensively utilized in the design of aspartyl protease inhibitors, particularly for renin and HIV-1 protease inhibitors.9–11 The present methodology will provide access to substituted statine derivatives in optically active form.
Scheme 4.
Synthesis of Styrene Derivative 21
We have utilized this methodology in the total synthesis of (—)-tetrahydrolipstatin (14). This natural product was isolated from Streptomyces toxytricini in 1987, and was approved by the FDA under the trade name Orlistat in 2006, for the treatment of obesity.12 The β-lactone moiety present in Orlistat inhibits gastric and pancreatic lipases, thus slowing the hydrolysis of triglycerides into free fatty acids and reducing their absorption in the gut.13 Futhermore, tetrahydrolipstatin was recently shown to be a potent fatty acid synthase (FAS) inhibitor. FAS is an enzyme responsible for the synthesis of fatty acids in many human carcinomas and is required for tumor cell survival, making FAS inhibitor a promising drug for the treatment of cancer.14 Tetrahydrolipstatin and its analogues have been of great interest in the past years, as they have shown many interesting biological activities. This has provided immense interest in the chemistry and biology of tetrahydrolipstatin.15
As shown in Figure 3, the structural element of the β-lactone functionality can be derived from the styrene derivative 15 by oxidative cleavage to provide the anti-aldol unit with appropriate stereochemistry. Styrene derivative 15 can be obtained from the tetrahydrofuran derivative 16 by a stereoselective reduction followed by a Lewis acid-catalyzed, acyloxonium ion-mediated ring-opening reaction. Stereochemically defined tetrahydrofuran derivative 16 would be derived from optically active lactone 1 by the sequence of reaction steps described above.
Figure 3.
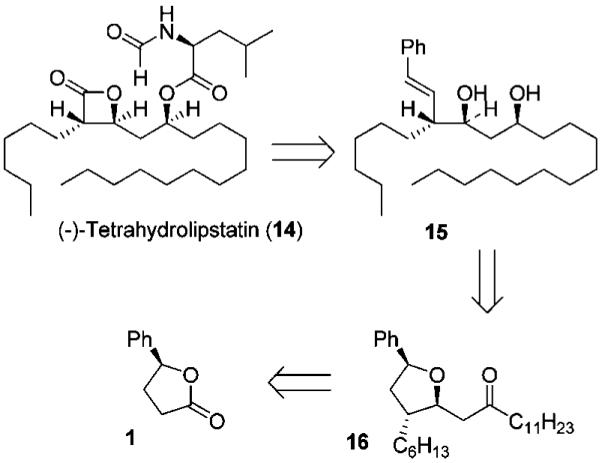
Retrosynthetic analysis for (—)-tetrahydrolipstatin.
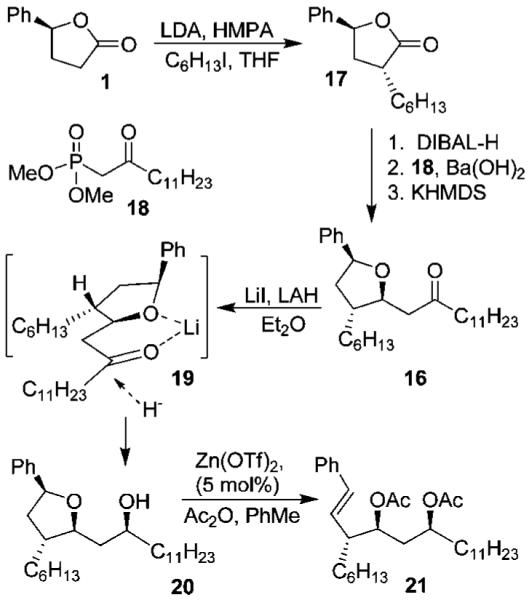
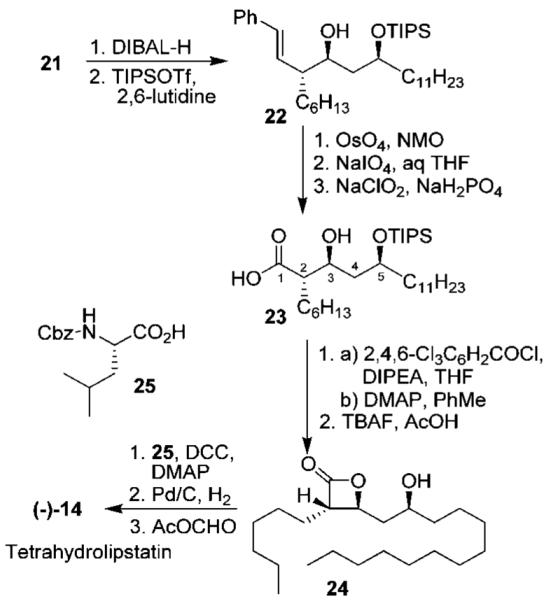
The synthesis of styrene 21 is shown in Scheme 4. Alkylation of lactone 1 with iodohexane using LDA in the presence of HMPA furnished the requisite lactone 17 in 84% yield with an excellent diastereomeric ratio (99:1 by 1H NMR). DIBAL-H reduction of lactone 17 afforded the corresponding lactol in quantitative yield. A Horner–Wadsworth–Emmons reaction of the resulting lactol with phosphonate 18 in the presence of Ba(OH)2 resulted in the formation of tetrahydrofuran derivative 16 in 83% yield with an excellent diastereomeric ratio (33:1 by 1H NMR).16,17
At this point, our attempt at the ring-opening reaction of substituted tetrahydrofuran 16 was unsuccessful resulting in recovery of the starting material. This was possibly due to the sequestering of the Lewis acid through the formation of a stable six-membered complex with the ketone. We therefore planned to reduce the ketone stereoselectively and then carry out the ring-opening reaction. For the stereoselective reduction of the ketone, we elected to carry out a chelation-controlled reduction described by us previously.18 Thus, reduction of ketone 16 with LAH in the presence of LiI resulted in the formation of alcohol 20 in 88% yield with excellent diastereoselectivity (17:1 dr). The high degree of diastereoselectivity can be explained by formation of a chelated rigid intermediate 19. Hydride attack presumably proceeded from the less hindered bottom face, providing the syn-alcohol 20 selectively. Subsequent ring-opening reaction of alcohol 20 with 5 mol % of Zn(OTf)2 and an excess of Ac2O in refluxing toluene proceeded smoothly, providing diacetoxy styrene derivative 21 in 85% yield.
The completion of the total synthesis of tetrahydrolipstatin is shown in Scheme 5. Treatment of diacetate 21 with DIBAL-H at −78 °C gave the corresponding diol. Selective protection of this diol with TIPSOTf in the presence of 2,6-lutidine at −78 °C gave silyl ether 22.15i Oxidative cleavage of the styrene moiety with OsO4 and NaIO4 resulted in an aldehyde that was oxidized to the acid by using sodium chlorite in 50% yield over three steps. For the formation of the β-lactone, we planned to utilize conditions reported by Adam and co-workers (PhSO2Cl/pyridine, p-NO2PhSO2Cl/pyridine)19. Our many attempts to form the β-lactone using acid 23 under the same conditions resulted in low yields (10–15%). Ortar and co-workers have reported a similar poor yield during their synthesis of tetrahydrolipstatin.14b Interestingly, in an earlier synthesis of (—)-tetrahydrolipstatin from our laboratory, we employed the above Adam’s conditions to the C-5 epimer of acid 23 and obtained the corresponding C-5 epimeric β-lactone in 74% yield.15i Presumably, the steric hindrance of the TIPS-protected syn-diol 23 is responsible for the sluggish reaction and poor yield of the corresponding β-lactone under Adam’s conditions. We then utilized Yamaguchi conditions and formed the desired β-lactone in 55% yield.20
Scheme 5.
Synthesis of (—)-Tetrahydrolipstatin 14
Removal of the TIPS protecting group was achieved by using TBAF and acetic acid to give alcohol 24 in 62% yield. To complete the synthesis, we attempted the esterification of 24 with N-formyl leucine in the presence of DCC/DMAP. These conditions resulted in substantial epimerization of tetrahydrolipstatin (1:1 ratio). Reduction of DMAP concentration to 0.2 equiv suppressed the formation of the epimer to 1.4:1. To circumvent this problem, we carried out a three-step procedure as described by Uskokovic and co-workers.21 This involved (1) esterification with Cbz-Leu-OH 25, using DCC and DMAP,22 (2) removal of the Cbz protection by hydrogenolysis, and (3) formylation of the amine by using the mixed anhydride at 23 °C to provide (—)-tetrahydrolipstatin (14, [α]20D −32 (c 0.05, CHCl3); lit.15v ([α]23D −33 (c 0.36, CHCl3)) in 48% yield over 2 steps. Spectral data (1H and 13C NMR) of synthetic (—)-tetrahydrolipstatin are identical with those reported for the natural product.
In conclusion, we have developed an alternative strategy to the formation of anti-aldol segments in optically active form via a nonaldol route. The strategy utilizes optically active 4-phenylbutyrolactone as the key starting material. Both stereogenic centers of the anti-aldol segments are derived from the 4-phenyltetrahydrofuran. The scope and utility of this methodology was demonstrated by the asymmetric synthesis of statine derivatives which are important scaffolds for aspartyl protease inhibitors. Furthermore, the methodology was utilized in the stereoselective synthesis of (—)-tetrahydrolipstatin. The key steps involved a highly diastereoselective chelation-controlled syn-reduction of a ketone, a catalytic asymmetric ring-opening reaction, and the formation of the β-lactone under Yamaguchi conditions. Further application of this methodology is in progress.
Experimental Section
General Experimental Methods.
1H NMR and 13C NMR spectra were recorded on Bruker Avance ARX- 400 and DRX-500 spectrometers. IR spectra were recorded on a Mattason Genesis II FT-IR spectrometer. Optical rotations were recorded on a Perkin-Elmer 341 polarimeter. Anhydrous solvents were obtained as follows: THF and diethyl ether by distillation from sodium and benzophenone; pyridine and dichloromethane from CaH2. All other solvents were reagent grade. All moisture-sensitive reactions were carried out in a flame-dried flask under argon atmosphere. Column chromatography was performed with Whatman 240–400 mesh silica gel under a low pressure of 3–5 psi. TLC was carried out with E. Merck silica gel 60-F-254 plates. HPLC data were collected using a system composed of an Agilent 1100 series degasser, quaternary pump, thermostatable column compartment, variable wavelength detector, and Agilent 1200 series auto sampler and fraction collector controlled by Chemstation software. All chromatographic reagents used were HPLC grade.
(3R,5S)-3-Methyl-5-phenyldihydrofuran-2(3H)-one (5a).
To a stirring solution of THF (5 mL) and diisopropylamine (490 μL, 3.7 mmol) at 0 °C under argon was added n-BuLi (2.1 mL, 1.6 M solution in hexanes, 3.39 mmol) dropwise; the mixture was then stirred for 15 min and cooled to −78 °C. Then lactone 1 (500 mg, 3.08 mmol) was dissolved in 15 mL of THF and added dropwise over a 0.5 h period. The reaction mixture was stirred for an additional 0.5 h at −78 °C. Iodomethane (210 μL, 3.4 mmol) was added dropwise and the reaction mixture was stirred for 1 h at −78 °C. The reaction was quenched with saturated aqueous NH4Cl and warmed to room temperature, then the aqueous layer was extracted with EtOAc (3×) and the organic layer was washed with water and brine and dried over anhydrous Na2SO4. The residue was chromatographed on silica gel (85:15 hexanes/EtOAc) to give 5a (407 mg, 75% yield) as an oil; dr (16:1), [α]20D −19.5 (c 1.1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.43–7.18 (m, 5H), 5.55 (dd, J = 8, 5 Hz, 1H), 2.71 (m, 1H), 2.46–2.31 (m, 2H), 1.33 (d, J = 4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 179.6, 139.8, 128.6, 128.0, 124.9, 78.2, 38.2, 33.5, 15.3; IR (NaCl) 3066, 1773, 1450, and 1171 cm−1.
(3R,5S)-3-Ethyl-5-phenyldihydrofuran-2(3H)-one (5b).
To a stirring solution of THF (5 mL) and diisopropylamine (450 μL, 3.39 mmol) at 0 °C under argon was added n-BuLi (2 mL, 1.6 M solution in hexanes, 3.24 mmol) dropwise, the mixture was stirred for 15 min, then DMPU (3 mL) was added dropwise and the reaction mixture was stirred at 0 °C for 15 min and then cooled to −78 °C. Lactone 1 (500 mg, 3.08 mmol) was dissolved in 15 mL of THF and added dropwise over a 0.5 h period. The reaction mixture was stirred for an additional 0.5 h at −78 °C. Then iodoethane (370 μL, 4.62 mmol) was added dropwise and the reaction mixture was stirred for 1 h at −78 °C. The reaction was quenched with saturated aqueous NH4Cl and warmed to room temperature. The aqueous layer was extracted with EtOAc (3×), then the combined organic layers were washed with water and brine and dried over anhydrous Na2SO4. The residue was chromatographed on silica gel (80:20 hexanes/EtOAc) to give 5b (393 mg, 67% yield) as an oil; dr (17:1); [α]20D −28.5 (c 2.1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.40–7.26 (m, 5H), 5.55 (t, J = 4 Hz, 1H), 2.64–2.56 (m, 1H), 2.45–2.34 (m, 2H), 1.95–1.87 (m, 1H), 1.64–1.55 (m, 1H), 1.03 (t, J = 8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 179.1, 139.9, 128.6, 128.1, 124.9, 78.5, 40.1, 35.9, 23.5, 11.6; IR (NaCl) 2955, 2872, 1773, 1453, and 1171 cm−1; CI-HRMS (m/z) calcd for C12H14O2 ([M]+) 190.0994, found 190.0992.
2-((3R,5S)-2-Oxo-5-phenyltetrahydrofuran-3-yl)acetonitrile (5c).
The title compound was prepared from 1 following the same procedure that was used for the synthesis of 5a, using bromoacetonitrile. Purification by flash chromatography (80:20 hexanes/EtOAc) afforded 5c as an oil (471 mg, 76% yield); dr (17:1); [α]20D+2.60 (c 1.2, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.43–7.28 (m, 5H), 5.73 (dd, J = 7, 4 Hz, 1H), 3.04–2.97 (m, 1H), 2.87 (dd, J = 17, 5 Hz, 1H), 2.73–2.65 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 175.4, 138.5, 128.9, 128.6, 124.6, 116.7, 78.3, 35.2, 35.2, 18.4; IR (NaCl) 3032, 2925, 2854, 2251, 1777, 1451, and 1171 cm−1; EI-HRMS (m/z) calcd for C12H11NO2 ([M + H]+) 202.9888, found 202.0866.
(3R,5S)-3-Benzyl-5-phenyldihydrofuran-2(3H)-one (5d).
The title compound was prepared from γ-lactone 1 following the same procedure that was used for the synthesis of 5a, using benzyl bromide. Purification by flash chromatography (90:10, hexanes/EtOAc) afforded 5d (116 mg, 63% yield) as an oil; dr (99:1); [α]20D+135.3 (c 0.5, CHCl3); 1H NMR (CDCl3; 400 MHz) δ 7.38–7.16 (m, 10H), 5.39 (dd, J = 7.8, 4.7 Hz, 1H), 3.25 (dd, J = 13.6, 4.3 Hz, 1H), 3.02–2.95 (m, 1H), 2.90–2.85 (m, 1H), 2.50–2.43 (m, 1H), 2.31–2.24 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 178.5, 139.6, 138.0, 128.8, 128.6, 128.6, 128.1, 126.8, 124.8, 78.6, 40.4, 36.1, 35.5; IR (NaCl) 3033, 2926, 1764, 1495, 1451, 1132, and 1011 cm−1; EI-HRMS (m/z) calcd for C17H16O2 ([M]+) 252.1150, found 252.1147.
(3S,5S)-3-(4-Nitrophenyl)-5-phenyldihydrofuran-2(3H)-one (5e).
The title compound was prepared from γ-lactone 1 following the same procedure that was used for the synthesis of 5a, using p-nitrobenzyl bromide. Purification by flash chromatography (95:5 toluene/MeOH) afforded 5e (917 mg, 71% yield) as an oil; dr (99: 1); [α]20D +29.6 (c 1.1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.19 (d, J = 8 Hz, 2H), 7.41–7.31 (m, 5H), 7.26–7.24 (m, 2H), 5.50 (dd, J = 12, 5 Hz, 1H), 3.32 (m, 1H), 3.04–2.95 (m, 2H), 2.48–2.41 (m, 1H), 2.35–2.30 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 177.6, 146.9, 145.7, 139.2, 129.8, 128.8, 128.3, 124.7, 123.9, 78.3, 39.7, 35.8, 35.4; IR (NaCl) 2998, 2885, 2837, 2362, 1776, 1602, 1516, and 1346 cm−1; EI-HRMS (m/z) calcd for C17H15NO4 ([M]+) 297.1001, found 297.1004.
(3R,5S)-3-(But-3-enyl)-5-phenyldihydrofuran-2(3H)-one (5f).
To a stirring solution of THF (5 mL) and diisopropylamine (485 μL, 3.71 mmol) at 0 °C under argon was added n-BuLi (2.2 mL, 1.6 M solution in hexanes, 3.54 mmol) dropwise; the mixture was stirred for 15 min, then HMPA (3 mL) was added dropwise and the reaction mixture was stirred at 0 °C for 15 min and then cooled to −78 °C. Then lactone 1 (546 mg, 3.37 mmol) was dissolved in 15 mL of THF and added dropwise over a 0.5 h period. The reaction mixture was stirred for another 0.5 h at −78 °C. Then a catalytic amount of TBAI (190 mg, 0.51 mmol) was added in one portion, and then 1-bromo-3-butene (380 μL, 3.71 mmol) was added dropwise; next the reaction mixture was stirred for 1 h at −78 °C and then warmed to 0 °C and stirred for 3 h. The reaction was quenched with saturated aqueous NH4Cl and warmed to room temperature. The aqueous layer was extracted with EtOAc (3×), then the combined organic layers were washed with water and brine and dried over anhydrous Na2SO4. The residue was chromatographed on silca gel (90:10 hexanes/EtOAc) to give 5f (233 mg, 32% yield) as an oil; dr (20:1); [α]20D −11.9 (c 0.9, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.40–7.29 (m, 5H), 5.84–5.74 (m, 1H), 5.56 (t, J = 6 Hz, 1H), 5.08–4.99 (m, 2H), 2.71–2.63 (m, 1H) 2.41–2.38 (m, 2H), 2.25–2.13 (m, 2H), 2.05–1.96 (m, 1H), 1.68–1.59 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 179.0, 139.7, 136.9, 128.6, 128.1, 124.8, 115.7, 78.5, 38.0, 36.5, 31.2, 29.5; IR (NaCl) 2969, 1775, and 1167 cm−1; EI-HRMS (m/z) calcd for C14H16O2 ([M]+) 216.1150, found 216.1152.
(E,S)-Ethyl 6-Hydroxy-6-phenylhex-2-enoate (6g).
To a solution of 1 (307 mg, 1.89 mmol) in 15 mL of CH2Cl2 under argon at −78 °C was added DIBAL-H (2.3 mL, 1 M solution in CH2Cl2, 2.28 mmol) dropwise. The reaction was stirred for 0.5 h at −78 °C and then quenched with 2 mL of methanol then warmed to room temperature, 15 mL of K–Na tartrate was added, and once the emulsion had separated, the aqueous layer was extracted with CH2Cl2 (3×) and the combined organic layers were washed with brine and dried over anhydrous Na2SO4. Without further purification the crude oil was dissolved in 20 mL of CH2Cl2 then Ph3PCHCO2Et (835 mg, 2.39 mmol) was added and the reaction mixture was refluxed for 12 h at 40 °C. The reaction mixture was then concentrated and chromatographed (90:10 hexanes/EtOAc) to give 6g (393 mg, 89% yield over two steps) as an oil; 1H NMR (400 MHz, CDCl3) δ 7.35–7.27 (m, 5H), 6.98–6.94 (m, 1H), 5.82 (d, J = 15 Hz, 1H), 4.70–4.66 (m, 1H), 4.18 (q, J = 7 Hz, 2H), 2.30–2.25 (m, 2H), 2.06 (s, 1H), 1.96–1.84 (m, 2H), 1.27 (t, J = 7 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 166.6, 148.4, 144.1, 128.5, 127.7, 125.8, 121.6, 73.6, 60.1, 37.0, 28.4, 14.2; IR (NaCl) 3300, 2982, 1716, 1652, 1451, and 1193 cm−1; EI-HRMS (m/z) calcd for C14H18O3 ([M]+) 234.1256, found 234.1255.
(4R,6S,E)-Ethyl 6-Hydroxy-4-methyl-6-phenylhex-2-enoate (6a).
The title compound was prepared from γ-lactone 5a following the same procedure that was used for the synthesis of 6g. Purification by flash chromatography (90:10 hexanes/EtOAc) afforded 6a (502 mg, 94% yield over two steps) as a single diastereomer; [α]20D −21.8 (c 1.1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.35–7.28 (m, 5H), 6.86 (dd, J = 16, 8 Hz, 1H), 5.86 (d, J = 16 Hz, 1H), 4.64–4.61 (m, 1H), 4.18 (q, J = 7 Hz, 2H), 2.63–2.60 (m, 1H), 2.03 (s, 1H), 1.89–1.82 (m, 1H), 1.70–1.63 (m, 1H), 1.29 (t, J = 7 Hz, 3H), 1.08 (d, J = 7 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 166.8, 153.4, 144.6, 128.5, 127.6, 125.6, 120.4, 72.1, 60.2, 45.3, 33.4, 20.1, 14.2; IR (NaCl) 3300, 2958, 1717, 1652, 1455, and 1272 cm−1.
(4R,6S,E)-Ethyl 4-Ethyl-6-hydroxy-6-phenylhex-2-enoate (6b).
The title compound was prepared from γ-lactone 5b following the same procedure that was used for the synthesis of 6g. Purification by flash chromatography (90:10 hexanes/EtOAc) afforded 6b (482 mg, 89% yield over two steps) as a single diastereomer; [α]20D −6.6 (c 2, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.34–7.23 (m, 5H), 6.75 (dd, J = 16, 10 Hz, 1H), 5.88 (d, J = 16 Hz, 1H), 4.59–4.55 (m, 1H), 4.18 (q, J = 7 Hz, 2H), 2.49–2.44 (m, 1H), 2.25 (s, 1H), 1.92–1.85 (m, 1H), 1.63–1.56 (m, 1H), 1.51–1.46 (m, 1H), 1.41–1.36 (m, 3H), 1.30 (t, J = 7 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 166.6, 152.3, 144.9, 128.4, 128.2, 127.4, 125.5, 122.1, 60.2, 43.6, 40.9, 27.7, 14.1, 11.5; IR (NaCl) 3136, 2961, 1719, 1649, 1452, and 1199 cm−1; CI-HRMS (m/z) calcd for C16H22O3 ([M]+) 262.1569, found 262.1570.
(4R,6S,E)-Ethyl 4-(Cyanomethyl)-6-hydroxy-6-phenylhex-2-enoate (6c).
The title compound was prepared from γ-lactone 5c following the same procedure that was used for the synthesis of 6g. Purification by flash chromatography (90:10 hexanes/EtOAc) afforded 6c (110 mg, 83% yield over two steps) as a single diastereomer; [α]20D −25.7 (c 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.39–7.27 (m, 5H), 6.81 (dd, J = 16, 9 Hz, 1H), 6.03 (d, J = 16 Hz, 1H), 4.65–4.62 (m, 1H), 4.21 (q, J = 7 Hz, 2H,), 3.03–2.97 (m, 1H), 2.52–2.49 (m, 2H), 2.19 (s, 1H), 2.03–1.95 (m, 1H), 1.81–1.74 (m, 1H), 1.31 (t, J = 7 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 165.8, 146.7, 143.9, 128.6, 127.9, 125.5, 124.1, 117.5, 71.5, 60.6, 42.5, 35.8, 22.8, 14.1; IR (NaCl) 3146, 3053, 2249, 1712, 1657, 1451, 1306, and 1223 cm−1; EI-HRMS (m/z) calcd for C16H19NO3 ([M]+) 273.1365, found 273.1370.
(4R,6S,E)-Ethyl 4-Benzyl-6-hydroxy-6-phenylhex-2-enoate (6d).
The title compound was prepared from γ-lactone 5d following the same procedure that was used for the synthesis of 6g. Purification by flash chromatography (80:20 hexanes:EtOAc) afforded 6d (944 mg, 84% yield over two steps) as a single diastereomer; oil; [α]20D +13.1 (c 0.9, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.35–7.11 (m, 10H), 6.84 (dd, J = 15.5, 9.4 Hz, 1H), 5.79 (d, J = 15.7 Hz, 1H), 4.61 (dd, J = 10.3, 3.1 Hz, 1H), 4.21–4.14 (m, 2H), 2.96–2.87 (m, 1H), 2.75–2.72 (m, 2H), 1.99–1.87 (m, 2H), 1.69–1.62 (m, 1H), 1.29 (t, J = 7.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 166.4, 151.3, 144.7, 138.9, 129.1, 128.4, 128.2, 127.5, 126.1, 125.5, 122.2, 71.7, 60.2, 43.1, 41.3, 40.8, 14.1; IR (NaCl) 3447, 3062, 1716, 1651, 1494, 1453, 1370, 1305, 1217, and 1030 cm−1; ESI-HRMS (m/z) calcd for C21H24O3 ([M]+) 324.1725, found 324.1728.
(4R,6S,E)-Ethyl 6-Hydroxy-4-(4-nitrobenzyl)-6-phenylhex-2-enoate (6e).
The title compound was prepared from γ-lactone 5e following the same procedure that was used for the synthesis of 6g. Purification by flash chromatography (80:20 hexanes/EtOAc) afforded 6e (531 mg, 96% yield over two steps) as a single diastereomer; [α]20D +21.04 (c 1.9, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.10 (d, J = 9 Hz, 2H), 7.34–7.24 (m, 7H), 6.76 (dd, J = 16, 10 Hz, 1H), 5.75 (d, J = 16 Hz, 1H), 4.62–4.59 (m, 1H), 4.18–4.12 (m, 2H), 2.99–2.94 (m, 1H), 2.89–2.84 (m, 1H), 2.79–2.74 (m, 1H), 2.20–2.19 (m, 1H), 1.96–1.89 (m, 1H), 1.69–1.63 (m, 1H), 1.26 (t, J = 7 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 166.1, 149.9, 146.9, 146.4, 144.5, 129.8, 128.5, 127.7, 125.4, 123.5, 122.9, 71.5, 60.4, 43.3, 41.1, 40.7, 14.1; IR (NaCl) 2900, 1709, 1516, and 1345 cm−1; EI-HRMS (m/z) calcd for C21H23NO5 ([M + H]+) 370.1654, found 370.1657.
(R,E)-Ethyl 4-((S)-2-Hydroxy-2-phenylethyl)octa-2,7-dienoate (6f).
The title compound was prepared from γ-lactone 5f following the same procedure that was used for the synthesis of 6g. Purification by flash chromatography (90:10 hexanes/EtOAc) afforded 6f (95 mg, quantitative yield over two steps) as a single diastereomer; [α]20D +1.6 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.39–7.23 (m, 5H), 6.74 (dd, J = 16, 10 Hz, 1H), 5.89 (d, J = 16 Hz, 1H), 5.81–5.71 (m, 1H), 5.01–4.90 (m, 2H), 4.59–4.55 (m, 1H), 4.18 (q, J = 7 Hz, 2H), 2.64–2.55 (m, 1H), 2.25 (s, 1H), 2.07–1.93 (m, 2H), 1.91–1.85 (m, 1H), 1.64–1.57 (m, 1H), 1.55–1.42 (m, 1H), 1.30 (t, J = 7 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 166.5, 152.0, 144.9, 138.0, 128.4, 127.4, 125.5, 122.2, 114.8, 71.7, 60.3, 43.9, 38.8, 33.9, 31.2, 14.1; IR (NaCl) 3265, 2852, 1709, 1641, 1303, and 1161 cm−1; EI-HRMS (m/z) calcd for C18H24O3 ([M]+) 287.1725, found 288.1722.
Ethyl 2-((5S)-5-Phenyltetrahydrofuran-2-yl)acetate (2g).
To a stirred solution of KHMDS (4.0 mL, 0.5 M solution in toluene, 2.01 mmol) in THF (5 mL) was added 6g (392 mg, 1.67 mmol) in THF (15 mL) at −78 °C dropwise over a 30 min period and the mixture was then stirred at −78 °C for an additional 1 h. Then the reaction was quenched with saturated aqueous NH4Cl (10 mL) and warmed to room temperature. The aqueous layer was extracted with EtOAc (3×), then the organic layer was washed with brine and dried over anhydrous Na2SO4. The organic layer was then concentrated and chromatographed (90:10 hexanes/EtOAc) to give 2g as a mixture of diastereomers (216 mg, 55% yield); 1H NMR (400 MHz, CDCl3) δ 7.40–7.22 (m, 10H), 5.04 (t, J = 7 Hz, 1H), 4.92 (t, J = 7 Hz, 1H), 4.65–4.59 (m, 1H), 4.51–4.44 (m, 1H), 4.21–4.14 (m, 4H), 2.81–2.71 (m, 2H), 2.62–2.52 (m, 2H), 2.42–2.09 (m, 4H), 1.95–1.72 (m, 4H), 1.28 (t, J = 7 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 171.1, 143.3, 142.8, 128.2, 128.2, 127.1, 125.6, 125.4, 81.1, 80.3, 75.9, 75.8, 60.4, 41.0, 40.9, 35.0, 34.2, 32.0, 31.1, 14.1; IR (NaCl) 2977, 1734, and 1188 cm−1; EI-HRMS (m/z) calcd for C14H18O3 ([M]+) 234.1256, found 234.1254.
Ethyl 2-((2S,3R,5S)-3-Methyl-5-phenyltetrahydrofuran-2-yl)acetate (2a).
The title compound was prepared from alcohol 6a following the same procedure that was used for the synthesis of 2g. Purification by flash chromatography (90:10 hexanes/EtOAc) afforded 2a (354 mg, 70% yield) as an oil; dr (11:1); [α]20D +53.3 (c 1.3, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.34–7.22 (m, 5H), 5.03 (t, J = 7 Hz, 1H), 4.22–4.16 (m, 2H), 4.05–4.00 (m, 1H), 2.72–2.61 (m, 2H), 2.09–1.99 (m, 3H), 1.29 (t, J = 7 Hz, 3H), 1.10 (d, J = 6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 171.3, 143.4, 128.1, 126.9, 125.1, 82.5, 79.3, 60.4, 42.6, 39.8, 38.1, 17.1, 14.1; IR (NaCl) 2961, 1736, 1029, and 699 cm−1; ESI-HRMS (m/z) calcd for C15H20O3Na ([M + Na]+) 271.1310, found 271.1303.
Ethyl 2-((2S,3R,5S)-3-Ethyl-5-phenyltetrahydrofuran-2-yl)acetate (2b).
The title compound was prepared from alcohol 6b following the same procedure that was used for the synthesis of 2g. Purification by flash chromatography (90:10 hexanes/EtOAc) afforded 2b (379 mg, 91% yield) as an oil; dr (60:1); [α]20D −39.2 (c 1.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.40–7.22 (m, 5H), 5.55 (t, J = 7 Hz, 1H), 4.23–4.15 (m, 2H), 4.14–4.10 (m, 1H), 2.72–2.62 (m, 2H), 2.10–2.00 (m, 2H), 1.96–1.87 (m, 1H), 1.61–1.53 (m, 1H), 1.44–1.33 (m, 1H), 1.29 (t, J = 7 Hz, 3H), 0.96 (t, J = 7 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 171.2, 143.3, 128.0, 126.9, 125.5, 81.0, 79.6, 60.3, 45.5, 40.3, 40.3, 25.4, 14.0, 12.4; IR (NaCl) 3011, 1736, and 1274 cm−1; CI-HRMS (m/z) calcd for C16H22O3 ([M]+) 262.1569, found 262.1568.
Ethyl 2-((2S,3R,5S)-3-(Cyanomethyl)-5-phenyltetrahydrofuran-2-yl)acetate (2c).
The title compound was prepared from alcohol 6c following the same procedure that was used for the synthesis of 2g. Purification by flash chromatography (80:20 hexanes/EtOAc) afforded 2c (62 mg, 82% yield) as an oil; dr (11:1); [α]20D −36.9 (c 3.1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.36–7.24 (m, 5H), 5.02 (t, J = 7 Hz, 1H), 4.21–4.14 (m, 3H), 2.84–2.40 (m, 6H), 2.29–2.13 (m, 2H), 1.28 (t, J = 7 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 170.5, 141.4, 128.3, 127.5, 125.5, 118.2, 79.8, 79.4, 60.7, 40.2, 39.9, 39.6, 20.4, 14.0; IR (NaCl) 2872, 2400, 1729, and 1240 cm−1; EI-HRMS (m/z) calcd for C16H19NO3 ([M]+) 273.1365, found 273.1369.
Ethyl 2-((2S,3R,5S)-3-Benzyl-5-phenyltetrahydrofuran-2-yl)acetate (2d).
The title compound was prepared from alcohol 6d following the same procedure that was used for the synthesis of 2g. Purification by flash chromatography (90:10 hexanes/EtOAc) afforded 2d (435 mg, 91% yield) as an oil; dr (49:1); [α]20D −32.7 (c 1.3, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.39–7.20 (m, 10H) 5.08 (t, J = 7.2 Hz, 1H), 4.28–4.22 (m, 1H), 4.18 (q, J = 7.1 Hz, 2H), 2.87 (dd, J = 13.7, 6.3 Hz, 1H), 2.76–2.62 (m, 2H), 2.53 (dd, J = 15.1, 4.9 Hz, 1H), 2.40–2.34 (m, 1H), 2.21–2.14 (m, 1H), 2.03–1.96 (m, 1H), 1.29 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 171.1, 143.1, 139.8, 128.7, 128.4, 128.1,127.1, 126.2, 125.5, 81.0, 79.5, 60.5, 45.3, 40.3, 40.3, 38.8, 14.1; IR (NaCl) 3027, 2978, 2928, 1734, 1603, 1494, 1453, 1368, 1304, 1158, and 1029 cm−1; ESI-HRMS (m/z) calcd for C21H24O3 ([M]+) 324.1725, found 324.1724.
Ethyl 2-((2S,3R,5S)-3-(4-Nitrobenzyl)-5-phenyltetrahydrofuran-2-yl)acetate (2e).
The title compound was prepared from alcohol 6e following the same procedure that was used for the synthesis of 2g. Purification by flash chromatography (80:20 hexanes/EtOAc) afforded 2e (344 mg, 70% yield) as an oil; dr (14:1); [α]20D −20.1 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.15 (d, J = 8 Hz, 2H), 7.37–7.17 (m, 7H), 5.06 (t, J = 7 Hz, 1H), 4.24–4.13 (m, 3H), 3.01–2.96 (m, 1H), 2.82–2.77 (m,1H), 2.71–2.65 (m, 1H), 2.58–2.53 (m, 1H), 2.42–2.37 (m, 1H), 2.14–2.08 (m, 1H), 2.04–1.92 (m, 1H), 1.27 (t, J = 7 Hz, 3H); 13C NMR (CDCl3; 100 MHz) δ 170.9, 147.7, 146.5, 142.5, 129.5, 128.2, 127.2, 125.4, 125.0, 123.7, 80.7, 79.4, 60.6, 44.9, 40.0, 38.6, 14.1; IR (NaCl) 2926, 1727, 1517, and 1346 cm−1; EI-HRMS (m/z) calcd for C21H23NO5 ([M]+) 369.1576, found 369.1573.
Ethyl 2-((2S,3R,5S)-3-(But-3-enyl)-5-phenyltetrahydrofuran-2-yl)acetate (2f).
The title compound was prepared from alcohol 6f following the same procedure that was used for the synthesis of 2g. Purification by flash chromatography (80:20 hexanes/EtOAc) afforded 2f (63 mg, 86% yield) as an oil; dr (100:1); [α]20D −51.2 (c 0.7, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.40–7.22 (m, 5H), 5.88–5.74 (m, 1H), 5.05–4.97 (m, 3H), 4.23–4.10 (m, 3H), 2.68–2.65 (m, 2H), 2.15–1.99 (m, 6H), 1.67–1.61 (m, 1H), 1.50–1.45 (m, 1H), 1.30–1.27 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 171.2, 143.2,138.0, 128.1, 127.0, 125.5, 114.8, 81.2, 79.7, 60.5, 43.4, 40.5, 40.3, 32.1, 31.9, 14.1; IR (NaCl) 2898, 1736, 1658, 1549, and 1449 cm−1; EI-HRMS (m/z) calcd for C18H24O3 ([M]+) 288.1725, found 288.1725.
(R,E)-Ethyl 3-Acetoxy-6-phenylhex-5-enoate (3g).
To a stirred suspension of Zn(OTf)2 (15 mg, 0.042 mmol) in toluene (2 mL) under argon was added 2g (197 mg, 0.84 mmol) dissolved in toluene (10 mL). Then Ac2O (1.58 mL, 16.7 mmol) was added and the reaction mixture was refluxed for 4 h until TLC indicated complete consumption of the starting material. The reaction was then cooled to room temperature and saturated aqueous NaHCO3 was added. The aqueous layer was extracted with EtOAc (3×), then the combined organic layers were washed with saturated aqueous NaHCO3 and brine and dried over anhydrous Na2SO4. The organic layer was then concentrated and chromatographed (90:10 hexane/EtOAc) to give 3g (181 mg, 78% yield) as an oil; 1H NMR (400 MHz, CDCl3) δ 7.35–7.20 (m, 5H), 6.45 (d, J = 16 Hz, 1H), 6.17–6.09 (m, 1H), 5.38–5.35 (m, 1H), 4.12 (q, J = 7 Hz, 2H), 2.61 (d, J = 7 Hz, 2H), 2.56–2.53 (m, 2H), 2.03 (s, 3H), 1.24 (t, J = 7 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 170.2, 170.1, 136.9, 133.5, 128.4, 127.3, 126.0, 124.1, 69.7, 60.5, 38.5, 37.5, 21.0, 14.0; IR (NaCl) 2933, 2857, 1739, 1455, 1372, 1237, and 1030 cm−1; CI-HRMS (m/z) calcd for C16H21O4 ([M + H]+) 277.1440, found 277.1442.
(3S,4R,E)-Ethyl 3-Acetoxy-4-methyl-6-phenylhex-5-enoate (3a).
The title compound was prepared from furan 2a following the same procedure that was used for the synthesis of 3g. Purification by flash chromatography (90:10 hexanes/EtOAc) afforded 3a (323 mg, 76% yield) as an oil; [α]20D +27.3 (c 1.9, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.37–7.20 (m, 5H), 6.44 (d, J = 16 Hz, 1H), 6.09 (dd, J = 16, 8 Hz, 1H), 5.36–5.32 (m, 1H), 4.14–4.08 (m, 2H), 2.68–2.56 (m, 3H), 2.04 (s, 3H), 1.23 (t, J = 7 Hz, 3H), 1.12 (d, J = 7 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 170.5, 170.2, 137.0, 131.4, 130.1, 128.4, 127.3, 126.1, 73.0, 60.6, 40.8, 36.9, 20.9, 16.0, 14.0; IR (NaCl) 2949, 2875, 1740, 1371, 1237, 1179, and 1028 cm−1; CI-HRMS (m/z) calcd for C17H22O4 ([M – H]+) 289.1440, found 289.1437.
(3S,4R,E)-Ethyl 3-Acetoxy-4-ethyl-6-phenylhex-5-enoate (3b).
The title compound was prepared from furan 2b following the same procedure that was used for the synthesis of 3g. Purification by flash chromatography (90:10 hexanes/EtOAc) afforded 3b (251 mg, 60% yield) as an oil; [α]20D −19.4 (c 2.7, CHCl3); 1H NMR (CDCl3 400 MHz) δ 7.37–7.19 (m, 5H), 6.41 (d, J = 16 Hz, 1H), 6.00 (dd, J = 16, 9 Hz, 1H), 5.42–5.38 (m, 1H), 4.15–4.05 (m, 2H), 2.63–2.52 (m, 2H), 2.34–2.28 (m, 1H), 2.03 (m, 3H), 1.61–1.55 (m, 1H), 1.40–1.36 (m, 1H) 1.23 (t, J = 7 Hz, 3H), 0.91 (t, J = 7 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 170.4, 170.1, 136.9, 133.1, 128.5, 128.3, 127.2, 126.0, 72.0, 60.4, 48.9, 37.5, 24.0, 20.8, 13.9, 11.7; IR (NaCl) 3097, 1742, 1640, 1370, 1236, and 1028 cm−1; ESI-HRMS (m/z) calcd for C18H24O4 ([M + Na]+) 327.1572, found 327.1569.
(3S,4R,E)-Ethyl 3-Acetoxy-4-(cyanomethyl)-6-phenylhex-5-enoate (3c).
The title compound was prepared from furan 2c following the same procedure that was used for the synthesis of 3g, using 3 equiv of Zn(OTf)2. Purification by flash chromatography (85:15 hexanes/EtOAc) afforded 3c (116 mg, 53% yield) as an oil; [α]20D −24.6 (c 1.1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.41–7.28 (m, 5H), 6.60 (d, J = 16 Hz, 1H), 6.10 (dd, J = 16, 9 Hz, 1H), 5.47–5.43 (m, 1H), 4.20–4.06 (m, 2H), 2.68–2.49 (m, 5H), 2.12 (s, 3H), 1.30–1.17 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 170.2, 169.7, 135.9, 128.6, 128.2, 126.5, 123.5, 117.7, 71.1, 60.9, 43.0, 37.0, 29.6, 20.9, 20.7, 14.0; IR (NaCl) 2876, 2130, 1544, 1370, and 1231 cm−1; CI-HRMS (m/z) calcd for C18H22NO4 ([M + H]+) 316.1549, found 316.1544.
(3S,4R,E)-Ethyl 3-Acetoxy-4-benzyl-6-phenylhex-5-enoate (3d).
To a stirred suspension of Zn(OTf)2 (3 mg, 0.008 mmol), 2d (50 mg, 0.15 mmol), and CH3CN (400 μL, 7.71 mmol) in toluene (3 mL) under argon was added Ac2O (730 μL, 7.71 mmol) then the reaction mixture was refluxed for 4 h. The reaction was then cooled to room temperature and saturated aqueous NaHCO3 was added. The aqueous layer was extracted with EtOAc (3×), then the combined organic layers were washed with saturated solution NaHCO3 brine and dried over Na2SO4. The organic layer was then concentrated and chromatographed (90:10 hexanes/EtOAc) to give 3d, 11a, and 11b (55.8 mg) as a complex mixture. The following mixture was separated by HPLC by using the following conditions: column, YMC-Pack ODS-A 250 × 10 mm, 5 μm coupled to Luna C18 250 × 10 mm, 5 μm; flow rate, 2.75 mL/min; solvents, isocratic 80:20 MeOH:H2O; UV, 210 nm; temperature, 25 °C. The yield was 9 mg (16% yield). [α]20D −102.28 (c 0.4, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.36–7.16 (m, 10H), 6.28 (d, J = 16 Hz, 1H), 6.13 (dd, J = 16, 9.1 Hz, 1H), 5.43 (m, 1H), 4.14 (m, 2H), 2.92 (dd, J = 13, 5.2 Hz, 1H), 2.84–2.78 (m, 1H), 2.76–2.57 (m, 3H), 2.12 (s, 3H), 1.25 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 170.3, 170.2, 139.2, 137.0, 133.6, 129.2, 128.4, 128.2, 127.7, 127.4, 126.2, 126.1, 72.0, 60.6, 48.6, 37.9, 37.5, 29.7, 21.0, 14.1; IR (NaCl) 3026, 2929, 2956, 1741, 1601, 1495, 1453, 1370, 1235, 1179, and 1029 cm−1; ESI-HRMS (m/z) calcd for C23H26O4 ([M + H]+) 367.1909, found 367.1913.
(R)-Ethyl 3-Acetoxy-3-((2S,4S)-4-phenyl-1,2,3,4-tetrahydronaphthalen-2-yl)propanoate (11a).
The title compound was obtained in 47% yield (26 mg); [α]20D −25.7 (c 0.7, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.37–7.04 (m, 8H), 6.79 (d, J = 7.8 Hz, 1H), 5.35 (dt, J = 7.3, 5.8 Hz, 1H), 4.21–4.15 (m, 2H), 4.09 (dd, J = 12.2, 5.5 Hz, 1H), 2.97–2.91 (m, 1H), 2.86–2.78 (m, 1H), 2.72–2.70 (m, 2H), 2.35–2.21 (m, 2H), 2.09 (s, 3H), 1.66 (dd, J = 25.3, 12.8 Hz, 1H), 1.29 (t, J = 7.2 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 170.4, 170.3, 146.4, 139.5, 135.6, 129.4, 128.9, 128.7, 128.5, 126,4, 126.0, 125.9, 73.0, 60.7, 46.8, 38.1, 36.7, 35.6, 32.5, 20.9, 14.1; IR (NaCl) 3061, 1735, 1599, 1493, 1371, 1178, and 1028 cm−1; ESI-HRMS (m/z) calcd for C23H26O4 ([M]+) 366.1831, found 366.1833.
(R)-Ethyl 3-Acetoxy-3-((2S,4R)-4-phenyl-1,2,3,4-tetrahydronaphthalen-2-yl)propanoate (11b).
The title compound was obtained in 20% yield (11 mg); [α]20D −60.9 (c 0.5, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.30–7.12 (m, 6H), 7.02–6.98 (m, 3H), 5.32 (dt, J = 5.6, 3.7 Hz, 1H), 4.37 (t, J = 4.3 Hz, 1H), 4.10–4.03(m, 2H), 2.96 (dd, J = 16.1, 5.1 Hz, 1H), 2.72–2.50 (m, 3H), 2.12–2.06 (m, 1H), 2.04 (s, 3H), 2.04–2.00 (m, 2H), 1.21 (t, J = 7.2 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 170.4, 170.3, 146.7, 137.6, 136.1, 130.3, 129.0, 128.5, 128.1, 126.3, 126.1, 125.9, 72.6, 60.6, 43.7, 37.1, 32.8, 32.7, 31.9, 20.9, 14.0; IR (NaCl) 3059, 1741, 1601, 1493, 1448, 1370, 1236, 1176, and 1028 cm−1; ESI-HRMS (m/z) calcd for C23H26O4 ([M + H]+) 367.1909, found 367.1904.
(3S,4R,E)-Ethyl 3-Acetoxy-4-(4-nitrobenzyl)-6-phenylhex-5-enoate (3e).
The title compound was prepared from furan 2e following the same procedure that was used for the synthesis of 3g. Purification by flash chromatography (90:10 hexanes/EtOAc) afforded 3e in 90% yield; [α]20D −142.8 (c 1.7, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.10 (d, J = 9 Hz, 2H), 7.31–7.21 (m, 7H), 6.22 (d, J = 16 Hz, 1H), 6.06 (dd, J = 16, 9 Hz, 1H), 5.49–5.32 (m, 1H), 4.15–4.04 (m, 2H), 3.05–2.98 (m, 1H), 2.85–2.79 (m, 2H), 2.69–2.55 (m, 2H), 2.11 (s, 3H), 1.22 (t, J = 7 Hz, 3H); 13CNMR (100 MHz, CDCl3) δ 170.1, 170.1, 147.2, 146.4, 136.3, 134.4, 129.9, 128.5, 127.7, 126.3, 126.1, 123.5, 71.8, 60.7, 48.4, 37.9, 37.3, 20.9, 14.0; IR (NaCl) 2918, 1740, 1600, 1345, and 1235 cm−1; ESI-HRMS (m/z) calcd for C23H25NO6Na ([M + Na]+) 434.1580, found 434.1577.
(3S,4R)-Ethyl 3-Acetoxy-4-styryloct-7-enoate (3f).
The title compound was prepared from furan 2f following the same procedure that was used for the synthesis of 3g. Purification by flash chromatography (95:5 hexanes/EtOAc) afforded 3f in 86% yield; dr (50:1); [α]20D −37.8 (c 2.4, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.39–7.24 (m, 5H), 6.41 (d, J = 16 Hz, 1H), 6.00 (dd, J = 16, 9 Hz, 1H), 5.83–5.73 (m, 1H), 5.41–5.37 (m, 1H), 5.03–4.96 (m, 2H), 4.18–4.06 (m, 2H), 2.63–2.52 (m, 2H), 2.51–2.43 (m, 1H), 2.17–2.11 (m, 1H), 2.06–1.98 (m, 4H), 1.65–159 (m, 1H), 1.54–1.45 (m, 1H), 1.24 (t, J = 7 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 170.4, 170.2, 138.0, 136.9, 133.4, 128.4, 128.3, 127.3, 126.1, 114.9, 72.3, 60.5, 46.4, 37.6, 31.2, 30.2, 20.9, 14.0; IR (NaCl) 3039, 2884, 1742, 1597,1449, 1369, and 1234 cm−1; ESI-HRMS (m/z) calcd for C20H27O4 ([M + H]+) 331.1909, found 331.1906.
(2S,3S)-5-Ethyl 1-Methyl 3-acetoxy-2-methylpentanedioate (7a).
To a stirring solution of 3a (113 mg, 0.62 mmol) in a mixture of CH3CN:CCl4:H2O (1:1:1.5, 3.5 mL) was added RuCl3•H2O (13 mg, 0.062 mmol) followed by NaIO4 (663 mg, 3.1 mmol). The resulting solution was allowed to stir overnight; after this time the solvents were evaporated under reduced pressure and saturated aqueous NaHCO3 was added and the mixture was washed with Et2O (2×). The layers were then separated and the aqueous layer was acidified with 1 N HCl to pH 3. The aqueous layer was then extracted with EtOAc (3×) and the combined organic layers were dried on anhydrous Na2SO4 and concentrated under reduced pressure to give a mixture of acids.
To the above crude mixture of acids in ether was added CH2N2 in Et2O until a yellow color persisted. The excess CH2N2 was quenched with AcOH until the solution turned colorless. The solvents were then evaporated under reduced pressure and the residue was purified by column chromatography (1:4 EtOAc/hexanes) to give ester 7a (99 mg, 64% yield over 2 steps) as colorless oil; [α]20D +13.4 (c 1, CHCl3); 1H NMR (CDCl3, 300 MHz) δ 5.37–5.43 (m, 1H), 4.02–4.12 (m, 2H), 3.64 (s, 3H), 2.81–2.90 (m, 1H), 2.50–2.66 (m, 2H), 1.97 (s, 3H), 1.19 (t, J = 7.2 Hz, 3H), 1.12 (d, J = 7.5 Hz, 3H); 13C NMR (CDCl3, 75 MHz) δ 173.0, 169.7, 169.5, 100.9, 70.5, 60.4, 51.6, 42.1, 35.9, 20.5, 13.8, 12.1; IR (NaCl) 1739, 1463, 1373, and 1200 cm−1; ESI-HRMS (m/z) calcd for C11H18 O6 ([M + H]+) 247.1182, found 247.1185.
(2S,3S)-5-Ethyl 1-Methyl 3-acetoxy-2-ethylpentanedioate (7b).
The title compound was obtained from 3b (216 mg, 0.71 mmol) as described for 7a after flash chromatography (1:4 EtOAc/hexanes) as a colorless oil (144 mg, 87% yield over 2 steps); [α]20D +1.4 (c 2.1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.37–5.43 (m, 1H), 4.06–4.12 (m, 2H), 3.65 (s, 3H), 2.63–2.69 (m, 2H), 2.55 (dd, J = 15.8, 7.7 Hz, 1H), 1.98 (s, 3H), 1.51–1.68 (m, 2H), 1.20 (t, J = 7.3 Hz, 3H), 0.86 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 172.7, 169.9, 169.7, 70.0, 60.7, 51.6, 50.3, 36.6, 20.9, 20.7, 14.0, 11.7; IR (NaCl) 1743, 1370, 1233, and 1177 cm−1; ESI-HRMS (m/z) calcd for C12H20O6 ([M + H]+) 261.1338, found 261.1339.
(3S,4S)-Ethyl 3-Acetoxy-4 (benzyloxycarbonylamino)-5-(4-nitrophenyl)pentanoate (13).
To a stirred solution of 3e (50 mg, 0.12 mmol) in DMF (0.6 mL) was added OsO4 in 2.5% tert-butyl alcohol (1 μL) and the mixture was stirred for 5 min. Then Oxone (300 mg, 0.48 mmol) was added and the reaction mixture was stirred for 3 h. After 3 h, Na2SO3 was added and the reaction was stirred until the mixture turned brown, then 1 M HCl was added, the aqueous layer was extracted with EtOAc (3×), and the combined organic layers were washed with 1 M HCl (3×) and brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure to give a mixture of the crude acid and benzoic acid.
To a stirred solution of the crude and benzoic acid (58 mg, 0.12 mmol) in PhMe (2.5 mL) under argon was added Et3N (38 μL, 0.27 mmol) and diphenylphosphoryl azide (59 μL, 0.27 mmol). The resulting mixture was heated at reflux for 3 h and then benzyl alcohol (51 μL, 0.49 mmol) was added. Stirring and refluxing were continued for an additional 12 h. After this period the reaction mixture was cooled to room temperature and concentrated under reduced pressure. The resulting residue was partitioned between EtOAc and saturated aqueous NaHCO3. The organic layer was then washed with brine, dried over anhydrous Na2SO4, concentrated under reduced pressure, and chromatographed (70:30 hexanes/EtOAc) to give protected amine 13 (37 mg, 67% yield over 2 steps) as an oil; [α]20D −47.2 (c 6, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.07 (d, J = 8.6 Hz, 2H), 7.34–7.21 (m, 7H), 5.35–5.31 (m, 1H), 5.10–4.93 (m, 3H), 4.26–4.20 (m, 1H), 4.09 (q, J = 7.1 Hz, 2H), 2.90 (dd, J = 6.2, 14 Hz, 1H), 2.78 (dd, J = 8.6, 13.9 Hz, 1H), 2.62 (t, J = 7.2 Hz, 2H), 2.07 (s, 3H), 1.20 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 169.7, 169.6, 155.8, 146.6, 144.7, 135.9, 129.9, 128.4, 128.2, 128.0, 123.5, 70.5, 66.8, 60.8, 54.0, 38.7, 36.45, 20.7, 13.9; IR (NaCl) 3344, 3034, 3066, 2981, 1740, 1604, 1520, 1347, 1228, and 1045 cm−1; EI-HRMS (m/z) calcd for C23H26N2O8 ([M + H]+) 459.1767, found 459.1765.
(3S,4S)-Ethyl 3-Acetoxy-4-(benzyloxycarbonylamino)hexanoate (12).
The title compound was prepared from styrene 3b following the same procedure that was used for the synthesis of 13. Purification by flash chromatography (80:20 to 70:30 hexanes/EtOAc) afforded protected amine 12 (48 mg, 99% yield over 2 steps) as an oil; [α]20D −22.6 (c 2, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.38–7.29 (m, 5H), 5.37 (dt, J = 8.8, 2.5 Hz, 2H), 5.11 (s, 2H), 4.80 (d, J = 10.1 Hz, 1H), 4.15–4.09 (m, 2H), 3.82–3.74 (m, 1H), 2.62–2.59 (m, 2H), 2.02 (s, 3H), 1.24 (t, J = 7.1 Hz, 3H), 0.95 (t, J = 7.3, 3H); 13C NMR (100 MHz, CDCl3) δ 170.0, 169.8, 156.3, 136.2, 128.5, 128.2, 128.1, 70.9, 66.9, 60.7, 55.0, 36.8, 25.7, 20.7, 14.0, 10.3; IR (NaCl) 3347, 1732, 1531, 1456, 1372, 1229, 1183, 1054, and 1028 cm−1; EI-HRMS (m/z) calcd for C18H25NO6 ([M]+) 351.1682, found 351.1682.
(3R,5S)-3-Hexyl-5-phenyldihydrofuran-2(3H)-one (17).
To a stirring solution of THF (7 mL) and diisopropylamine (0.9 mL, 6.78 mmol) at 0 °C under argon was added n-BuLi (4.1 mL, 1.6 M solution in hexanes, 6.48 mmol) dropwise; the mixture was then stirred for 15 min, HMPA (4 mL) was added dropwise, and the reaction mixture was stirred at 0 °C for 15 min and then cooled to −78 °C. Then lactone 1 (1.0 g, 6.17 mmol) in THF (15 mL) was added dropwise over a 0.5 h period. The reaction mixture was stirred for an additional 0.5 h at −78 °C. Then iodohexane (2.7 mL, 7.41 mmol) was added dropwise and the reaction mixture was stirred for 6 h at −78 °C. The reaction was quenched with saturated aqueous NH4Cl and warmed to room temperature. The aqueous layer was extracted with EtOAc (3×), then the combined organic layers were washed with water, brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was chromatographed on silica gel (95:5 to 90:10 hexanes/EtOAc) to give 17 (1.21 g, 84% yield) as an amorphous solid; [α]20D −11.5 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.40–7.29 (m, 5H), 5.55 (t, J = 6.3 Hz, 1H), 2.64–2.60 (m, 1H), 2.41–2.37 (m, 2H), 1.92–1.85 (m, 1H), 1.58–1.49 (m, 1H), 1.42–1.23 (m, 9H), 0.88 (t, J = 6.9 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 179.3, 139.9, 128.6, 128.1, 124.9, 78.6, 38.8, 36.4, 31.5, 30.4, 28.9, 27.2, 22.5, 14.0; IR (NaCl) 2947, 2855, 1865, 1649, 1454, and 1166 cm−1; CI-HRMS (m/z) calcd for C16H22O2 ([M]+) 246.1620, found 246.1617.
1-((2S,3R,5S)-3-Hexyl-5-phenyltetrahydrofuran-2-yl)tridecan-2-one (16).
To a stirring solution of CH2Cl2 (35 mL) and 17 (1.04 g, 4.22 mmol) at −78 °C under argon was added DIBAL-H (5.10 mL, 1 M solution in dichloromethane, 5.07 mmol) dropwise. The solution was stirred for a 0.5 h at −78 °C and then quenched with a saturated solution of Na–K tartrate and allowed to warm to room temperature. Once the emulsion had separated the aqueous layer was then extracted with CH2Cl2 (3×), then the combined organic layers were washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. Without further purification the crude lactol was used in the next reaction.
To a stirring suspension of THF (5 mL) and Ba(OH)2 (1.07 g, 3.38 mmol) at room temperature under argon was added 18 (9.05 g, 29.6 mmol) dissolved in THF (20 mL) dropwise and the mixture was stirred for 15 min. Then the crude lactol in THF (40 mL) was added dropwise and the mixture was stirred for 72 h. The reaction was quenched with H2O and the aqueous layer was extracted with EtOAc (3×). The combined organic layers were washed with H2O and brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was chromatographed on silica gel (95:5 hexanes/EtOAc) to give 16 (1.5 g, 83% yield over two steps) as an oil; dr (33:1); [α]20D −45.8 (c 1.1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.37–7.22 (m, 5H), 4.97 (t, J = 7 Hz, 1H), 4.08–4.03 (m, 1H), 2.79 (dd, J = 15, 8 Hz, 1H), 2.65 (dd, J = 15, 4 Hz, 1H), 2.54–2.51 (m, 2H), 2.02 (t, J = 7 Hz, 2H), 1.94–1.86 (m, 1H), 1.60 (t, J = 7 Hz, 2H,), 1.50–1.47 (m, 1H), 1.40–1.11 (m, 30H), 0.89 (t, J = 7 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 209.6, 143.5, 128.1, 127.0, 125.5, 125.2, 81.2, 79.5, 48.0, 44.2, 43.8, 40.6, 32.6, 31.8, 31.7, 29.5, 29.4, 29.3, 29.1, 28.1, 23.5, 22.6, 22.5, 14.0, 14.0; IR (NaCl) 2924, 2853, 1715, 1651, and 1457 cm−1; ESI-HRMS (m/z) calcd for C29H48O2Na ([M + Na]+) 451.3552, found 451.3554.
(S)-1-((2S,3R,5S)-3-Hexyl-5-phenyltetrahydrofuran-2-yl)tridecan-2-ol (20).
To a stirring solution of ether (25 mL) and 16 (530 mg, 1.24 mmol) at −40 °C under argon was added LiI (1.66 g, 12.4 mmol). The reaction was stirred for 20 min at −40 °C and then cooled to −78 °C, then LiAlH4 (469 mg, 12.4 mmol) was added in three portions and the reaction was then stirred for 30 min. The reaction was quenched with Na–K tartrate and allowed to warm to room temperature. Once the emulsion had separated the aqueous layer was extracted with EtOAc (3×). The combined organic layers were washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was chromatographed on silica gel (95:5 hexanes/EtOAc) to give 20 (464 mg, 88% yield) as an oil; dr (17:1); [α]20D −52.4(c 1.1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.39–7.22 (m, 5H), 5.01 (t, J = 7 Hz, 1H), 3.91–3.84 (m, 2H), 3.77–3.71 (m, 1H), 2.05–1.96 (m, 2H), 1.89–1.82 (m, 2H), 1.69–1.61 (m, 2H), 1.46–1.37 (m, 3H), 1.10–1.40 (m, 30H), 0.88 (t, J = 7 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 143.3, 128.2, 127.1, 125.5, 86.3, 80.1, 72.1, 44.9, 41.5, 40.6, 37.5, 32.5, 31.9, 31.7, 29.7, 29.6, 29.4, 29.3, 28.2, 25.5, 22.6, 22.5, 14.1, 14.0; IR (NaCl) 3148, 2924, 2853, 1643, 1457, and 1092 cm−1; ESI-HRMS (m/z) calcd for C29H51O2 ([M + H]+) 431.3889, found 431.3892.
(7R,8S,10S)-7-Styrylhenicosane-8,10-diyl Diacetate (21).
To a stirring suspension of Zn(OTf)2 (19.5 mg, 0.054 mmol) in toluene (3 mL) under argon was added furan 20 (462 mg, 1.1 mmol) dissolved in toluene (10 mL). Then Ac2O (2.03 mL, 21.5 mmol) was added and the reaction mixture was refluxed for 4 h. The reaction was quenched with a saturated solution of aqueous NaHCO3. The aqueous layer was extracted with EtOAc (3×), then the combined organic layers were washed with saturated aqueous NaHCO3 (2×) and brine and dried over anhydrous Na2SO4. The organic layer was then concentrated and chromatographed (95:5 hexanes/EtOAc) to give 21 (460 mg, 85% yield) as an oil; [α]20D −19.3 (c 1.1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.42–7.20 (m, 5H), 6.40 (d, J = 16 Hz, 1H), 6.00 (dd, J = 16, 10 Hz, 1H), 5.04–5.00 (m, 1H), 4.95–4.85 (m, 1H), 2.39–2.32 (m, 1H), 2.05 (s, 3H), 2.03 (s, 3H), 1.83–1.79 (m, 2H), 1.52–1.41 (m, 3H), 1.31–1.19 (m, 31H), 0.88 (t, J = 7 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 170.7, 170.6, 137.2, 132.6, 129.4, 128.5, 127.2, 126.1, 73.1, 71.3, 47.0, 36.7, 34.0, 31.8, 31.7, 31.2, 29.5, 29.5, 29.3, 29.3, 29.2, 27.2, 22.6, 22.6, 21.2, 21.1, 14.1, 14.0; IR (NaCl) 2923, 2853, 1737, and 1238 cm−1; ESI-HRMS (m/z) calcd for C33H54O4Na ([M + Na]+) 537.3920, found 537.3921.
(7R,8S,10S)-7-Styryl-10-(triisopropylsilyloxy)henicosan-8-ol (22).
To a stirring solution of CH2Cl2 (12 mL) and diacetate 21 (448 mg, 0.87 mmol) at −78 °C under argon was added DIBAL-H (1.83 mL, 1 M in CH2Cl2, 1.82 mmol) dropwise and then the reaction mixture was stirred for 0.5 h. The reaction was then quenched with Na–K tartrate and allowed to warm to room temperature. Once the emulsion had separated the aqueous layer was extracted with CH2Cl2 (3×) and the combined organic layers were washed with brine and dried over Na2SO4. The organic layer was then concentrated and chromatographed (85:15 hexanes/EtOAc) to give the diol (303 mg, 81% yield) as an oil; [α]20D −7.2 (c 0.9, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.46–7.19 (m, 5H), 6.42 (d, J = 16 Hz, 1H), 6.07 (dd, J = 16, 9 Hz, 1H), 3.84–3.81 (m, 2H), 3.13 (s, 1H), 2.93 (s, 1H), 2.24–2.11 (m, 1H), 1.66–1.36 (m, 6H), 1.33–1.19 (m, 29H), 0.90–0.85 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 137.1, 132.8, 130.3, 128.5, 127.2, 126.1, 75.5, 73.0, 50.5, 40.7, 38.1, 31.8, 31.7, 31.0, 29.6, 29.3, 27.4, 25.4, 22.6, 22.6, 14.1, 14.0; IR (NaCl) 3018, 2925, and 1215 cm−1; ESI-HRMS (m/z) calcd for C29H50O2Na ([M + Na]+) 453.3709, found 453.3706.
To a stirring solution of CH2Cl2 (5 mL) and the diol (138 mg, 0.321 mmol) at −78 °C under argon were added 2,6-lutidine (75 μL, 0.64 mmol) and TIPSOTf (91 μL, 0.34 mmol) dropwise, then the reaction mixture was stirred at −78 °C for 1 h. The reaction was then quenched with H2O and allowed to warm to room temperature, the aqueous layer was extracted with CH2Cl2 (3×), then the combined organic layers were washed with brine and dried over anhydrous Na2SO4. The organic layer was then concentrated and chromatographed (98:2 hexanes/EtOAc) to give 22 (131 mg, 70% yield) as an oil; [α]20D +4.5 (c 1.1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.44–7.19 (m, 5H), 6.39 (d, J = 15 Hz, 1H), 6.15 (dd, J = 15, 9 Hz, 1H), 4.08–4.05 (m, 1H), 3.83–3.80 (m, 1H), 2.99 (s, 1H), 2.17–2.10 (m, 1H), 1.67–1.45 (m, 7H), 1.30–1.02 (m, 33H), 1.19–1.04 (m, 20H), 0.90–0.85 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 137.6, 131.9, 131.0, 128.4, 126.9, 126.1, 73.5, 50.1, 41.0, 37.9, 31.9, 31.8, 31.2, 29.8, 29.6, 29.4, 29.3, 27.5, 24.9, 22.6, 18.3, 18.1, 14.1, 12.9; IR (NaCl) 3018, 2927, and 1215 cm−1; ESI-HRMS (m/z) calcd for C38H71O2Si ([M + H]+) 587.5223, found 587.5228.
(2S,3S,5S)-2-Hexyl-3-hydroxy-5-(triisopropylsilyloxy)hexadecanoic Acid (23).
To a stirring solution of 22 (1.13 g, 1.92 mmol) in acetone:H2O (24 mL:3 mL) were added OsO4 (1.21 mL, 2.5% in tert -butyl alcohol, 0.095 mmol) and NMO (451 mg, 3.84 mmol) at room temperature, then the reaction was stirred for 48 h. Then Na2SO3 (500 mg) was added and once the reaction mixture turned brown H2O (20 mL) was added. The aqueous layer was extracted with EtOAc (3×), then the combined organic layers were washed with H2O and brine, dried over anhydrous Na2SO4, and concentrated under vacuo to give the crude triol.
To a solution of the crude triol in THF:H2O (16 mL:2 mL) was added NaIO4 (617 mg, 2.88 mmol) at room temperature and the reaction mixture was stirred for 24 h. Water was added and the aqueous layer was extracted with EtOAc (3×), then the combined organic layer was washed with brine, dried over anhydrous Na2SO4, and concentrated under vacuo to give the crude aldehyde.
To a solution of the crude aldehyde in tert-butyl alcohol:H2O (10 mL:10 mL) was added NaClO2 (1.74 g, 19.2 mmol), NaH2PO4 (2.65 g, 19.2 mmol), and 2-methyl-2-butene (3 mL, 28.8 mmol) at room temperature and then the reaction was stirred for 12 h. Then 10 mL of H2O was added and the reaction mixture was acidified with 1 M HCl to pH 3. The aqueous layer was extracted with EtOAc (3×) then the combined organic layers were washed with brine and dried over anhydrous Na2SO4. The organic layer was then concentrated and chromatographed (90:10 to 80:20 hexanes/EtOAc) to give acid 23 (507 mg, 50% yield over three steps) as an oil; [α]20D +3.4 (c 1.3, CHCl3); 1H NMR (400 MHz, CDCl3) δ 4.14–4.08 (m, 1H), 4.00–3.97 (m, 1H), 2.42–2.38 (m, 1H), 1.79–1.72 (m, 2H), 1.64–1.52 (m, 4H), 1.36–1.16 (m, 33H), 1.12–1.01 (m, 20H), 0.89–0.82 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 176.8, 74.1, 71.5, 51.7, 40.4, 37.9, 31.8, 31.5, 29.7, 29.5, 29.5, 29.3, 29.0, 27.2, 24.9, 22.6, 22.5, 18.1, 18.0, 14.0, 14.0, 12.9; IR (NaCl) 3565, 2924, 1713, 1463, and 1090 cm−1; ESI-HRMS (m/z) calcd for C31H64O4SiNa ([M + Na]+) 551.4472, found 551.4478.
(3S,4S)-3-Hexyl-4-((S)-2-hydroxytridecyl)oxetan-2-one (24).
To a solution of acid 23 (69 mg, 0.13 mmol) and diisopropylethylamine (57 μL, 0.32 mmol) in THF (2 mL) was added 2,4,6-trichlorobenzoyl chloride (31 μL, 0.19 mmol) at room temperature, then the mixture was stirred for 3 h. Then THF was removed under vacuum and the reaction mixture was dissolved in toluene (2 mL) and added dropwise over a period of 3 h to a solution of DMAP in toluene (2 mL) with stirring for 36 h. The reaction mixture was concentrated in vacuo and purified by column chromatography (98:2 hexanes/EtOAc) to give the lactone (37 mg, 55% yield) as an oil; [α]20D −15.6 (c 0.6, CHCl3); 1H NMR (400 MHz, CDCl3) δ 4.48 (dt, J = 6.2, 4.3 Hz, 1H), 3.99–3.97 (m, 1H), 3.29–3.24 (m, 1H), 2.10–2.04 (m, 1H), 1.99–1.94 (m, 1H), 1.80–1.72 (m, 2H), 1.58–1.54 (m, 4H), 1.37–1.19 (m, 36H), 1.08–0.99 (m, 26H), 0.93–0.86 (m, 6H); 13C NMR (125 MHz, CDCl3) δ 171.8, 74.7, 69.5, 56.7, 40.5, 36.2, 31.8, 31.4, 29.7, 29.6, 29.5, 29.3, 29.0, 27.7, 26.6, 25.1, 22.6, 22.4, 18.1, 18.1, 14.1, 14.0, 12.5; IR (NaCl) 2924, 2855, 1827, 1464, 1116, and 1062 cm−1; ESI-HRMS (m/z) calcd for C31H63O3Si ([M + H]+) 511.4547, found 511.4540.
To a solution of lactone (12 mg, 0.024 mmol) and AcOH (10 μL, 0.19 mmol) in THF (2 mL) at 0 °C under argon was added TBAF (0.26 mL, 1 M in THF, 0.26 mmol) and the resulting mixture was stirred for 4 h at 0 °C. The reaction was quenched with pH 7 buffer and the aqueous layer was extracted with CH2Cl2 (3×) and the combined organic layers were washed with brine and dried over anhydrous Na2SO4. The organic layer was then concentrated and chromatographed (85:15 hexanes/EtOAc) to give β-lactone 24 (5 mg, 62% yield) as an oil; [α]20D −10.4 (c 0.1, CHCl3); 1H NMR (500 MHz, CDCl3) δ 4.48–4.44 (m, 1H), 3.82–3.76 (m, 1H), 3.33–3.29 (m, 1H), 2.05–1.98 (m, 1H), 1.93–1.86 (m, 1H), 1.85–1.79 (m, 1H), 1.78–1.70 (m, 1H), 1.54–1.49 (m, 2H), 1.47–1.38 (m, 2H), 1.35–1.21 (m, 27H), 0.90–0.86 (m, 6H); 13C NMR (125 MHz, CDCl3) δ 171.2, 76.2, 69.3, 56.8, 56.8, 41.2, 37.6, 31.9, 31.5, 29.6, 29.5, 29.3, 28.6, 27.7, 26.8, 25.4, 22.6, 22.5, 14.1, 14.0; IR (NaCl) 3325, 2925, 2854, 1817, 1704,1526, 1467, and 1263 cm−1; CI-HRMS (m/z) calcd for C22H43O3 ([M + H]+) 355.3212, found 355.3207.
(—)-Tetrahydrolipstatin (14).
To a stirred solution of Cbz-Leu-OH 25 (48 mg, 0.18 mmol) in CH2Cl2 (1 mL) at 0 °C under argon was added DCC (38 mg, 0.18 mmol). The reaction mixture was stirred for 20 min at 0 °C, then the solvent was removed under vacuum and the reaction mixture was dissolved in DMF (1 mL). Then the reaction mixture was added dropwise to a solution of β-lactone 24 (32 mg, 0.1 mmol) and DMAP (0.13 mg, 0.01 mmol) in DMF (1 mL) and was stirred for 12 h at room temperature. The reaction was quenched with H2O (3 mL), the aqueous layer was extracted with EtOAc (3×), and the combined organic layers were washed with H2O and brine and dried over anhydrous Na2SO4. The organic layer was then concentrated under vacuum and chromatographed (95:5 to 90:10 hexanes/EtOAc) to give the ester (35 mg, 63% yield) as an oil; [α]20D −20 (c 1.3, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.41–7.28 (m, 5H), 5.17–5.06 (m, 3H), 5.02–4.97 (m, 1H), 4.37–4.26 (m, 2H), 3.22–3.18 (m, 1H), 2.17–2.11 (m, 1H), 1.99–1.94 (m, 1H), 1.77–1.44 (m, 10H), 1.36–1.15 (m, 33H), 1.01–0.94 (m, 6H), 0.89–0.85 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 172.5, 170.8, 155.9, 136.1, 128.5, 128.1, 128.0, 74.5, 72.2, 66.9, 56.9, 52.7, 41.5, 38.6, 34.0, 31.8, 31.5, 31.4, 29.6, 29.5, 29.4, 29.3, 29.2, 29.0, 28.9, 27.6, 26.6, 25.0, 24.7, 24.6, 22.9, 22.8, 22.6, 22.4, 21.6, 14.1, 14.0; IR (NaCl) 3363, 2926, 1824, 1727, 1522, 1467, 1261, and 1048 cm−1; ESI-HRMS (m/z) calcd for C36H59NO6Na ([M + Na]+) 624.4240, found 624.4244.
A stirred solution of the ester (93 mg, 0.15 mmol) in ethanol (1.5 mL) was hydrogenated in the presence of 10% Pd/C (20 mg) at room temperature and atmospheric pressure for 4 h. The suspension was filtered through a short pad of Celite and the filtrate was evaporated under reduced pressure to give the crude amine, which was used without further purification in the next reaction.
A solution of crude amine was stirred in THF (1 mL) then freshly generated formic acetic anhydride was added (0.14 mL, 1.85 mmol) dropwise and the reaction was stirred for 20 min. The reaction was quenched with saturated aqueous NaHCO3, the aqueous layer was extracted with EtOAc (3×), and the combined organic layers were washed with H2O, saturated aqueous NaHCO3, and brine and dried over anhydrous Na2SO4. The organic layer was then concentrated under vacuum and chromatographed (7:3 to 1:1 pentanes/ether) to give (—)-tetrahydrolipstatin (37 mg, 48% yield over two steps) as an oil; [α]20D −32 (c 0.05, CHCl3); 1H NMR (500 MHz, CDCl3) δ 8.22 (s, 1H), 5.91 (d, J = 8.1 Hz, 1H), 5.05–5.0 (m, 1H), 4.71–4.66 (m, 1H), 4.29 (dt, J = 7.3, 4.7 Hz, 1H), 3.22 (dt, J = 7.1, 3.2 Hz, 1H), 2.15–2.10 (m, 1H), 2.08–2.00 (m, 1H), 1.85–1.51 (m, 7H), 1.41–1.15 (m, 26H), 0.96 (t, J = 6.3 Hz, 6H), 0.89 (t, J = 6.5 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 171.9, 170.7, 160.5, 74.7, 72.7, 57.0, 49.6, 41.5, 38.7, 34.0, 31.8, 31.4, 29.6, 29.4, 29.3, 28.9, 27.6, 26.6, 25.8, 25.0, 24.8, 22.8, 22.6, 22.5, 21.7, 14.1, 14.0; IR (NaCl) 3328, 2925, 2854, 1825, 1717, 1708, 1457, 1377, 1258, and 1186 cm−1; ESI-HRMS (m/z) calcd for C29H53NO5Na ([M + Na]+) 518.3821, found 518.3824.
Formic Acetic Anhydride.
To a solution of formic acid 88% (1 mL) at 0 °C was added acetic anhydride (2 mL) dropwise. The reaction was then refluxed at 60 °C for 1 h. The resulting solution was then directly used in the synthesis of 14.
Scheme 3.
Oxidative Cleavage and Synthesis of Statines
Acknowledgment.
Financial support of this work was provided in part by the National Institutes of Health and Purdue University. We thank Mr. David D. Anderson of Purdue University for his help with the HPLC analysis.
Footnotes
Supporting Information Available: Copies of 1H and 13C NMR spectra of selected compounds and HPLC chromatograms for 3d, 11a, and 11b. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1) (a).Staunton J, Weissman KJ. Nat. Prod. Rep. 2001;18:380. doi: 10.1039/a909079g. [DOI] [PubMed] [Google Scholar]; (b) Hoffmann RW. Angew Chem., Int. Ed. 1987;26:489. [Google Scholar]; (c) Heathcock CH, Morrison JD. Asymmetric Synthesis. Vol. 3. Academic Press; New York: 1984. p. 111. [Google Scholar]; (d) Masamune S, Choy W, Peterson JS, Sita LR. Angew. Chem., Int. Ed. 1985;24:1. [Google Scholar]; (e) Franklin AS, Paterson I. Contemp. Org. Synth. 1994:317. [Google Scholar]
- (2) (a).Meyers AI, Yamamoto Y. Tetrahedron. 1984;40:2309. [Google Scholar]; (b) Helmchen G, Leikauf U, Taufer-Knopfel I. Angew. Chem., Int. Ed. 1985;24:874. [Google Scholar]; (c) Gennari C, Bernardi A, Colombo L, Scolastico C. J. Am. Chem. Soc. 1985;107:5812. [Google Scholar]; (d) Masamune S, Sato T, Kim BM, Wollman TA. J. Am. Chem. Soc. 1986;108:8279. [Google Scholar]; (e) Corey EJ, Kim SS. J. Am. Chem. Soc. 1990;112:4976. [Google Scholar]; (f) Meyers AG, Widdowson KL. J. Am. Chem. Soc. 1990;112:9672. [Google Scholar]; (g) Walker MA, Heathcock CH. J. Org. Chem. 1991;56:5747. [Google Scholar]; (h) Braun M, Sacha H. Angew. Chem., Int. Ed. 1991;30:1318. [Google Scholar]; (i) Oppolzer W, Lienard P. Tetrahedron Lett. 1993;34:4321. [Google Scholar]; (j) Paterson I, Wren SP. J. Chem. Soc., Chem. Commun. 1993:1790. [Google Scholar]; (k) Abiko A, Liu J-F, Masamune S. J. Am. Chem. Soc. 1997;119:2586. [Google Scholar]; (l) Ghosh AK, Onishi M. J. Am. Chem. Soc. 1996;118:2527. doi: 10.1021/ja9539148. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Ghosh AK, Fidanze S, Onishi M, Hussain KA. Tetrahedron Lett. 1997;38:7171. doi: 10.1016/S0040-4039(97)01765-6. [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Kurosu M, Lorca M. J. Org. Chem. 2001;66:1209. doi: 10.1021/jo001293h. [DOI] [PubMed] [Google Scholar]; (o) Evans DA, Tedrow JS, Shaw JT, Downey CW. J. Am. Chem. Soc. 2002;124:392. doi: 10.1021/ja0119548. [DOI] [PubMed] [Google Scholar]; (p) Denmark SE, Wynn T, Beutner GL. J. Am. Chem. Soc. 2002;124:13405. doi: 10.1021/ja0282947. [DOI] [PubMed] [Google Scholar]; (q) Ghosh AK, Kim J-H. Org. Lett. 2003;5:1063. doi: 10.1021/ol034086n. [DOI] [PubMed] [Google Scholar]; (r) Crimmins MT, McDougall PJ. Org. Lett. 2003;5:591. doi: 10.1021/ol034001i. [DOI] [PubMed] [Google Scholar]; (s) Fanjul S, Hulme AN, White JW. Org. Lett. 2006;8:4219. doi: 10.1021/ol0614774. [DOI] [PubMed] [Google Scholar]; (t) Gnas Y, Glorius F. Synthesis. 2006:1899. [Google Scholar]
- (3) (a).Jung ME, D’Amico DC. J. Am. Chem. Soc. 1993;115:12208. [Google Scholar]; (b) Jung ME, D’Amico DC. J. Am. Chem. Soc. 1995;117:7379. [Google Scholar]; (c) Jung ME, D’Amico DC. J. Am. Chem. Soc. 1997;119:12150. [Google Scholar]; (d) Jung ME, Marquez R. Tetrahedron Lett. 1999;40:3129. [Google Scholar]; (e) Jung ME, Lee WS, Sun D. Org. Lett. 1999;1:307. doi: 10.1021/ol990619+. [DOI] [PubMed] [Google Scholar]; (f) Jung ME, Sun D. Tetrahedron Lett. 1999;40:8343. [Google Scholar]; (g) Jung ME, Marquez R. Org. Lett. 2000;2:1669. doi: 10.1021/ol005675l. [DOI] [PubMed] [Google Scholar]; (h) Jung ME, Lee CP. Tetrahedron Lett. 2000;41:9719. [Google Scholar]; (i) Jung ME, Lee CP. Org. Lett. 2001;3:333. doi: 10.1021/ol000329p. [DOI] [PubMed] [Google Scholar]; (j) Jung ME, van den Heuvel A. Tetrahedron Lett. 2002;43:8169. [Google Scholar]; (k) Jung ME, Hoffmann B, Rausch B, Contreras JM. Org. Lett. 2003;5:3159. doi: 10.1021/ol035295a. [DOI] [PubMed] [Google Scholar]; (l) Jung ME, van den Heuvel A, Leach AG, Houk KN. Org. Lett. 2003;5:3375. doi: 10.1021/ol0350801. [DOI] [PubMed] [Google Scholar]; (m) Jung ME, van den Heuvel A. Org. Lett. 2003;5:4705. doi: 10.1021/ol0358760. [DOI] [PubMed] [Google Scholar]; (n) Jung ME, Yoo D. Org. Lett. 2007;9:3543. doi: 10.1021/ol0714038. [DOI] [PubMed] [Google Scholar]; (o) Jung ME, Zhang T. Org. Lett. 2008;10:137. doi: 10.1021/ol702729u. [DOI] [PubMed] [Google Scholar]; (p) Jung ME, Yoo D. Tetrahedron Lett. 2008;49:816. doi: 10.1016/j.tetlet.2008.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]; (q) Jung ME, Allen DA. Org. Lett. 2008;10:2039. doi: 10.1021/ol800423m. [DOI] [PubMed] [Google Scholar]
- (4).Ghosh AK, Swanson L. J. Org. Chem. 2003;68:9823. doi: 10.1021/jo035077v. [DOI] [PubMed] [Google Scholar]
- (5).Corey EJ, Bakshi RK, Shibata S, Chen C, Singh VK. J. Am. Chem. Soc. 1987;109:7925. [Google Scholar]
- (6) (a).Hori N, Nagasawa K, Shimizu T, Nakata T. Tetrahedron Lett. 1999;40:2145. [Google Scholar]; (b) Nakata T, Nomura S, Matsukura H. Tetrahedron Lett. 1996;37:213. [Google Scholar]
- (7).Travis RB, Narayan RS, Borhan B. J. Am. Chem. Soc. 2002;124:3824. doi: 10.1021/ja017295g. [DOI] [PubMed] [Google Scholar]
- (8).Ghosh AK, Liu W. J. Org. Chem. 1996;61:6175. doi: 10.1021/jo960670g. [DOI] [PubMed] [Google Scholar]
- (9) (a).Baxter EW, Reitz AB. Annu. Rep. Med. Chem. 2005;40:35. [Google Scholar]; (b) Baeck M, Nyhlen J, Kvarnstroem I, Appelgren S, Borkakoti N, Jansson K, Lindberg J, Nystroem S, Hallberg A, Rosenquist A, Samuelsson B. Bioorg. Med. Chem. 2008;16:9471. doi: 10.1016/j.bmc.2008.09.041. [DOI] [PubMed] [Google Scholar]; (c) Barazza A, Gotz M, Cadamuro SA, Goettig P, Willem M, Steuber H, Kohler T, Jestel A, Reinemer P, Renner C, Bode W, Moroder L. ChemBioChem. 2007;8:2078. doi: 10.1002/cbic.200700383. [DOI] [PubMed] [Google Scholar]
- (10) (a).Billich A, Fricker G, Mueller I, Donatsch P, Ettmayer P, Gstach H, Lehr P, Peichl P, Scholz D, Rosenwirth B. Antimicrob. Agents Chemother. 1995;39:1406. doi: 10.1128/aac.39.7.1406. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sakurai M, Sugano M, Handa H, Komai T, Yagi R, Nishigaki T, Yabe Y. Chem. Pharm. Bull. 1993;41:1369. doi: 10.1248/cpb.41.1369. [DOI] [PubMed] [Google Scholar]
- (11) (a).Jones MD, Sueiras-Diaz J, Szelke M, Leckie BJ, Beattie SR, Morton J, Neidle S, Kuroda R. J. Pept. Res. 1997;50:109. doi: 10.1111/j.1399-3011.1997.tb01176.x. [DOI] [PubMed] [Google Scholar]; (b) Hom RK, Fang LY, Mamo S, Tung JS, Guinn AC, Walker DE, Davis DL, Gailunas AF, Thorsett ED, Sinha S, Knops JE, Jewett NE, Anderson JP, Varghese J. J. Med. Chem. 2003;46:1799. doi: 10.1021/jm025619l. [DOI] [PubMed] [Google Scholar]
- (12).Weibel EK, Hadvary P, Hochuli E, Kupfer E, Lengsfeld H. J. Antibiot. 1987;40:1081. doi: 10.7164/antibiotics.40.1081. [DOI] [PubMed] [Google Scholar]
- (13).Hochuli E, Kupfer E, Maurer R, Meister W, Mercadel Y, Schmidt K. J. Antibiot. 1987;40:1086. doi: 10.7164/antibiotics.40.1086. [DOI] [PubMed] [Google Scholar]
- (14) (a).Richardson RD, Yatsandra Oyola G, Zancanella M, Knowles LM, Cieplak P, Romo D, Jeffrey W, Smith JW. J. Med. Chem. 2008;51:5285. doi: 10.1021/jm800321h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ortar G, Bisogno T, Ligresti A, Morera E, Nalli M, Di Marzo V. J. Med. Chem. 2008;51:6970. doi: 10.1021/jm800978m. and references cited therein. [DOI] [PubMed] [Google Scholar]
- (15) (a).Kumaraswamy G, Markondaiah B. Tetraherdon Lett. 2008;49:327. [Google Scholar]; (b) Yadav JS, Rao KV, Prasad AR. Synthesis. 2006:3888. [Google Scholar]; (c) Polkowska J, Lukaszewicz E, Kiegiel J, Jurczak J. Tetrahedron Lett. 2004;45:3873. [Google Scholar]; (d) Yadav JS, Vishweahwar Rao K, Sridhar Reddy M, Prasad AR. Tetrahedron Lett. 2006;47:4393. [Google Scholar]; (e) Yadav J, Sridhar Reddy M, Prasad AR. Tetrahedron Lett. 2006;47:4995. [Google Scholar]; (f) Thadani AN, Batey RA. Tetrahedron Lett. 2003;44:8051. [Google Scholar]; (g) Bodkin JA, Humphries EJ, McLeod MD. Aust. J. Chem. 2003;56:795. [Google Scholar]; (h) Bodkin JA, Humphries EJ, McLeod MD. Tetrahedron Lett. 2003;44:2869. [Google Scholar]; (i) Ghosh AK, Fidanze S. Org. Lett. 2000;2:2405. doi: 10.1021/ol000070a. [DOI] [PubMed] [Google Scholar]; (j) Dirat O, Kouklovsky C, Langlois Y. Org. Lett. 1999;1:753. doi: 10.1021/ol990734k. [DOI] [PubMed] [Google Scholar]; (k) Ghosh AK, Liu C. Chem. Commun. 1999:1743. doi: 10.1039/A904533C. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Paterson I, Doughty VA. Tetrahedron Lett. 1999;40:393. [Google Scholar]; (m) Fleming I, Lawrence NJ. J. Chem. Soc., Perkin Trans. 1998;1:2679. [Google Scholar]; (n) Giese B, Roth MJ. J. Braz. Chem. Soc. 1996;7:243. [Google Scholar]; (o) Pommier A, Pons JM, Kocienski PJ, Wong L. Synthesis. 1994:1294. [Google Scholar]; (p) Hanessian S, Tehim A, Chen P. J. Org. Chem. 1993;58:7768. [Google Scholar]; (q) Case-Green SC, Davies SG, Hedgecock CJR. Synlett. 1991:781. [Google Scholar]; (r) Chadha NK, Batcho AD, Tang PC, Courtney LF, Cook CM, Wovkulich PM, Uskokovic MR. J. Org. Chem. 1991;56:4714. [Google Scholar]; (s) Fleming I, Lawrence NJ. Tetrahedron Lett. 1990;31:3645. [Google Scholar]; (t) Pons J-M, Kocienski P. Tetrahedron Lett. 1989;30:1833. [Google Scholar]; (u) Barbier P, Schneider F. J. Org. Chem. 1988;53:1218. [Google Scholar]; (v) Barbier P, Schneider F, Widmer U. Helv. Chim. Acta. 1987;70:1412. [Google Scholar]; (w) Barbier P, Schneider F. Helv. Chim. Acta. 1987;70:196. [Google Scholar]
- (16) (a).Grimes KD, Lu Y-J, Zhang Y-M, Luna VA, Hurdle JG, Carson EI, Qi J, Kudrimoti S, Rock CO, Lee RE. ChemMedChem. 2008;3:1936. doi: 10.1002/cmdc.200. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gurjar MK Pramanik C Bhattasali D Ramana CV Mohapatra DK J. Org. Chem 2007. 72 6591–6594 [DOI] [PubMed] [Google Scholar]
- (17).Paterson I, Yeung KS, Smaill JB. Synlett. 1993:774. [Google Scholar]
- (18) (a).Ghosh AK, Lei H. J. Org. Chem. 2002;67:8783. doi: 10.1021/jo020402k. [DOI] [PubMed] [Google Scholar]; (b) Mori Y, Kuhara M, Takeuchi A, Suzuki M. Tetrahedron Lett. 1988;29:5419. [Google Scholar]; (c) Mori Y, Takeuchi A, Kageyama H, Suzuki M. Tetrahedron Lett. 1988;29:5423. [Google Scholar]
- (19).Adam W, Baeza J, Liu J-C. J. Am. Chem. Soc. 1972;94:2000. [Google Scholar]
- (20).Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull. Chem. Soc. Jpn. 1979;52:1989. [Google Scholar]
- (21).Chadha NK, Batcho AD, Tang PC, Courtney LF, Cook CM, Wovkulich PM, Uskokovic MR. J. Org. Chem. 1991;56:4714. [Google Scholar]
- (22).Frelek J, Fryszkowska A, Kwit M, Ostaszewski R. Tetrahedron: Asymmetry. 2006;17:2469. [Google Scholar]