

Abstract
Introduction
Comprehensive genetic testing for dystrophinopathy can detect ~95% of pathogenic variants in DMD and is often the preferred diagnostic approach.
Methods
We reviewed pathology reports for muscle biopsies evaluated at the University of Iowa with a pathological diagnosis of dystrophinopathy based on dystrophic histopathology and abnormal immunofluorescence staining: reduced to absent dystrophin, expression of utrophin, and loss of nNOS.
Results
The percentage of muscle biopsies with dystrophinopathy has been stable since 1997. Of 2298 biopsies evaluated between 2011 and 2016, 72 (3.1%) had pathologic features of dystrophinopathy. Median age at biopsy was 8 years (range 0.66–84). Half had undergone DMD genetic testing prior to biopsy. Clinical phenotypes recorded on requisitions were typical of muscular dystrophy for 57 (79%) biopsies.
Discussion
Muscle biopsy continues to play an important role in the diagnosis of dystrophinopathy, particularly in patients with later symptom onset, comorbidities, or normal DMD genetic testing.
Keywords: Duchenne-Becker muscular dystrophy, dystrophinopathy, muscle biopsy, pathology, diagnosis, indication
Introduction
Dystrophinopathy (Duchenne-Becker muscular dystrophy) is an X-linked recessive disorder with a prevalence of 1.38 per 10,000 males ages 5–24 in the United States.1 Patients with Duchenne muscular dystrophy (DMD) typically present between the ages of 1.2 and8 years, with muscle weakness and delayed motor development.2 In contrast, the mean age of onset in Becker muscular dystrophy (BMD) is 12 years, and patients with BMD have slower progression.3
The preferred diagnostic approach to a patient with probable dystrophinopathy is genetic testing followed by muscle biopsy if genetic testing is normal.4 Deletion/duplication testing and sequencing of DMD can detect 95–98% of pathogenic DMD variants.5, 6 The University of Iowa (UIHC) is a referral center for muscle biopsy evaluation. We evaluated how the role of muscle biopsy in dystrophinopathy has changed with the improvements in and availability of genetic testing.
Materials/Methods
This study was approved by the University of Iowa Institutional Review Board. The total number of muscle biopsies (male and female) evaluated at UIHC and number of biopsies diagnosed with dystrophinopathy was determined by searching the past 20 years of anatomic pathology cases in the laboratory information system. Complete pathology reports issued for dystrophinopathy cases between 2011 and 2016 were reviewed to verify there was dystrophic histopathology and an abnormal pattern of immunofluorescence staining characteristic of a dystrophinopathy: reduced to absent dystrophin, expression of utrophin, and loss of nNOS. Prior to 2005, the anti-dystrophin (NCL-DYS1, NCL-DYS2, and NCL-DYS3) mouse monoclonal antibodies were from Novocastra/Leica Biosystems. Beginning around 2005, the expression of dystrophin was initially evaluated with four antibodies: the rabbit polyclonal anti-C-terminus antibody ab15277 (Abcam) and mouse monoclonal anti-dystrophin antibodies [Developmental Studies Hybridoma Bank (DSHB), The University of Iowa] directed at the rod domain exon 50 (MANEX50), rod domain exon 46 (MANEX46B), and amino terminus exons 7/8 (MANEX7B). Additional anti-dystrophin antibodies directed at exons 1, 8, 10–12, 20/21, 27, 31/32, 38/39, 43, 45, 47, 48, and 48–50 (all from DSHB) were utilized as needed to better characterize dystrophin expression. Anti-utrophin (NCL-DRP2) and anti-nNOS (NCL-NOS1) antibodies were purchased from Novocastra/Leica Biosystems. Data was abstracted from these reports, including the indication(s) for biopsy, referring physician, clinical information, and genetic testing results. For each of the last 20 years, we determined the percent of total biopsies with a pathological diagnosis of a dystrophinopathy together with 95% confidence intervals.
We grouped biopsies based on whether DMD genetic testing was done prior to biopsy, and subdivided groups by clinical information abstracted from the reports. Typical DMD presentation was classified as age at biopsy (presumed to be near the time of presentation to a neurologist) between 1.2–8 years2 with weakness, delayed motor development, elevated CK, and/or hypertrophic calves.2, 3 We attempted to supplement the biopsy report-derived information by contacting referring physicians by mail. Of the 55 requests for information, 3 responded. Their responses are included in our results.
Results
20-year trend
The University of Iowa evaluated 5999 muscle biopsies (56% male) from 1997–2016, and 247 (4.1%) of those were diagnosed with dystrophinopathy by muscle pathology and immunofluorescence. The absolute number of muscle biopsies diagnosed annually with dystrophinopathy has been relatively stable (Figure 1a), and the annual percentage of dystrophinopathy biopsies has generally been between 2–5%. Confidence intervals overlap with the exception of the year 2000 (12.1%) (Figure 1b).
Figure 1.
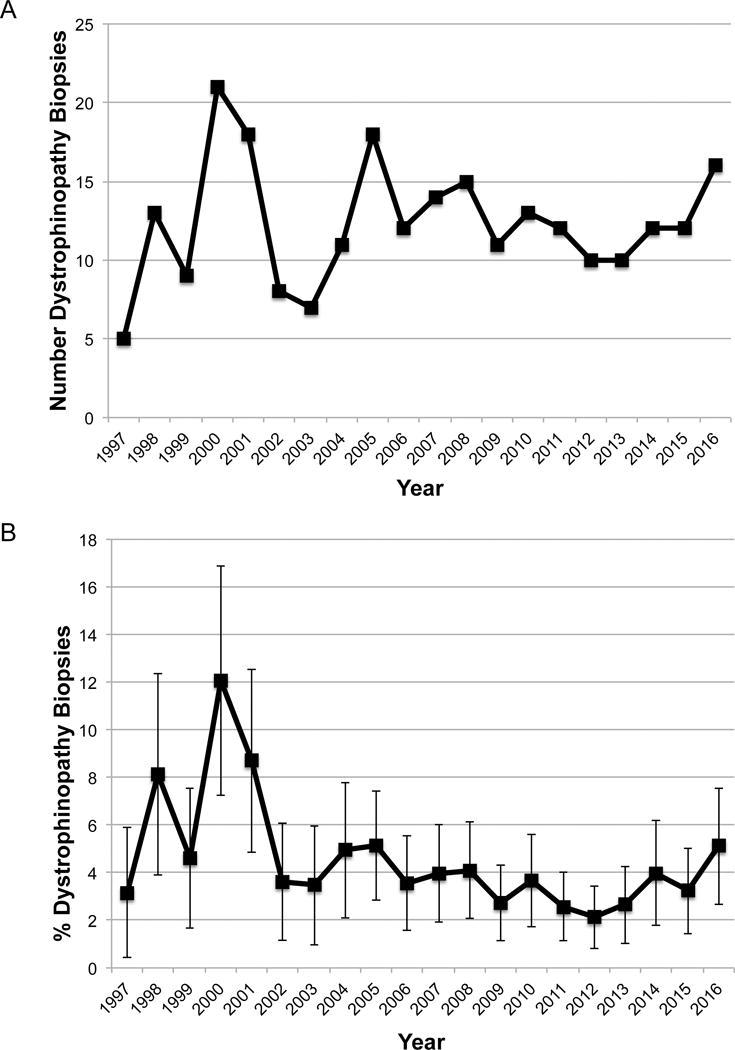
A: The number of dystrophinopathy biopsies evaluated at UIHC per year since 1997. B: The percentage of dystrophinopathy muscle biopsies evaluated at UIHC per year since 1997, with error bars representing 95% confidence intervals.
2011–2016 Cohort
There were 2298 muscle biopsies evaluated at UIHC from 2011–2016, and 72 (3.1%) had pathologic features of dystrophinopathy. Of these, half had DMD genetic testing prior to biopsy. Four biopsied patients had non-DMD genetic testing prior to biopsy (CAPN3 sequencing, LAMA2 sequencing, myotonic dystrophy panel, and limb-girdle muscular dystrophy (LGMD) panel without including DMD). Eleven biopsied patients had normal DMD genetic testing prior to biopsy. In 25 patients, a DMD variant was identified prior to biopsy. See Figure 2a for details.
Figure 2.
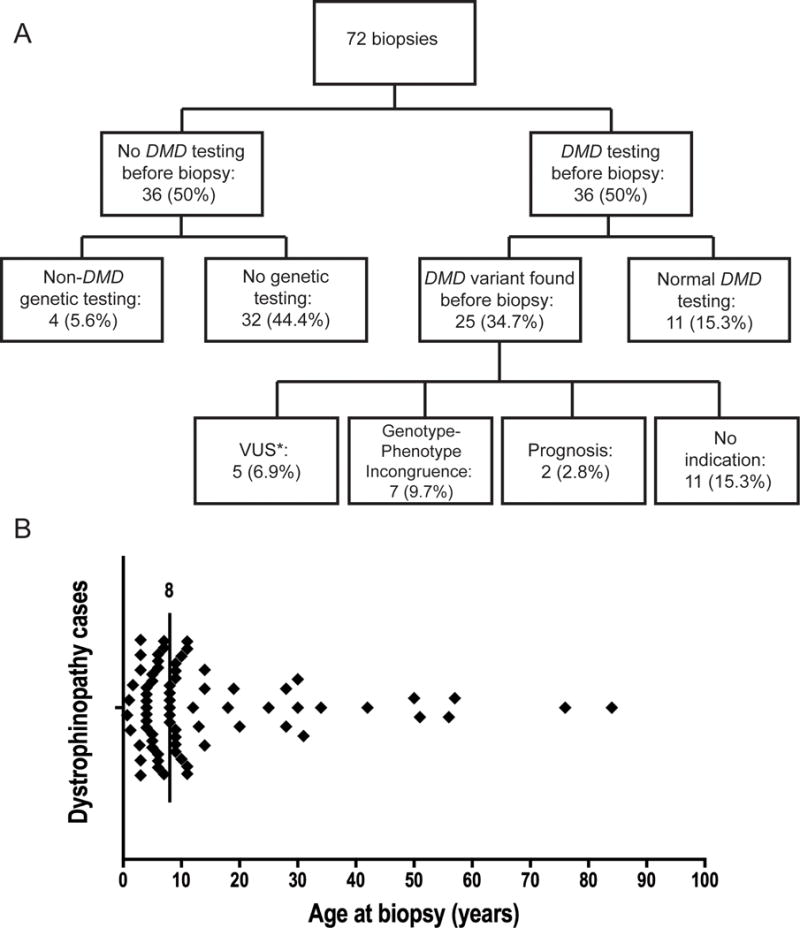
A. Distribution of 72 dystrophinopathy biopsies diagnosed from 2011–2016. B. Distribution of the 72 patients’ ages at biopsy. The vertical line is drawn at the median age at biopsy: 8 years.
*see supplemental table
DMD: dystrophin gene; VUS: variant of unknown significance
Clinical information was available in 70 (97%) dystrophinopathy pathology reports from 2011–2016. Fifty-seven (79%) had a clinical phenotype reported that suggested dystrophinopathy. Of these, 32 reports had a DMD phenotype; 22 (69%) had DMD genetic testing prior to biopsy. The other 25 reports described a later onset presentation of typical muscular dystrophy; 12 (48%) had DMD genetic testing prior to biopsy. The median age of biopsy was 8 years (range 8 months-84 years), and 16 (22%) biopsies were done when the patient was > 18 years (Figure 2b). Clinical features that are not typical of dystrophinopathy based on sex or comorbid conditions were reported for 13 biopsied patients, summarized in Table 1. Neuropathy was the most common comorbid condition in this series.
Table 1.
Summary of atypical presentations of dystrophinopathy
| Age at biopsy (years) | Sex | Genes tested | Variant(s) found | Comorbidities | Symptoms reported |
|---|---|---|---|---|---|
| 3 | F | CAPN3 | None | – | Elevated CK, constipation, elevated “liver enzymes” |
| 9 | F | – | – | – | Elevated CK, family history MD, calf hypertrophy, muscle weakness, + Gowers’ maneuver |
| 56 | F | – | – | Peripheral neuropathy | Family history MD, myalgia, mildly elevated CK |
| 0.67 | M | – | – | Feeding disorder | Elevated CK, delayed motor development |
| 1.25 | M | LAMA2 | None | Delayed myelination on MRI | Global developmental delay, FTT, muscle weakness, elevated CK |
| 1 | M | – | – | Elevated CK discovered during evaluation of GI illness | – |
| 9 | M | – | – | Non-ambulatory since age 5 | Muscle weakness |
| 9 | M | – | – | Cerebral palsy, peripheral neuropathy | Elevated CK, global developmental delay, muscle weakness |
| 13 | M | Congenital muscular dystrophy panel, with DMD testing | Xp22.31 duplication, paternally inherited COL6A variant, and an RYR2 variant | Microcephaly and congenital heart defects | Hypotonia, normal CK |
| 25 | M | LGMD panel | None | Scapular winging | Muscle weakness, calf hypertrophy, abnormal gait |
| 30 | M | – | – | Neuropathy | Clinical childhood MD diagnosis with new symptoms (cardiomyopathy, neuropathy, asthma) |
| 57 | M | – | – | – | Fatigue and muscle cramping |
| 76 | M | – | – | Difficulty swallowing, neuropathy | Elevated CK, muscle weakness, abnormal gait |
F= female, M= male, CK= creatine kinase, MD= muscular dystrophy, LGMD= limb-girdle muscular dystrophy, FTT= failure to thrive, GI= gastrointestinal, “-“ indicates no information provided
DMD genetic testing results were received after the biopsy for 8 patients: 3 were pathological DMD variants, 1 was a DMD variant of unknown significance (see supplemental table), and DMD intronic variants were identified by RNA analysis for the remaining four.
Referring health professionals named on requisitions were predominantly neurologists and pathologists. With rare exceptions, referral biopsies did not have immunohistochemistry done locally; biopsies were sent to UIHC for this testing.
Discussion
Our results show that muscle biopsy continues to be a part of patient diagnosis and care for a subset of individuals with dystrophinopathy. The annual absolute number and percentage of muscle biopsies evaluated at the University of Iowa with a pathological diagnosis of dystrophinopathy has not changed substantially over the past 20 years, despite the advances in genetic testing. From 2011–2016, over two-thirds of the biopsy reports suggesting a typical DMD presentation also reported DMD testing prior to biopsy. However, less than half of those reporting a later onset of typical muscular dystrophy had DMD testing prior to biopsy, suggesting dystrophinopathy was not the suspected diagnosis. This is consistent with the broader range of diagnostic possibilities outside of early childhood.7 Nearly 25% of patients with a dystrophinopathy pathologic diagnosis were older than 18 years when biopsied. Dystrophin abnormalities have previously been reported in 17% of all subjects and 31% of male subjects with a limb-girdle muscular dystrophy (LGMD) presentation.8 The clinical findings in these older individuals often do not allow distinction between BMD and LGMD, particularly if there is no known family history of muscular dystrophy. According to the LGMD practice parameter, muscle biopsy is the preferred diagnostic approach in this population.7 However with the increasing availability of next generation sequencing panels, this diagnostic approach might be in evolution.
Muscle biopsy was sometimes requested to provide prognostic information or to understand the phenotype by quantifying dystrophin in muscle. For example, a referral biopsy case had an out-of-frame DMD deletion so was expected to have a DMD phenotype,9–11 but his clinical progression was slower than expected. Biopsy was done to reconcile these findings. As genotype-phenotype relationships and the role of modifier genes are better defined,6, 12–14 we predict that fewer dystrophinopathy biopsies will be ordered for prognostic information.
Finally, 2–5% of individuals with dystrophinopathy have DMD mutations that cannot be detected by deletion/duplication analysis or sequencing.5, 6 Eleven (15%) biopsies in our series had normal DMD testing prior to muscle biopsy, and 5 subjects had a variant of unknown significance in DMD prior to biopsy. Muscle biopsies are still required for the diagnosis of dystrophinopathy in these patients both to determine dystrophin expression and to allow research-based analysis of RNA for identification of mutations in noncoding regions. Establishing a genetic diagnosis for patients with dystrophinopathy remains important for genetic counseling and patient management, particularly in the era of emerging genetic therapies for dystrophinopathy.6, 9
Some patients with typical presentation of DMD had a muscle biopsy prior to any genetic testing. We hypothesize that barriers to genetic testing, such as cost or insurance restrictions on genetic testing might explain some of these biopsies. In others, physicians might simply prefer to do the biopsy to guide genetic testing, particularly if the clinician is less comfortable with ordering and interpreting genetic tests.
Limitations of our study include sparse clinical history and lack of a detailed rationale for performing the biopsy available for some muscle biopsies. Biopsies are often referred by a pathologist, and requisitions are completed by the pathologist or support personnel. The brevity of clinical summaries may well be the result of this practice. We had little success in contacting the referring clinicians for additional information.
While the preferred diagnostic approach to a patient with probable dystrophinopathy is genetic testing followed by muscle biopsy if genetic testing is normal,4 the UIHC experience reported here indicates that muscle biopsies still play a role in diagnosis or management of dystrophinopathy. Muscle pathologists should use continuing vigilance for dystrophinopathy in their assessment of biopsies. Immunostaining for dystrophin in dystrophic-appearing biopsies from male patients of any age will often pay diagnostic dividends.
Supplementary Material
Acknowledgments
This study was supported by the Paul D. Wellstone Muscular Dystrophy Cooperative Research Center (MDCRC) grant from the National Institutes of Health (Bethesda, MD) (NIH U54 NS053672). We appreciate the discussions with Amber Gedlinske and Kristin Conway.
Author Katherine Mathews has received research is support by NIH (NINDS), CDC, and FARA. In addition, she is a site PI for industry sponsored trials: Sarepta Therapeutics, Horizon Therapeutics, Pfizer, FibroGen, PTC. She has received meals, and/or reimbursements, or honoraria to the University of Iowa within the last year from the following: Sarepta Therapeutics, Marathon, BMS, Genzyme/Sanofi. She has no personal financial interest in any of the above companies.
Abbreviations
- BMD
Becker muscular dystrophy
- CK
creatine kinase
- DMD
Duchenne muscular dystrophy
- LGMD
limb-girdle muscular dystrophy
- UIHC
University of Iowa Hospitals and Clinics
Footnotes
This work was presented at the American Academy of Neurology annual meeting, Boston, April 23, 2017.
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
The remaining authors have no conflicts of interest.
References
- 1.Romitti PA, Zhu Y, Puzhankara S, James KA, Nabukera SK, Zamba GK, et al. Prevalence of Duchenne and Becker muscular dystrophies in the United States. Pediatrics. 2015;135(3):513–521. doi: 10.1542/peds.2014-2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ciafaloni E, Fox DJ, Pandya S, Westfield CP, Puzhankara S, Romitti PA, et al. Delayed diagnosis in duchenne muscular dystrophy: data from the Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet) The Journal of pediatrics. 2009;155(3):380–385. doi: 10.1016/j.jpeds.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chelly J, Desguerre I. Progressive muscular dystrophies. Handbook of clinical neurology. 2013;113:1343–1366. doi: 10.1016/B978-0-444-59565-2.00006-X. [DOI] [PubMed] [Google Scholar]
- 4.Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9(1):77–93. doi: 10.1016/S1474-4422(09)70271-6. [DOI] [PubMed] [Google Scholar]
- 5.Stockley TL, Akber S, Bulgin N, Ray PN. Strategy for comprehensive molecular testing for Duchenne and Becker muscular dystrophies. Genetic testing. 2006;10(4):229–243. doi: 10.1089/gte.2006.10.229. [DOI] [PubMed] [Google Scholar]
- 6.Flanigan KM, Dunn DM, von Niederhausern A, Soltanzadeh P, Gappmaier E, Howard MT, et al. Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Human mutation. 2009;30(12):1657–1666. doi: 10.1002/humu.21114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Narayanaswami P, Weiss M, Selcen D, David W, Raynor E, Carter G, et al. Evidence-based guideline summary: diagnosis and treatment of limb-girdle and distal dystrophies: report of the guideline development subcommittee of the American Academy of Neurology and the practice issues review panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology. 2014;83(16):1453–1463. doi: 10.1212/WNL.0000000000000892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arikawa E, Hoffman EP, Kaido M, Nonaka I, Sugita H, Arahata K. The frequency of patients with dystrophin abnormalities in a limb-girdle patient population. Neurology. 1991;41(9):1491–1496. doi: 10.1212/wnl.41.9.1491. [DOI] [PubMed] [Google Scholar]
- 9.Aartsma-Rus A, Ginjaar IB, Bushby K. The importance of genetic diagnosis for Duchenne muscular dystrophy. Journal of medical genetics. 2016;53(3):145–151. doi: 10.1136/jmedgenet-2015-103387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. 1988;2(1):90–95. doi: 10.1016/0888-7543(88)90113-9. [DOI] [PubMed] [Google Scholar]
- 11.Vengalil S, Preethish-Kumar V, Polavarapu K, Mahadevappa M, Sekar D, Purushottam M, et al. Duchenne Muscular Dystrophy and Becker Muscular Dystrophy Confirmed by Multiplex Ligation-Dependent Probe Amplification: Genotype-Phenotype Correlation in a Large Cohort. Journal of clinical neurology (Seoul, Korea) 2017;13(1):91–97. doi: 10.3988/jcn.2017.13.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kesari A, Pirra LN, Bremadesam L, McIntyre O, Gordon E, Dubrovsky AL, et al. Integrated DNA, cDNA, and protein studies in Becker muscular dystrophy show high exception to the reading frame rule. Human mutation. 2008;29(5):728–737. doi: 10.1002/humu.20722. [DOI] [PubMed] [Google Scholar]
- 13.Tuffery-Giraud S, Beroud C, Leturcq F, Yaou RB, Hamroun D, Michel-Calemard L, et al. Genotype-phenotype analysis in 2,405 patients with a dystrophinopathy using the UMD-DMD database: a model of nationwide knowledgebase. Human mutation. 2009;30(6):934–945. doi: 10.1002/humu.20976. [DOI] [PubMed] [Google Scholar]
- 14.Vo AH, McNally EM. Modifier genes and their effect on Duchenne muscular dystrophy. Current opinion in neurology. 2015;28(5):528–534. doi: 10.1097/WCO.0000000000000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.