

We identified gene networks linking sleep and affect, highlighting those oppositely perturbed by sleep loss and depression.
Abstract
To understand the transcriptomic organization underlying sleep and affective function, we studied a population of (C57BL/6J × 129S1/SvImJ) F2 mice by measuring 283 affective and sleep phenotypes and profiling gene expression across four brain regions. We identified converging molecular bases for sleep and affective phenotypes at both the single-gene and gene-network levels. Using publicly available transcriptomic datasets collected from sleep-deprived mice and patients with major depressive disorder (MDD), we identified three cortical gene networks altered by the sleep/wake state and depression. The network-level actions of sleep loss and depression were opposite to each other, providing a mechanistic basis for the sleep disruptions commonly observed in depression, as well as the reported acute antidepressant effects of sleep deprivation. We highlight one particular network composed of circadian rhythm regulators and neuronal activity–dependent immediate-early genes. The key upstream driver of this network, Arc, may act as a nexus linking sleep and depression. Our data provide mechanistic insights into the role of sleep in affective function and MDD.
INTRODUCTION
Sleep and affective behaviors are highly interrelated phenotypes, commonly altered in a variety of neuropsychiatric diseases (1, 2). Major depressive disorder (MDD) is a devastating illness and often involves dysregulated sleep, including insomnia and altered sleep architecture, while acute total sleep deprivation (SD) has been used clinically to induce a temporary improvement of mood in patients with MDD (3). Detailed mechanisms underlying this complex interaction remain largely unknown, although it is believed that the two interactive processes of sleep drive―circadian timing and sleep homeostasis―are both altered in MDD (3, 4). Recent large-scale transcriptomic analyses have revealed many molecular correlates associated with sleep, affective function, or MDD. These efforts have highlighted some common pathways, such as the circadian clock system, synaptic transmission and plasticity, and lipid metabolism and myelination, suggesting potential molecular processes linking sleep, affective function, and MDD (5, 6). Despite these advances, we still do not fully understand how these molecular underpinnings operate in coordinated networks across multiple brain regions to control complex behaviors or how these functional organizations are altered in MDD.
To examine gene networks associated with different domains of behavioral functions, we comprehensively investigated 20 categories of phenotypes that included 283 cognitive, affective, and sleep traits in an F2 mouse population derived from C57BL/6J (B6) and 129S1/SvImJ (129) inbred strains (Table 1, data file S1, and fig. S1). We assessed genotypes at 2458 informative single-nucleotide polymorphism (SNP) markers and, in a subset of animals, measured gene expression by microarray across four brain regions key to sleep and affective functions (frontal cortex, hippocampus, thalamus, and hypothalamus; n = 83 to 108 samples for each region). Phenotypic and transcriptomic segregations in this F2 population allowed us to systemically interrogate the transcriptomic network organization across brain regions in relation to a variety of behaviors, including sleep, thus providing a foundation to study how functional gene networks may be altered in MDD. Multiple links between sleep and affective functions were uncovered at both the single-gene and gene-network levels.
Table 1. Experimental schedule.
A timetable displaying the mouse’s age at which each experimental manipulation occurred. EEG, electroencephalography; EMG, electromyography.
| Age (weeks) | Activity |
| 4 | Arrival |
| 5–6 | Undisturbed acclimatization |
| 7 | Elevated plus maze, open field arena, novel object recognition* |
| 8 | Fear conditioning, tail suspension test |
| 9 | Forced swim test† |
| 10 | EEG/EMG implant surgery |
| 11 | Habituation to sleep-recording equipment |
| 12 | Baseline sleep (BL), SD, and recovery sleep (SDR)‡ |
| 13 | Restraint stress and recovery sleep (Rst)‡ |
| 14–15 | Undisturbed |
| 16 | Euthanasia and tissue dissection |
*Novel object recognition was only performed for the first 90 mice and was discontinued.
†Forced swim test was only performed on the latter 106 mice.
‡Sleep was recorded in a subset of 121 mice, drawn from the later cohorts, of which gene expression was studied. See Materials and Methods for details.
We then used publicly available transcriptomic datasets to demonstrate how cortex-specific gene networks are differentially affected by MDD in humans and acute SD in mouse models. We identify an Arc-regulated gene network containing circadian clock and cyclic adenosine 3′,5′-monophosphate (cAMP)–responsive immediate-early genes (IEGs), which may provide a mechanistic basis for the role of sleep in MDD. Together, our findings add to our understanding of the molecular underpinnings underlying the interactions between sleep, affective functions, and neuropsychiatric disorders.
RESULTS
Genetic control of gene expression is conserved across brain regions linking sleep and affect
To investigate naturally occurring genetic variations of transcriptional networks underlying sleep and affective behaviors, we first identified genetic loci that regulate the expression of genes [expression quantitative trait locus (eQTL)] in the F2 population. Initially, we examined each brain region separately and focused our analysis on eQTLs acting on nearby genes (cis-eQTLs). We used a permutation-based approach (7) to control false discovery rate (FDR) for testing multiple linked SNPs per gene and across a total of ~18,500 genes per brain region. We found that the expression levels of 2417 genes in the cortex, 2301 genes in the hippocampus, 2802 genes in the hypothalamus, and 2629 genes in the thalamus were significantly associated with at least one cis-SNP at FDR < 0.05 (data file S2). Since previous work has demonstrated that multitissue eQTL approaches can increase the statistical power of cis-eQTL detection and determine eQTL conservation across tissues (7), we next used an empirical Bayes approach for multitissue eQTL analysis (8) to investigate the genetic landscape of gene expression across brain regions. This analysis revealed that 95.6% of identified cis-eQTLs were shared by all four brain regions (Fig. 1A; example genes in Fig. 1B), providing compelling evidence for eQTL conservation throughout the brain. These results suggest that DNA variation has system-wide effects on gene expression, implicating global regulatory mechanisms for both intraregional and interregional function.
Fig. 1. Genetic landscape of gene expression and its functional significance across brain regions.
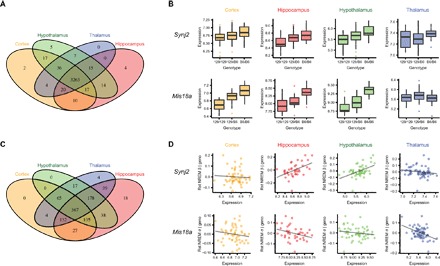
(A) Venn diagram showing the number of eQTL genes identified in each brain region using the empirical Bayes–based multitissue method. (B) Examples of two eQTL-regulated genes that showed brain region–conserved and brain region–specific genetic regulations. Synj2 showed brain region–conserved genetic correlations, while the cis-genetic regulation of Mis18a appeared to be brain region–specific (no cis-regulation in the thalamus). (C) Venn diagram showing the number of significant correlations between the expression of eQTL-controlled genes and phenotypes. Correlations were conditioned on eQTL genotype. (D) Examples of conditional correlations between phenotypes and the expression of eQTL genes, showing brain region–conserved and brain region–specific relationships with phenotypes. Phenotypes were regressed on eQTL genotype data, and the residuals were plotted against the expression of the gene that was regulated by the eQTL. Geno, eQTL genotype; Rst NREM β, changes in the EEG β power during non-rapid eye movement (NREM) sleep after Rst; Rst NREM σ, changes in the EEG σ power during NREM sleep after Rst. The association between Synj2 and Rst NREM β appeared to be specific to hippocampus and hypothalamus, while the association between Mis18a and Rst NREM σ can be observed across all four brain regions.
To understand the relevance of these eQTL-regulated genes to sleep and affective behavior, we computed partial correlations between the expression of eQTL genes and sleep/behavioral phenotypes. The expression-phenotype correlations were conditioned on eQTL genotype to remove spurious relationships between genes and phenotypes due to associations to the same or linked genetic loci. We then used the same empirical Bayes approach to investigate whether phenotypic relations of eQTL genes were also conserved across brain regions. Among the 3414 eQTL-controlled genes, we found 516 genes associated with at least one phenotype in at least one brain region, involving 994 pairs of gene-phenotype associations at FDR < 0.05. Since different brain regions are involved in different behavioral functions, we predicted that eQTL-linked genes would be associated with affective and sleep phenotypes in a region-specific manner despite their brain region–conserved genetic control. Surprisingly, although not as large as the proportion of regionally conserved eQTL, a significant portion (36.9%) of the identified gene-phenotype associations were conserved in all four brain regions (Fig. 1, C and D). Cross-region–conserved gene-phenotype associations were observed regardless of whether the phenotypes were sleep or affective behaviors. To ensure that the empirical Bayes approach does not bias toward cross-region conservation, we randomly permuted the gene-phenotype associations in each brain region and found that the occurrence of cross-region conservations on randomized data was extremely rare (0.166% ± 0.002% of all the significant gene-phenotype pairs; 1000 permutations). Furthermore, to exclude the possibility that the observed regional conservation in gene-phenotype associations is an artifact of correlated gene expression among brain regions, we further conditioned the partial correlation model on the expression of the gene in the other three brain regions, and a similar fraction (39.4%) of gene-phenotype associations persisted to be conserved across all brain regions. Therefore, our observations suggest that the functional relevance of genetically regulated gene expression to complex neurobehaviors may be conserved across multiple brain regions.
We next focused on eQTL genes associated with both sleep and behavioral phenotypes, as they could provide a molecular link between sleep and affective functions. We sought to determine whether the sleep and behavioral measurements share regulation at the genetic level or, conversely, are independently regulated. We found that 460 eQTL-regulated genes were associated with at least one sleep phenotype and 82 eQTL genes were associated with at least one affective phenotype. Among them, 26 were associated with both (data file S2), which is significantly higher than expected by random chance if sleep and affect are genetically independent (odds ratio = 3.10, P = 1.28 × 10−5). This observation thus suggests that genetically controlled gene expression across brain regions may in part underlie the interactions between sleep and affective functions. In particular, 11 eQTL genes were associated with both sleep phenotypes and measurements from the forced swim or tail suspension tests, which are often interpreted as signs of behavioral despair or depressive behavior (data file S2). Although the relationship between these behavioral tests and human depression is debated (9), these associations are still reminiscent of the sleep disturbances observed in patients with MDD. Many of the highlighted genes (Grm7, Kcnj10, and Tspo) have indeed been implicated in MDD (10–12), suggesting clinical relevance of these genetically regulated genes linking sleep and behavioral despair. In addition, we found that the amount of struggling activity during the first minute of forced swim and the amount of sleep rebound after SD were associated with the expression of Cdk5rap1, which encodes a regulator protein of CDK5, a neuronal cyclin-dependent kinase involved in the regulation of synaptic plasticity (13), circadian clock (14), and depression-like behavior (15, 16). This observation is consistent with the interaction between sleep homeostasis and MDD observed in humans, hinting that the molecular mechanisms of such interaction may involve Cdk5rap1. Together, our observations suggest that sleep is linked to mood regulation through a shared genetic landscape for gene expression across multiple brain regions.
The transcriptome is segregated genetically and functionally into networks across brain regions
While correlating DNA and transcriptional variation revealed a fraction of the transcriptome that is under genetic control, this analysis did not explicitly account for the organization of genes into higher-level networks involving larger portions of the transcriptome. To identify coregulated gene networks, we performed weighted gene coexpression network analysis (WGCNA) (Fig. 2A) (17). We identified 26 network modules in the cerebral cortex, 23 modules in the hippocampus, 27 modules in the thalamus, and 33 modules in the hypothalamus, ranging in size from 30 to 929 genes. Each module from each region was named by an arbitrary color; modules in different regions may share a color name, but similarly named modules do not imply equivalence or any biological relationship. We confirmed that many of these modules were overrepresented by genes involved in known biological pathways and Gene Ontology (GO) terms, which suggested that they were functionally coherent (data file S3). We also determined that each module was robust through resampling methods (18). Furthermore, specific modules in each region were also overrepresented by regional eQTL-controlled genes (data file S3), suggesting that shared genetic control can contribute to transcriptional coregulation of genes.
Fig. 2. Organizations of gene coexpression networks in multiple brain regions.
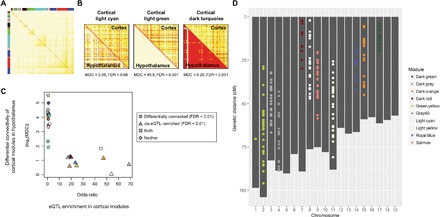
(A) Identification of gene coexpression network modules in the cortex, as an example. The colors in the heat map represent topological overlap measures (TOMs) of neighborhood similarity between a pair of genes (rows and columns) in the coexpression network; red indicates higher similarity. Colors in the column and row side bars label the assignment of network modules. (B) TOM plots showing differential network connectivities of three cortical modules compared to their gene counterparts in the hypothalamus, showing no change, loss of connectivity, and gain of connectivity, respectively. (C) eQTL-enriched modules were brain region–conserved. For each cortical module, its enrichment of eQTL genes (represented by odds ratios) was plotted against the absolute value of log2-transformed modular differential connectivity (MDC) comparing the cortical modules to their gene counterparts in the hypothalamus. Each point represents a module, colored according to their module names. (D) Genetic location of eQTL that regulates the genes in the eQTL-enriched cortical modules, colored according to their module names. While cortical modules were used here as an example, similar results were found across all four brain regions.
To investigate whether gene networks were conserved between brain regions, we compared the average connectivity of a module in one brain region to their gene counterparts in the other brain regions (Fig. 2B). This MDC (19) measure revealed a stark dichotomy. Modules that are highly enriched with cis-eQTL genes tended to show small or no differential connectivity across brain regions, whereas network modules not enriched with eQTL-controlled genes tended to exhibit large changes in connectivity across brain regions (Fig. 2C and data file S3). Within each eQTL-enriched module, the involved eQTLs were located predominantly on the same chromosome (Fig. 2D and data file S3). Thus, the coexpression of genes in these eQTL-enriched modules may be driven by genetic linkage among these closely located eQTLs, and not functionally concordant, as supported by the lack of GO and pathway enrichment in these modules (data file S3). However, genetic linkage may not be the sole reason for gene coexpression in eQTL-enriched modules. For example, the cortical dark gray module is an eQTL-enriched module (module genes located on chromosome 3) but is also highly enriched with target genes of transcription factor (TF) GATA6 (located on chromosome 18), as determined in chromatin immunoprecipitation studies (odds ratio = 13.97, adjusted P = 2.26 × 10−10). Similar enrichments of TF targets were observed in several other eQTL-enriched modules (data file S3), suggesting that coexpression of genes in these modules may also be driven by shared transcriptional regulation and, thus, functionally relevant. Nevertheless, since we have systematically characterized the functional relevance of eQTL-controlled genes at the single-gene level, we report here only results of non–eQTL-enriched modules from the network-level analyses for simplicity, although the eQTL-enriched modules were still included in these analyses.
Transcriptional networks underlie the interaction between sleep and affective behaviors
We have previously demonstrated that transcriptomic network organization in the mouse striatum is associated with multiple sleep, cognitive, and affective functions under chronic stress (20); in this study, we expanded our analysis to four brain regions. To investigate the association between gene networks and neurobehavioral functions, we computed correlations between each module’s eigengene (that is, the first principal component) and each phenotype. Each brain region was evaluated separately for these analyses, and all significant module-trait relationships were provided in data file S3. The relevance of a network module to a phenotypic category was determined by the percentage of phenotypes in the category that is significantly associated with the module (Fig. 3A). Consistent with our previous findings, network modules in each brain region were extensively associated with phenotypes across different behavioral domains. All non–eQTL-enriched network modules that were associated with behavioral phenotypes were also associated with sleep phenotypes, providing sets of functionally coordinated molecular correlates for the interactions between affective and sleep functions. For example, the saddle brown module in the hypothalamus was the most relevant module for behavioral despair phenotypes of the forced swim test. Saddle brown was enriched with genes involved in myelination (data file S3), abnormalities of which have been implicated in MDD by transcriptomic and imaging studies (21, 22). Coexpression of this module may be partially due to variation in cell composition between neurons and glia, especially considering that corpus callosum dysgenesis is seen in the 129 family of mouse strains (23). This module was also the most relevant for the anxiety-like behavior in the elevated plus maze and was associated with changes in the EEG power spectra and sleep fragmentation during recovery after SD (Fig. 3A). These findings suggest that multiple aspects of affective behavior and sleep changes after stress may interact with each other via a network of myelination genes in the hypothalamus.
Fig. 3. Functional significance of gene coexpression network modules.
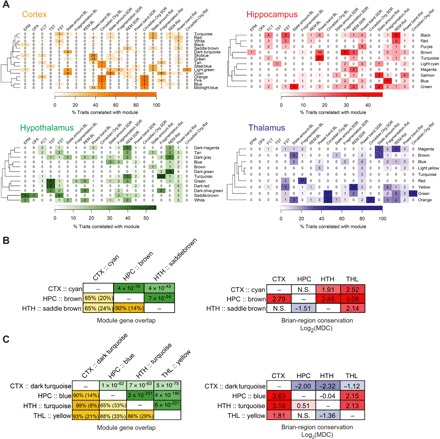
(A) Heat maps describing the relevance of modules (rows) to phenotypic categories (columns). Categories of affective phenotypes included those measured in the elevated plus maze (EPM), open field arena (OFA), fear conditioning test (FCT), tail suspension test (TST), and forced swim test (FST). Sleep phenotypic categories included sleep/wake state amount, sleep fragmentation, REM sleep, EEG power band, and circadian organization measured under undisturbed baseline conditions (BL), as well as the changes in these phenotypes after a 6-hour SD or 1-hour Rst. Only non–eQTL-enriched modules are shown. Heat colors indicate the percentage of phenotypes in a category that are significantly correlated with module eigengene. Numbers in each cell indicate the number of phenotypes in the category that are significantly associated with module eigengene. For clarity, the heat colors for cortex and thalamus are saturated at 60%. (B and C) Organization of myelination network modules (B) and extracellular matrix modules (C) across brain regions. Left: Gene overlap among modules of the same cellular function. The lower triangles of the matrices show the percentage of overlapping genes relative to the size of the smaller module (or relative to the combined size of two modules in parentheses). The upper triangles of the matrices show the P values from Fisher’s exact tests evaluating the enrichment of module in one another. Right: Differential connectivity of modules with the same cellular function across brain regions. Cells were colored according to their values. CTX, cortex; HPC, hippocampus; THL, thalamus; HTH, hypothalamus.
Myelination modules were notable in three brain regions: cortical cyan, hippocampal brown, and hypothalamic saddle brown. These modules shared a significant portion of member genes, but they were extensively reorganized and differentially connected across brain regions (Fig. 3B), with each region having unique sets of network hub genes (see module statistics in data file S3). In addition to the differential network organization, their associations to sleep and affective phenotypes were also altered across brain regions. The cortical cyan module was primarily correlated with sleep/wake state amount, the hippocampal brown module was primarily correlated with EEG activity during baseline conditions and changes in sleep fragmentation during recovery from SD, and as described above, the hypothalamic saddle brown module was mostly associated with anxiety and depressive behaviors. Similar region-specific connectivity and phenotypic relevance were observed for networks of extracellular matrix genes (cortical dark turquoise, hippocampal blue, thalamic yellow, and hypothalamic turquoise; Fig. 3C). Together, these results describe how differential connectivity of gene networks with similar cellular or molecular functions may be linked to their brain region–specific roles in sleep and affective behaviors.
Acute SD affects the expression of sleep and neurobehavioral networks
Since gene expression in our study was profiled at only one time point after the completion of all phenotypic measurements, the observed gene networks across brain regions provided a static snapshot of the functional transcriptomic organizations relevant to sleep and affective behaviors. Altered physiological states, such as sleep and wake, are known to associate with profound and dynamic changes in the transcriptome (6). To evaluate how these gene networks may be altered across sleep and wake, we leveraged publicly available transcriptomic datasets collected from multiple brain regions of sleep-deprived mice to integrate with our data. A search of the Gene Expression Omnibus (GEO) database identified three datasets in the cerebral cortex (accession numbers GSE6514, GSE78215, and GSE33491) (24–26), one dataset in the hippocampus (accession number GSE33302) (27), and one dataset in the hypothalamus (accession number GSE6514) (24). Focusing our analysis on acute SD of 5 to 6 hours, we first performed a gene-level analysis and identified 2080 differentially expressed genes (15% of all tested genes) in the cortex and 1567 genes (7%) in the hippocampus at FDR < 0.05. Although no differential expression was found at FDR < 0.05 in the hypothalamus, the expression levels of 5480 genes (24%) were altered at FDR < 0.10. The identified genes and highlighted pathways are comparable to previous reports examining these datasets (data file S4).
To evaluate differential expression at the gene-network level, we combined the gene coexpression network modules reconstructed using our (B6 × 129) F2 data with the SD differential expression signatures. We used gene set enrichment analysis (GSEA) (28) to identify coexpression network modules in which the overall distribution of SD-induced changes in gene expression was skewed or shifted toward increased expression (positive GSEA scores) or decreased expression (negative GSEA scores). Significant GSEA scores thus suggest network-level differential gene expression in coexpression modules. We found five modules in the cortex, eight modules in the hippocampus, and five modules in the hypothalamus that were differentially expressed after SD (Fig. 4). A number of these sleep/wake-affected network modules were also highly relevant to sleep phenotypes in our dataset, supporting their connection to sleep. For example, network-level gene expression in the previously mentioned cortical myelination module (cortical cyan) and a hippocampal module associated with nicotinic acetylcholine receptor signaling (hippocampal green) was down-regulated after acute SD (Fig. 4). Both modules were highly relevant to sleep/wake amount under baseline conditions, and cortical cyan was also highly relevant to SD-induced changes in sleep/wake amount and restraint-induced changes in sleep fragmentation (Fig. 3A). This is consistent with previously characterized roles of acetylcholine signaling and myelination in sleep and wake (29, 30). In addition, several sleep/wake-affected network modules were also highly relevant to affective phenotypes. SD reduced the expression of a cortical synaptic transmission module (cortical turquoise; Fig. 4), which was highly relevant to behavioral despair phenotypes measured in the forced swim test (Fig. 3A). The enriched GO terms of this module also included genes influencing behavior (data file S3). Therefore, these observations suggest that sleep loss may influence neurobehavioral output by perturbing sleep/wake-dependent synaptic transmission gene networks.
Fig. 4. SD-induced module-level differential expression in each brain region.
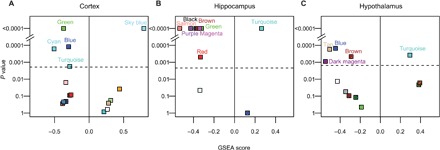
GSEA was used to test whether the gene coexpression network modules reconstructed using the (B6 × 129) F2 data were enriched with genes whose expression were up-regulated (positive GSEA score) or down-regulated (negative GSEA score) by SD in the cortex (A), hippocampus (B), and hypothalamus (C). For each module, the permutation-determined P values are plotted in log scale against GSEA scores. The dashed line indicates the Bonferroni-corrected P value threshold. Only non–eQTL-enriched modules are shown. Each module is colored according to the module name.
MDD alters the expression of sleep-affected cortical networks in the opposite directions
Since sleep disruptions are core symptoms in a range of psychiatric conditions, particularly MDD, we tested the hypothesis that MDD, like sleep loss, perturbs sleep and affective gene networks. To evaluate this, we investigated publicly available transcriptomic datasets collected from postmortem brain samples of MDD patients. Thirteen datasets were available in the cerebral cortex (GSE35977, GSE54562, GSE54563, GSE54565, GSE54567, GSE54568, GSE54570, GSE54571, GSE54572, GSE54575, GSE53987, and two sets from GSE92538), along with one dataset in the hippocampus (GSE53987; further dataset summaries are provided in data file S5) (31–34). Both male and female subjects were included in each of these datasets. While differential expression analysis did not reveal significantly changed genes in the hippocampus (FDR < 0.1), a meta-analysis of the 13 datasets in the cerebral cortex identified 107 differentially expressed genes at FDR < 0.05 (Fig. 5A). The differentially expressed genes in the cerebral cortex of depressed patients highlighted pathways known to be involved in MDD (Fig. 5B), such as elevated expression of nitric oxide synthase regulator genes and decreased gene expression in serotonin signaling (35, 36). Although single datasets might be strongly biased by factors such as postmortem interval, medication, length of the agonal state, time of death, and population heterogeneity, a consensus signature obtained from a meta-analysis of multiple datasets is advantageous as these dataset–specific biases are expected to be diluted or canceled out.
Fig. 5. MDD-induced differential expression at the gene level and at the module level in the cerebral cortex.
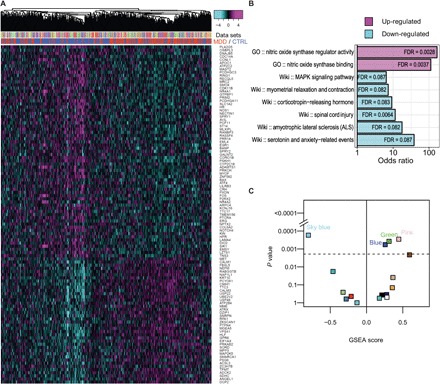
(A) The heat map shows expression of genes (rows) that were significantly changed in MDD in all samples (columns) across 13 datasets. Expression values were scaled within each dataset. Samples were hierarchically clustered on the basis of the scaled expression value and were color-labeled by datasets (upper column side color bar) and case/control (lower column side color bar). (B) Pathway and GO enrichment of the 107 significantly changed genes in MDD. GO, GO database; Wiki, WikiPathways database; MAPK, mitogen-activated protein kinase. (C) Module-level differential expression induced by MDD in the cerebral cortex, as determined by GSEA. For each module, the permutation-determined GSEA P values are plotted in log scale against GSEA scores. The dashed line indicates the Bonferroni-corrected P value threshold. A significant positive GSEA score indicates enrichment of genes up-regulated in MDD, while a significant negative GSEA score indicates enrichment of genes down-regulated in MDD. Only non–eQTL-enriched modules are shown. Each module is colored according to the module name.
We then used GSEA to test which of the mouse coexpression modules were enriched with this consensus differential expression signature of MDD in humans to understand how MDD may alter the network-level gene expression in functional modules associated with sleep and affective phenotypes. We found that network-level gene expression in four cortical modules was altered in depression (Fig. 5C). These include three of the modules (blue, green, and sky blue) that were also enriched with genes differentially expressed after SD (Fig. 4A), and the GSEA scores of these networks had opposite signs for SD and MDD signatures, indicating that SD and MDD oppositely altered the network-level gene expression in these networks (Figs. 4A and 5C). The opposite effects of SD and MDD on network-level gene expression were most clearly visualized in the sky blue module, in which gene expression was up-regulated after SD and down-regulated in MDD (Fig. 6B). The sky blue module was enriched with cAMP-responsive IEGs that are known molecular markers of neuronal activity (data file S3). The module was correlated with a large number of sleep EEG power spectra phenotypes during baseline conditions (Fig. 3A). In particular, sky blue’s eigengene was positively correlated with NREM sleep δ power, the primary marker of the sleep homeostatic pressure that accumulates during wake and dissipates during sleep. The sky blue module was also enriched with genes regulating circadian rhythms (data file S3), a process known to be involved in MDD and the regulation of sleep (37–39). In addition to the sky blue module, SD and MDD also oppositely altered gene expression in the blue and green modules (Figs. 4A and 5C). The blue module, whose network-level gene expression was up-regulated in MDD and down-regulated by SD, was also linked to circadian rhythms. The module was enriched with diurnally regulated genes and tricarboxylic acid (TCA) cycle genes (data file S3) and was associated with the timing of NREM sleep under baseline conditions, and its changes were induced by SD. The other MDD–up-regulated and SD–down-regulated network module (green) was enriched with genes associated with pre-mRNA processing and the mitochondrial inner membrane (data file S3). This module was also widely associated with EEG power spectra phenotype under baseline conditions but was mostly associated with EEG activities during the wake. Together, given their opposite expression patterns in SD and MDD, as well as their associated cellular functions and sleep/affective phenotypes, these cortical gene networks may suggest plausible mechanisms underlying the reduced sleep drive in MDD and the well-known antidepressant effects of acute SD (40).
Fig. 6. Regulatory network analysis revealed intermodular interactions and identified a key network regulator of both sleep and MDD.
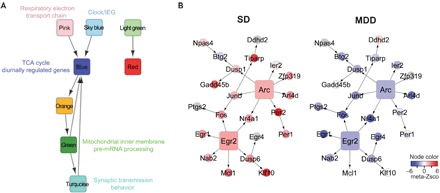
(A) A module-level regulatory network. Each node represents a coexpression network module in the frontal cortex of the (B6 × 129) F2 mice. A directed edge, representing a regulatory relationship between two modules, is drawn if (i) the source node module is enriched in the TF-PPI network of the target node module and (ii) the gene-level regulatory edges defined by Bayesian network reconstruction are observed significantly more than random chance between the two modules. GO and cellular pathways associated with SD-affected or MDD-affected modules are noted. (B) Key driver analysis identified Arc and Egr2 as the upstream regulators in a subnetwork resulting from the intersection of sky blue module genes and the Bayesian regulatory network. Each node represents a gene, and each regulatory edge indicates regulatory relationships predicted by the Bayesian network reconstruction. Nodes are colored according to the meta–z score from meta-analysis of differential gene expression of SD (left) and MDD (right) datasets. Gray nodes represent genes that were not included in the meta-analysis of SD or MDD transcriptomic datasets.
Regulatory relationships among cortical functional networks suggest a pathway underlying the interactions between sleep and MDD
To better understand the regulatory relationships among modules affected by MDD and SD, we evaluated potential transcriptional regulatory relationships among all cortical coexpression network modules by constructing a TF protein-protein interaction (PPI) network for each module. We tested enrichment of TF-PPI networks in cortical coexpression modules as an indication that a module may be involved in the transcriptional regulation of another module (or itself). In the resulting module-level transcriptional regulatory network, the top four most interconnected modules were the ones that were differentially expressed in MDD (fig. S2A), highlighting a central role of depression-affected gene networks in cortical transcriptomic organization relevant to sleep and affective function.
In a second approach to study cortical intermodular regulatory relationships, we reconstructed a causal Bayesian network using our cis-eQTL data as a causal anchor to infer gene-gene regulatory relationships from transcriptomic data (41). We intersected the Bayesian network with the coexpression network modules and counted the number of causal relationships among genes within a module and between modules. As expected, regulatory interactions recovered by the Bayesian network were observed mostly among genes within the same module, although many intermodular relationships were also seen. A module-level regulatory relationship between two models was identified if their intermodular causal edges were significantly more than expected by random chance. Similar to the transcriptional regulatory network, significant regulatory relationships were observed in this module-level Bayesian network among sleep-affected and/or depression-affected functional modules, particularly the blue, green, pink, and turquoise modules (fig. S2B).
We then combined these two intermodular regulatory networks and produced a consensus network (Fig. 6A), which revealed high-confidence intermodular interactions involving five of the six SD-affected and/or MDD-affected coexpression modules. This analysis revealed that the circadian rhythm and cAMP-responsive IEG module (sky blue) and a depression–up-regulated module involved in respiratory electron transport chain (pink) were the primary drivers of this complex network. Downstream of these networks were the TCA cycle and diurnally regulated gene module (blue), the pre-mRNA processing and mitochondrial inner membrane module (green), and the synaptic transmission module (turquoise). Together, these data suggest that a highly interactive pathway involving cellular energy metabolism, sleep/wake-dependent neuronal activity, circadian timing, and synaptic transmission underlies the interactions between sleep and MDD.
Analysis of network drivers identifies Arc as a key regulator of sleep and affective function
To identify key genes regulating sleep-affected and/or depression-affected networks, we performed key driver analysis in the subnetworks resulting from intersecting the cortical Bayesian network by the blue, cyan, green, pink, sky blue, and turquoise modules (data file S6). We ranked the key network drivers by the percentage of their respective module genes that are downstream of the driver. The top two key drivers, Arc and Egr2, were both found in the sky blue module, regulating 41 and 26% of the genes in the module, respectively (Fig. 6B). In our mouse data, Arc was correlated with NREM δ power, the electrographic hallmark of sleep homeostatic drive, during baseline sleep (r = 0.28, P = 0.005). We then investigated the functional relevance of sky blue genes that are downstream of Arc by searching their mutant phenotypes noted in the Mammalian Phenotype Ontology of the Mouse Genome Informatics (MGI) database (42). The Arc-regulated subnetwork is overrepresented by genes that are annotated in the MGI database to influence affective behavior (Arc, Dusp1, Fos, and Ptgs2; odds ratio = 7.56, P = 0.0042) and sleep pattern (Fos, Per1, and Per2; odds ratio = 22.66, P = 6.02 × 10−4). The circadian clock gene Per2 itself is also involved in both homeostatic regulation of sleep (39) and the expression of behavioral despair, as Per2 mutant mice show decreased immobility in the forced swim test (43). Being the upstream driver of these sky blue network genes, Arc therefore is likely a key regulator of both sleep and affective functions.
DISCUSSION
Sleep/wake and affective behaviors are interactive and complex. Transcriptomic alterations that are associated with changes in sleep/wake and affective states have provided important clues to the mechanisms underlying sleep, affective functions, and neuropsychiatric disorders. Here, we performed transcriptional profiling across four brain regions in a (B6 × 129) F2 mouse population that was extensively phenotyped for sleep and affective behaviors, elucidating how interactions between sleep and affective functions emerged from multilevel transcriptomic organizations across brain regions. At the single-gene level, we showed that genetically regulated genes were associated with sleep and behavioral phenotypes across brain regions and that the number of eQTL genes associated with both sleep and affective behaviors was higher than expected by random chance, indicating a convergence of genes that are relevant to different domains of neurobehavioral functions. In particular, we highlighted a number of genes (for example, Grm7, Kcnj10, and Tspo) relevant to MDD in humans and a gene (Cdk5rap1) that was associated with sleep and depressive phenotypes key to the clinical manifestations of MDD. At the network level, we demonstrated that brain region–specific gene networks were extensively associated with both sleep and behavioral phenotypes, suggesting that network-level transcriptomic organizations are key to the interactions between sleep and affective functions. Finally, by integrating our dataset with publicly available transcriptomic datasets, we showed that SD in mice and MDD in humans affected a set of cortical networks in opposite directions and identified Arc as a potential network driver of this relationship, providing a mechanistic basis for the role of sleep/wake in MDD.
Our analyses also revealed insights into how the transcriptome is organized across brain regions. We demonstrated that genetic regulation of gene expression acted in a “global” fashion across the frontal cortex, hippocampus, thalamus, and hypothalamus in mice. A previous study of diverse human tissues suggested that eQTLs tend to be either tissue-specific or act universally across the body, with few eQTLs regulating in some but not all tissues (7). Our data suggest that eQTLs across brain regions are much more likely to be universal rather than region-specific, which may reflect the relative anatomical and functional similarity of different brain tissues, as opposed to a wide range of diverse tissues throughout the body. We also demonstrated that such conserved genetic regulation across brain regions had a strong impact on the network-level organization in our F2 mouse population, producing brain region–conserved coexpression network modules. These genetically coregulated gene networks are likely due to extensive linkage across relatively large chromosomal regions in our F2 mice, although there were hints that some of these networks might also be transcriptionally coregulated by TFs.
Functionally, we show that phenotypic associations of genetically regulated gene expression were significantly conserved across brain regions, even after conditioning the phenotype-expression correlation on the expression of the gene in other brain regions, indicating that these genes regulate or respond to sleep and affective functions in a similar manner in different brain regions. This is a surprising finding, as we expected brain region specificity in gene functions. However, it is interesting to note that a previous study of SD in rats has uncovered substantial overlap (~40 to 50%) of gene expression changes between the cerebral cortex and the cerebellum, a brain region that lacks electrographic characteristic of sleep (44). This suggests that expression variations of individual genes may be involved in neurobehavioral functions in a regionally conserved fashion. Since this conservation of associations between eQTL genes and phenotypes was found indiscriminately for sleep and affective behaviors, our observations suggest that the regulation of affective functions, in addition to sleep, may also involve altering the expression of individual genes in a region-conserved fashion across multiple brain regions. On the other hand, this conservation may also be partially due to the fact that we analyzed relatively large brain regions, each of which is composed of many diverse subunits. If one instead analyzes expression in individual nuclei or specific cell populations, less conservation of eQTL behavior associations may be seen.
In contrast to the gene-level conservation of functional relevance, gene networks were associated with diverse profiles of phenotypes in different brain regions. We demonstrated that gene networks involved in the same molecular or cellular processes (exemplified by the myelination and extracellular matrix modules) exhibited distinctive network connectivity across brain regions, which was accompanied by brain region–specific phenotypic associations. These observations suggest an intriguing hypothesis that functional specificity of various brain regions may arise from network reorganization and “rewiring” of cellular and molecular pathways, which may represent a previously underappreciated mechanism of functional differentiation of brain regions.
A prominent feature of our study is the integrated analysis combining mouse functional gene networks and differential expression signatures observed for SD and MDD. Such integrated analysis overcomes the limitations of a static snapshot of gene-network organization in our mouse data, as well as the lack of insights into the gene regulatory relationships in typical differential expression studies. Our analysis identified six cortical network modules that were differentially expressed in sleep-deprived mice and/or patients with MDD. Notably, three modules were affected by both SD and MDD, and these were altered in opposite directions by the two conditions. These modules were associated with relevant sleep and affective phenotypes in our mouse dataset and were involved in specific molecular and cellular processes, including circadian rhythms, neuronal activity–dependent IEGs, mitochondrial respiration, myelination, and synaptic transmission. While these molecular and cellular processes have been previously implicated in sleep, affective functions, or MDD, our network analysis provided novel insights into the regulatory relationships among these network functionalities by unbiasedly reconstructing module-level regulatory networks. We show that the mitochondrial electron transport chain module (pink) and the clock/IEG module (sky blue) were most upstream in the module-level regulatory network, highlighting a central role of mitochondrial function, IEGs, and the circadian clock in multiple neuronal processes involved in sleep and MDD (37, 45, 46).
It has been hypothesized that circadian timing and sleep homeostatic processes, which interact with each other to drive sleep propensity and intensity, are both impaired in MDD, contributing to the disease pathology (3, 4). The clock/IEG module provides a potential molecular basis for this hypothesis. In this module, the circadian clock genes and regulators were expressed in concordance with molecular markers of neuronal activity. The module expression was correlated with electrographic hallmarks of sleep pressure and was sleep/wake-dependent. Thus, given its gene composition, phenotypic associations, and changes in response to SD, the clock/IEG module may function as an integrator of the homeostatic and circadian components of sleep drive. It has indeed been established that the circadian and homeostatic sleep drives are entangled with each other at the molecular level, involving the core circadian clock genes, including Per1 and Per2 in this module (39). Our findings suggest another layer of such molecular integration via coregulated expression of circadian regulators and neuronal activity markers.
It is important to note that MDD is associated with sex-specific characteristics, and recent studies have revealed interesting sex specificity in MDD-associated gene expression changes, which, in some cases, can even be in opposite directions in male and female patients (47, 48). Our MDD meta-analysis, however, focused on transcriptomic changes that are general in both male and female patients by including data from both sexes and using sex as a covariate. By linking this non–sex-dependent MDD signature with mouse sleep gene networks and SD signatures, our analysis identified molecular candidates underlying aspects of sleep-MDD interactions that are not prominently sex-specific, such as the antidepressant effects of SD (49). Our analysis particularly highlighted the clock/IEG module, and its role in linking sleep and MDD was supported by multiple observations. In addition to its cellular functions and associated sleep phenotypes, we demonstrated that the network-level expression of the clock/IEG module was up-regulated by SD and down-regulated in MDD. This observation is consistent with the hypothesis that sleep homeostatic drive is reduced in MDD, leading to nocturnal insomnia, and that acute SD leads to mood improvement due to increased sleep homeostatic pressure (4). A recent study of gene networks in multiple postmortem brain regions of MDD patients has identified a very similar clock/IEG network, which is enriched with genes showing decreased expression in female MDD patients (47). While this study did not evaluate whether this human clock/IEG network was also enriched with genes down-regulated in male MDD patients, our analysis combining mouse functional gene networks and human non–sex-specific transcriptomic signatures of MDD suggests that decreased expression in the clock/IEG network is likely generalizable in both male and female MDD patients. The human clock/IEG network identified in female MDD patients did not show significant structural differences (measured by MDC) between male and female MDD patients or between MDD and control subjects (47), and this human network bears remarkable resemblance to the sky blue module identified in our mouse data. Eight of the 18 genes in the human network module have homologs in our sky blue module. Since this very recent dataset was not included in our meta-analysis, our results provide an independent confirmation of this key MDD gene network, and, more importantly, our analysis linked the function of this network to the altered sleep regulation in MDD. Finally, we showed that the clock/IEG module appeared to be upstream of other sleep-affected or MDD-affected network modules, including the behavioral output module turquoise, which was enriched with genes known to influence various behaviors according to the GO analysis. Together, our results strongly support a role of the clock/IEG network at the center of sleep and MDD interactions.
We identified Arc as a key network driver of the clock/IEG module, regulating a large proportion of the module genes. Given the functional importance of the module, its primary network driver represents an interesting candidate nexus linking sleep and MDD. Arc is an IEG and a crucial regulator of synaptic plasticity (50), a process influenced by sleep and perhaps an important component of sleep need (51, 52). One popular hypothesis posits that impaired synaptic plasticity is a major mechanistic cause of MDD, and this is supported by recent studies showing that ketamine rapidly alleviates MDD symptoms while triggering synaptogenesis (53, 54); perhaps the up-regulation of Arc transcription by SD could affect mood via a similar mechanism. In humans, a polymorphism mapped to the Arc gene region has been associated with commonalities in schizophrenia and bipolar disorder in a genome-wide association study (55), and it is a network hub gene in the clock/IEG network found in postmortem brains of MDD patients (47). During the preparation of this paper, a preliminary study (56) has found that deletion of Arc in mice impairs the homeostatic sleep rebound after SD, accompanied by a lack of SD-induced elevation in the expression of IEGs, many of which indeed overlap with the Arc downstream network identified in our study. Whether Arc also contributes to MDD pathogenesis in humans and depressive behaviors in animal models remains an interesting question for future studies.
MATERIALS AND METHODS
Animals and phenotyping procedures
All animal studies were approved in advance by the Institutional Animal Care and Use Committee at Northwestern University and were in compliance with the Federal Animal Welfare guideline. Male (C57BL/6J × 129S1/SvImJ) F2 mice (n = 232) were purchased from The Jackson Laboratory. The parental strains used in the present study were chosen on the basis of prior knowledge of strain differences in multiple phenotypes associated with sleep, affective behavior, and stress reactivity. Mice arrived at Northwestern University in 13 separate cohorts at 4 weeks of age and were group-housed (two to four per cage) until sleep implant surgery at 10 weeks of age, after which animals were individually housed until necropsy. Mice were housed under a 12-hour light/12-hour dark cycle at room temperature (23° ± 2°C) with food and water available ad libitum.
All animals were subjected to a battery of behavioral tests and sleep measurements as outlined in Table 1, with three exceptions. First, the novel object recognition test was only applied to the first five cohorts (n = 90), after which the test was discontinued, and the data were not included in analyses because of the lack of robust object preferences demonstrated under our test settings. Second, only the last six cohorts of animals (n = 106) were tested for forced swim. Last, a subset of 121 animals in the last nine cohorts was studied for sleep phenotypes. Detailed phenotyping procedures for each test are included in the Supplementary Methods. Affective behavioral phenotypes were categorized by the behavioral test (five categories), and sleep phenotypes were categorized into sleep/wake state amount, sleep fragmentation, REM sleep, EEG power band, and circadian organization based on prior knowledge (20, 57). Sleep phenotypes were measured under undisrupted baseline conditions, after 6 hours of SD (ZT2 to ZT8), and after 1 hour of restraint stress (ZT5 to ZT6), resulting in 15 sleep phenotypic categories (data file S1).
Phenotypic data preparation
All phenotypic data were carefully inspected for outliers and covariates. Outliers were defined as those falling outside of six interquartile ranges from the first and third quartile, and up to two outlier values for each phenotype were removed. We then used the R/car package to inspect each measurement for normality, and when the data distribution is severely deviated from normal (P < 0.001 in a likelihood ratio test), a Yeo-Johnson power transformation was used to transform the distribution to normal (fig. S1B). The influence of potential experimental covariates, such as animal cohorts and housing condition variations, was carefully evaluated by computing analysis of variance (ANOVA) P values of the covariate across all phenotypes and comparing the distribution of the P values against theoretical quantiles from a uniform distribution in a q-q plot. The animal cohort was found to have significant effects across phenotypes, and thus, all phenotypic data were adjusted for this covariate by fitting the data in a robust linear model and taking the intercept + residuals as the adjusted values.
Genotyping
DNA was extracted from small tail biopsies using the DNeasy Kit (Qiagen) and then stored at −20°C before genotyped using the Affymetrix MegAllele genotyping mouse 5K SNP Panel. Quality control and processing of the genotyping data were performed according to the manufacturer’s guidelines. Of the ~5000 SNPs included on the chip, 2458 were polymorphic between the C57BL/6J and 129/SvImJ inbred strains.
Expression profiling
For gene expression profiling, mice completing the behavioral and sleep phenotyping (that is, 121 animals in the last nine cohorts) were euthanized by decapitation at ZT6-7 (that is, 6 to 7 hours after light onset), and four brain regions (thalamus/midbrain, hypothalamus, frontal cortex, and hippocampus) were carefully dissected by hand. Tissues were immediately flash-frozen in liquid nitrogen and stored at −80°C before being shipped to Rosetta Inpharmatics in a single batch for microarray assays and data processing. At the Rosetta Gene Expression Laboratory, total RNA was isolated from homogenized brain tissue samples using TRIzol reagent (Invitrogen), and gene expression profiling was performed using the Affymetrix GeneChip Mouse Genome 430 2.0 Array according to the manufacturer’s guidelines. The data were processed separately for each tissue. Briefly, raw data were normalized using robust multiarray averaging with quantile normalization and log2-transformed. Using the same covariate investigation procedure as that used for the phenotypic data, we investigated and adjusted data for effects of technical covariates (such as RNA processing and array batches) and animal cohorts. We also used principal components analysis to capture the unknown source of variations in each tissue. For any principal component that is not associated with any genomic loci [logarithm of the odds (LOD) < 3], we further adjusted the data by treating those principal components as covariates. The covariate-adjusted expression data at the probeset level were then collapsed to the gene level, using the median of all probesets that map to the same gene (58). We further trimmed the expression data in each brain region by removing the bottom 10% genes with least variance and any sample whose interarray correlation was two standard deviations away from the mean (59). The final set of expression data contains 98 samples from the frontal cortex, 83 samples from the hippocampus, 94 samples from the hypothalamus, and 108 samples from the thalamus.
eQTL analysis and phenotypic associations
We used Matrix eQTL to identify genes regulated by genetic variations in each brain region (60), focusing only on cis-SNP located in proximity to the genes (<1 Mb from the transcriptional start site). Testing multiple SNPs per gene was controlled using a permutation-based approach (7). We permuted samples in the gene expression matrix 1000 times and computed an empirical P value for each gene, as the fraction that the minimum nominal P value of its associations with all SNPs in the permuted set was lower than the true minimum nominal P value. Finally, we controlled FDR by calculating q values (61) from these empirical P values and called eQTL-regulated genes using a cutoff of minimum P value < 0.05 and q values < 0.05. To identify eQTL shared across brain regions, we used a multivariate Bayesian hierarchical model (8). This approach considers SNP-probe pairs as the unit of analysis and uses the vector of Fisher-transformed SNP-probe correlations across brain regions for each SNP-probe pair. Local FDRs were estimated for SNP-probe pairs, and we provide all significant pairs (local FDR < 0.05) in data file S2, which also contains the (1, 0) configuration of presence or absence of an eQTL in each brain region that yielded the maximum likelihood, as well as the Bayesian posterior probability of eQTL not present in a brain region. The same multiple brain region approach was used to identify associations between eQTL-regulated genes and phenotypes. Genes identified in the multiregion eQTL analysis were included in this analysis, and the associations were conditioned on eQTL genotypes. Significant (local FDR < 0.05) gene-phenotype pairs, as well as the multiregion presence/absence configuration and Bayesian posterior probabilities, were included in data file S2.
Gene-network analysis
We reconstructed two types of gene networks using the expression data from each brain region. We first used WGCNA to identify transcriptional coexpression networks of genes (17). In each brain region, we used Pearson correlations to capture linear relationships between genes and raised the Pearson correlation matrix to a positive power, β, to define the “scale-free” adjacency matrix. The adjacency matrix was quadratically transformed into a TOM to account for nearest-neighbor links in the gene expression graph. Hierarchical clustering with dynamic branch cutting defined coexpression modules from the TOM. Modules were named by arbitrary colors, with “gray” denoting genes that failed to segregate into a specific module. In each brain region, we captured associations between modules and phenotypes by correlating the first principal component of each module (that is, the module eigengene) with each phenotype. Sample labels of the data were permuted 10,000 times, and a step-down procedure was used to calculate family-wise error rate (FWER) (62). Module-trait associations were considered significant when P < 0.05 and FWER < 0.05. Pathway and GO enrichment analysis of each module was performed using Fisher’s exact test using gene sets from multiple databases downloaded from Enrichr (63) (last accessed on 22 March 2017).
To infer regulatory relationships among genes in each brain region, we reconstructed a Bayesian regulatory network (19, 41). Briefly, we used significant cis-eQTL (FDR < 0.05) identified in each brain region as causal anchors to break Markov equivalent structures and infer causality. Monte Carlo Markov Chain simulations were used to introduce random changes into a null network, and a change will be accepted only if it improved the overall fit as assessed by Bayesian information criterion. The final network was generated as the consensus from 1000 reconstructions, containing edges that were found in >30% of the reconstructions. To retain the directed acyclic graph from the consensus network, we removed network edges that were in a loop and found in the least number of reconstructions.
Key network drivers of the coexpression network modules were identified by intersecting the Bayesian network by coexpression modules. In each of the resulting subnetworks, all nodes (genes) belong to the same coexpression module, and network edges represent regulatory relationships inferred by the global Bayesian network reconstruction. We then searched through each gene in the subnetwork for their downstream of the h-layer neighborhood (HLN), and key driver genes were defined as those whose HLN sizes (μ) were greater than mean(μ) + σ(μ) (64).
Intermodular regulatory networks in the cortex were constructed using two different approaches. First, we constructed a module-level transcriptional regulatory network. We downloaded TF target gene sets compiled by the ChEA (ChIP-X Enrichment Analysis) and ENCODE (Encyclopedia of DNA Elements) databases from Enrichr (63) (last accessed on 22 March 2017), and we constructed a global PPI network by combining human and mouse interacting protein pairs downloaded from BioGRID (65) (accessed on 20 March 2017). For each cortical module, we identified TFs whose target genes were enriched in the module at Benjamini-Hochberg FDR < 0.05 and constructed a TF-PPI network by intersecting the global PPI network using the identified TFs and their immediate binding partners to include cofactor and regulators of the TFs. A transcriptional regulatory edge between two modules was established if a module was found to be enriched in the TF-PPI network of another module. As a second approach, we collapsed the global Bayesian network to the module level. A regulatory edge between two modules was established when the number of gene-level–directed edges from one module to another in the Bayesian network was significantly larger than random (Benjamini-Hochberg FDR < 0.05) as determined by randomly permuting the module-gene assignments 1000 times.
Differential expression signature of SD and MDD
In all three mouse SD expression datasets, the data were collected from adult male C57BL/6J mice (24–27). Each of the human MDD dataset included data from both male and female subjects (31–34). The expression datasets were downloaded from GEO, and background-corrected, quantile-normalized, and log2-transformed expression data from each dataset were used for differential expression analysis. For each dataset, data were adjusted for known covariates (such as sex in human MDD data) using an empirical Bayes method, ComBat, implemented in the R/sva (surrogate variable analysis) package (66). Hidden covariates were estimated using sva (66). Each of the estimated hidden covariates and known numeric covariates (such as age and postmortem interval in human data) was evaluated for their effects on expression using robust linear models. Those that had widespread effects, as visualized on a q-q plot, were selected for further adjustment by fitting the expression data with a robust linear model and taking intercept + residuals as the adjusted expression values.
For mouse SD and human MDD datasets in the cortex, we used meta-analyses to combine multiple datasets (67). Briefly, for each dataset, we computed the effect size (Hedges’ g) of MDD or SD and a z statistic for each probeset. We condensed the dataset and kept the probeset with the most extreme z score for each gene, as it was the least likely by chance (68). Genes that were assessed in all datasets (12,572 genes for human cortical MDD datasets and 13,444 genes for mouse cortical SD datasets) were included in the meta-analysis. We linearly combined the dataset–specific z scores to compute a meta–z statistic and estimated FDR using 1000 permutations. For brain regions with only one SD or MDD dataset, dataset specific z statistics were computed.
To identify coexpression networks whose gene expression were concordantly affected at the network level by SD or MDD in a brain region, we rank-ordered the differential expression profile based on the z statistics and performed GSEA (28), which computes a Kolmogorov-Smirnov–like statistic to test whether the gene set (for example, a module) was enriched at the top or bottom of the rank-ordered gene list. Genes in the differential expression profile were permuted 10,000 times to estimate FDR.
Enrichment of sleep and affective genes in the Arc-regulated clock/IEG subnetwork
To evaluate the functional relevance of the clock/IEG subnetwork regulated by Arc, we downloaded the full phenotype ontology annotation from MGI (42) (accessed 14 July 2017), which included mutant phenotypes of 11,298 genes. For genes that alter affective behavior, we searched for genes annotated with the ontology term “abnormal emotion/affect behavior” (MP:0002572) or its child ontology terms and found 125 genes. For genes that influence sleep, we searched for genes annotated with the ontology term “abnormal sleep behavior” (MP:0011396) or its child ontology terms and found 544 genes. We then used Fisher’s exact test to investigate whether the Arc-regulated clock/IEG subnetwork (15 genes) was overrepresented by these sleep or affective genes. To avoid biases related to gene coverage, we limited the test to consider only genes whose expression was measured by microarray in our (B6 × 129) F2 dataset and whose mutant phenotypes were available in MGI. This gives a background of 8911 genes, and under this background, there were 486 affective genes, 119 sleep genes, and 13 genes in the Arc-regulated clock/IEG subnetwork.
Supplementary Material
Acknowledgments
The views, opinions, and/or findings contained in this work are those of the authors and should not be interpreted as representing the official views or policies, either expressed or implied, of the Defense Advanced Research Projects Agency or the Department of Defense. Funding: This work was supported by a research grant from Merck Research Laboratories, by the Defense Advanced Research Projects Agency, and by the U.S. Army Research Office (contract no. W911NF-10-1006). J.R.S. was supported by the National Institute of Mental Health of the NIH (F30MH106293), and V.D.G. was supported by the National Heart, Lung, and Blood Institute of the NIH (T32HL007909). Author contributions: J.R.S., P.J., and V.D.G. contributed equally to data analysis and interpretation and were the primary writers of this manuscript. K.F. and C.O. contributed primarily to the initial analysis of the phenotypic data. J.M. processed the microarray data. A.G., C.J.W., and J.J.R. supervised the generation of genotypic and microarray data and advised the design of the project. M.H.V., A.K., and F.W.T. contributed equally to the project’s conception and design. M.H.V. and F.W.T. supervised all animal experiments, and A.K. supervised the data analysis. Competing interests: F.W.T. has received grants from Merck & Co. Inc. A.G., C.J.W., and J.J.R. were former employees of Merck & Co. Inc. A.G. is currently employed by Biogen, C.J.W. is currently employed by Ironwood Pharmaceuticals, and J.J.R. is currently employed by Purdue Pharma. J.M. was a former employee of Sage Bionetworks. All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper are available through GEO (accession number GSE109112).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/7/eaat1294/DC1
Fig. S1. Distributions of phenotypic measurements.
Fig. S2. Regulatory relationships among gene coexpression network modules.
Data file S1. Phenotypic categories and descriptions.
Data file S2. eQTL and phenotypic associations.
Data file S3. Coexpression network modules.
Data file S4. SD signatures.
Data file S5. MDD signatures.
Data file S6. Bayesian network and key drivers.
Supplementary Methods
REFERENCES AND NOTES
- 1.Walker M. P., The role of sleep in cognition and emotion. Ann. N. Y. Acad. Sci. 1156, 168–197 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Wulff K., Gatti S., Wettstein J. G., Foster R. G., Sleep and circadian rhythm disruption in psychiatric and neurodegenerative disease. Nat. Rev. Neurosci. 11, 589–599 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Nutt D., Wilson S., Paterson L., Sleep disorders as core symptoms of depression. Dialogues Clin. Neurosci. 10, 329–336 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borbély A. A., Daan S., Wirz-Justice A., Deboer T., The two-process model of sleep regulation: A reappraisal. J. Sleep Res. 25, 131–143 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Mehta D., Menke A., Binder E. B., Gene expression studies in major depression. Curr. Psychiatry Rep. 12, 135–144 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.B. F. O’Hara, P. Jiang, F. W. Turek, P. Franken, Genetics and genomic basis of sleep in rodents, in Principles and Practice of Sleep Medicine, M. H. Kryger, T. Roth, W. C. Dement, Eds. (Elsevier, 2017), chap. 29, pp. 296–309.e5. [Google Scholar]
- 7.GTEx Consortium , The genotype-tissue expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 348, 648–660 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li G., Shabalin A. A., Rusyn I., Wright F. A., Nobel A. B., An empirical Bayes approach for multiple tissue eQTL analysis. Biostatistics 19, 391–406 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nestler E. J., Hyman S. E., Animal models of neuropsychiatric disorders. Nat. Neurosci. 13, 1161–1169 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shyn S. I., Shi J., Kraft J. B., Potash J. B., Knowles J. A., Weissman M. M., Garriock H. A., Yokoyama J. S., McGrath P. J., Peters E. J., Scheftner W. A., Coryell W., Lawson W. B., Jancic D., Gejman P. V., Sanders A. R., Holmans P., Slager S. L., Levinson D. F., Hamilton S. P., Novel loci for major depression identified by genome-wide association study of sequenced treatment alternatives to relieve depression and meta-analysis of three studies. Mol. Psychiatry 16, 202–215 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Medina A., Watson S. J., Bunney W. Jr, Myers R. M., Schatzberg A., Barchas J., Akil H., Thompson R. C., Evidence for alterations of the glial syncytial function in major depressive disorder. J. Psychiatr. Res. 72, 15–21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holmes S. E., Hinz R., Conen S., Gregory C. J., Matthews J. C., Anton-Rodriguez J. M., Gerhard A., Talbot P. S., Elevated translocator protein in anterior cingulate in major depression and a role for inflammation in suicidal thinking: A positron emission tomography study. Biol. Psychiatry 83, 61–69 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Cheung Z. H., Fu A. K. Y., Ip N. Y., Synaptic roles of Cdk5: Implications in higher cognitive functions and neurodegenerative diseases. Neuron 50, 13–18 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Kwak Y., Jeong J., Lee S., Park Y.-U., Lee S.-A., Han D.-H., Kim J.-H., Ohshima T., Mikoshiba K., Suh Y.-H., Cho S., Park S. K., Cyclin-dependent kinase 5 (cdk5) regulates the function of CLOCK protein by direct phosphorylation. J. Biol. Chem. 288, 36878–36889 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhong P., Liu X., Zhang Z., Hu Y., Liu S. J., Lezama-Ruiz M., Joksimovic M., Liu Q.-s., Cyclin-dependent kinase 5 in the ventral tegmental area regulates depression-related behaviors. J. Neurosci. 34, 6352–6366 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu W.-L., Shi H.-S., Wang S.-J., Xu C.-M., Jiang W.-G., Wang X., Wu P., Li Q.-Q., Ding Z.-B., Lu L., Increased Cdk5/p35 activity in the dentate gyrus mediates depressive-like behaviour in rats. Int. J. Neuropsychopharmacol. 15, 795–809 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Zhang B., Horvath S., A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 4, Article17 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Langfelder P., Luo R., Oldham M. C., Horvath S., Is my network module preserved and reproducible? PLOS Comput. Biol. 7, e1001057 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang B., Gaiteri C., Bodea L.-G., Wang Z., McElwee J. J., Podtelezhnikov A. A., Zhang C., Xie T., Tran L., Dobrin R., Fluder E., Clurman B. E., Melquist S., Narayanan M., Suver C., Shah H., Mahajan M., Gillis T., Mysore J. S., MacDonald M. E., Lamb J. R., Bennett D. A., Molony C., Stone D. J., Gudnason V., Myers A. J., Schadt E. E., Neumann H. A. M., Zhu J., Emilsson V., Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 153, 707–720 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang P., Scarpa J. R., Fitzpatrick K., Losic B., Gao V. D., Hao K., Summa K. C., Yang H. S., Zhang B., Allada R., Vitaterna M. H., Turek F. W., Kasarskis A., A systems approach identifies networks and genes linking sleep and stress: Implications for neuropsychiatric disorders. Cell Rep. 11, 835–848 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aston C., Jiang L., Sokolov B. P., Transcriptional profiling reveals evidence for signaling and oligodendroglial abnormalities in the temporal cortex from patients with major depressive disorder. Mol. Psychiatry 10, 309–322 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Sacchet M. D., Gotlib I. H., Myelination of the brain in major depressive disorder: An in vivo quantitative magnetic resonance imaging study. Sci. Rep. 7, 2200 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wahlsten D., Deficiency of corpus callosum varies with strain and supplier of the mice. Brain Res. 239, 329–347 (1982). [DOI] [PubMed] [Google Scholar]
- 24.Mackiewicz M., Shockley K. R., Romer M. A., Galante R. J., Zimmerman J. E., Naidoo N., Baldwin D. A., Jensen S. T., Churchill G. A., Pack A. I., Macromolecule biosynthesis: A key function of sleep. Physiol. Genomics 31, 441–457 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Gerstner J. R., Koberstein J. N., Watson A. J., Zapero N., Risso D., Speed T. P., Frank M. G., Peixoto L., Removal of unwanted variation reveals novel patterns of gene expression linked to sleep homeostasis in murine cortex. BMC Genomics 17 (suppl. 8), 727 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hinard V., Mikhail C., Pradervand S., Curie T., Houtkooper R. H., Auwerx J., Franken P., Tafti M., Key electrophysiological, molecular, and metabolic signatures of sleep and wakefulness revealed in primary cortical cultures. J. Neurosci. 32, 12506–12517 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vecsey C. G., Peixoto L., Choi J. H. K., Wimmer M., Jaganath D., Hernandez P. J., Blackwell J., Meda K., Park A. J., Hannenhalli S., Abel T., Genomic analysis of sleep deprivation reveals translational regulation in the hippocampus. Physiol. Genomics 44, 981–991 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., Mesirov J. P., Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pace-Schott E. F., Hobson J. A., The neurobiology of sleep: Genetics, cellular physiology and subcortical networks. Nat. Rev. Neurosci. 3, 591–605 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Bellesi M., Pfister-Genskow M., Maret S., Keles S., Tononi G., Cirelli C., Effects of sleep and wake on oligodendrocytes and their precursors. J. Neurosci. 33, 14288–14300 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen C., Cheng L., Grennan K., Pibiri F., Zhang C., Badner J. A. Members of the Bipolar Disorder Genome Study (BiGS) Consortium, Gershon E. S., Liu C., Two gene co-expression modules differentiate psychotics and controls. Mol. Psychiatry 18, 1308–1314 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hagenauer M. H., Li J. Z., Walsh D. M., Vawter M. P., Thompson R. C., Turner C. A., Bunney W. E., Myers R. M., Barchas J. D., Schatzberg A. F., Watson S. J., Akil H., Inference of cell type composition from human brain transcriptomic datasets illuminates the effects of age, manner of death, dissection, and psychiatric diagnosis. bioRxiv, 10.1101/089391 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang L.-C., Jamain S., Lin C.-W., Rujescu D., Tseng G. C., Sibille E., A conserved BDNF, glutamate- and GABA-enriched gene module related to human depression identified by coexpression meta-analysis and DNA variant genome-wide association studies. PLOS ONE 9, e90980 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lanz T. A., Joshi J. J., Reinhart V., Johnson K., Grantham L. E. II, Volfson D., Step levels are unchanged in pre-frontal cortex and associative striatum in post-mortem human brain samples from subjects with schizophrenia, bipolar disorder and major depressive disorder. PLOS ONE 10, e0121744 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kudlow P., Cha D. S., Carvalho A. F., McIntyre R. S., Nitric oxide and major depressive disorder: Pathophysiology and treatment implications. Curr. Mol. Med. 16, 206–215 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Köhler S., Cierpinsky K., Kronenberg G., Adli M., The serotonergic system in the neurobiology of depression: Relevance for novel antidepressants. J. Psychopharmacol. 30, 13–22 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Bunney B. G., Li J. Z., Walsh D. M., Stein R., Vawter M. P., Cartagena P., Barchas J. D., Schatzberg A. F., Myers R. M., Watson S. J., Akil H., Bunney W. E., Circadian dysregulation of clock genes: Clues to rapid treatments in major depressive disorder. Mol. Psychiatry 20, 48–55 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Orozco-Solis R., Montellier E., Aguilar-Arnal L., Sato S., Vawter M. P., Bunney B. G., Bunney W. E., Sassone-Corsi P., A circadian genomic signature common to ketamine and sleep deprivation in the anterior cingulate cortex. Biol. Psychiatry 82, 351–360 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Franken P., A role for clock genes in sleep homeostasis. Curr. Opin. Neurobiol. 23, 864–872 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Giedke H., Schwarzler F., Therapeutic use of sleep deprivation in depression. Sleep Med. Rev. 6, 361–377 (2002). [PubMed] [Google Scholar]
- 41.Zhu J., Wiener M. C., Zhang C., Fridman A., Minch E., Lum P. Y., Sachs J. R., Schadt E. E., Increasing the power to detect causal associations by combining genotypic and expression data in segregating populations. PLOS Comput. Biol. 3, e69 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith C. L., Eppig J. T., The mammalian phenotype ontology: Enabling robust annotation and comparative analysis. Wiley Interdiscip. Rev. Syst. Biol. Med. 1, 390–399 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hampp G., Ripperger J. A., Houben T., Schmutz I., Blex C., Perreau-Lenz S., Brunk I., Spanagel R., Ahnert-Hilger G., Meijer J. H., Albrecht U., Regulation of monoamine oxidase a by circadian-clock components implies clock influence on mood. Curr. Biol. 18, 678–683 (2008). [DOI] [PubMed] [Google Scholar]
- 44.Cirelli C., Gutierrez C. M., Tononi G., Extensive and divergent effects of sleep and wakefulness on brain gene expression. Neuron 41, 35–43 (2004). [DOI] [PubMed] [Google Scholar]
- 45.Manji H., Kato T., Di Prospero N. A., Ness S., Beal M. F., Krams M., Chen G., Impaired mitochondrial function in psychiatric disorders. Nat. Rev. Neurosci. 13, 293–307 (2012). [DOI] [PubMed] [Google Scholar]
- 46.Scharf M. T., Naidoo N., Zimmerman J. E., Pack A. I., The energy hypothesis of sleep revisited. Prog. Neurobiol. 86, 264–280 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Labonté B., Engmann O., Purushothaman I., Menard C., Wang J., Tan C., Scarpa J. R., Moy G., Loh Y.-H. E., Cahill M., Lorsch Z. S., Hamilton P. J., Calipari E. S., Hodes G. E., Issler O., Kronman H., Pfau M., Obradovic A. L. J., Dong Y., Neve R. L., Russo S., Kazarskis A., Tamminga C., Mechawar N., Turecki G., Zhang B., Shen L., Nestler E. J., Sex-specific transcriptional signatures in human depression. Nat. Med. 23, 1102–1111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seney M. L., Huo Z., Cahill K., French L., Puralewski R., Zhang J., Logan R. W., Tseng G., Lewis D. A., Sibille E., Opposite molecular signatures of depression in men and women. Biol. Psychiatry (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Armitage R., Hoffmann R. F., Sleep EEG, depression and gender. Sleep Med. Rev. 5, 237–246 (2001). [DOI] [PubMed] [Google Scholar]
- 50.Plath N., Ohana O., Dammermann B., Errington M. L., Schmitz D., Gross C., Mao X., Engelsberg A., Mahlke C., Welzl H., Kobalz U., Stawrakakis A., Fernandez E., Waltereit R., Bick-Sander A., Therstappen E., Cooke S. F., Blanquet V., Wurst W., Salmen B., Bösl M. R., Lipp H.-P., Grant S. G. N., Bliss T. V. P., Wolfer D. P., Kuhl D., Arc/Arg3.1 is essential for the consolidation of synaptic plasticity and memories. Neuron 52, 437–444 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Tononi G., Cirelli C., Sleep and the price of plasticity: From synaptic and cellular homeostasis to memory consolidation and integration. Neuron 81, 12–34 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abel T., Havekes R., Saletin J. M., Walker M. P., Sleep, plasticity and memory from molecules to whole-brain networks. Curr. Biol. 23, R774–R788 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Duman R. S., Aghajanian G. K., Sanacora G., Krystal J. H., Synaptic plasticity and depression: New insights from stress and rapid-acting antidepressants. Nat. Med. 22, 238–249 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zunszain P. A., Horowitz M. A., Cattaneo A., Lupi M. M., Pariante C. M., Ketamine: Synaptogenesis, immunomodulation and glycogen synthase kinase-3 as underlying mechanisms of its antidepressant properties. Mol. Psychiatry 18, 1236–1241 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang K.-S., Liu X.-F., Aragam N., A genome-wide meta-analysis identifies novel loci associated with schizophrenia and bipolar disorder. Schizophr. Res. 124, 192–199 (2010). [DOI] [PubMed] [Google Scholar]
- 56.Suzuki A., Greene R., Arc gene function in sleep. Sleep 40, A6 (2017). [Google Scholar]
- 57.Winrow C. J., Williams D. L., Kasarskis A., Millstein J., Laposky A. D., Yang H. S., Mrazek K., Zhou L., Owens J. R., Radzicki D., Preuss F., Schadt E. E., Shimomura K., Vitaterna M. H., Zhang C., Koblan K. S., Renger J. J., Turek F. W., Uncovering the genetic landscape for multiple sleep-wake traits. PLOS ONE 4, e5161 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miller J. A., Cai C., Langfelder P., Geschwind D. H., Kurian S. M., Salomon D. R., Horvath S., Strategies for aggregating gene expression data: The collapseRows R function. BMC Bioinformatics 12, 322 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oldham M. C., Konopka G., Iwamoto K., Langfelder P., Kato T., Horvath S., Geschwind D. H., Functional organization of the transcriptome in human brain. Nat. Neurosci. 11, 1271–1282 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shabalin A. A., Matrix eQTL: Ultra fast eQTL analysis via large matrix operations. Bioinformatics 28, 1353–1358 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Storey J. D., Tibshirani R., Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. U.S.A. 100, 9440–9445 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.P. H. Westfall, S. S. Young, Resampling-Based Multiple Testing: Examples and Methods for P-Value Adjustment (John Wiley, 1993). [Google Scholar]
- 63.Kuleshov M. V., Jones M. R., Rouillard A. D., Fernandez N. F., Duan Q., Wang Z., Koplev S., Jenkins S. L., Jagodnik K. M., Lachmann A., McDermott M. G., Monteiro C. D., Gundersen G. W., Ma’ayan A., Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.B. Zhang, J. Zhu, Identification of key causal regulators in gene networks, in Proceedings of the World Congress on Engineering (WCE’13), London, UK, 3 to 5 July 2013. [Google Scholar]
- 65.Stark C., Breitkreutz B.-J., Reguly T., Boucher L., Breitkreutz A., Tyers M., BioGRID: A general repository for interaction datasets. Nucleic Acids Res. 34, D535–D539 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Leek J. T., Johnson W. E., Parker H. S., Jaffe A. E., Storey J. D., The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Choi J. K., Yu U., Kim S., Yoo O. J., Combining multiple microarray studies and modeling interstudy variation. Bioinformatics 19, i84–i90 (2003). [DOI] [PubMed] [Google Scholar]
- 68.Ramasamy A., Mondry A., Holmes C. C., Altman D. G., Key issues in conducting a meta-analysis of gene expression microarray datasets. PLOS Med. 5, e184 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chesler E. J., Wilson S. G., Lariviere W. R., Rodriguez-Zas S. L., Mogil J. S., Identification and ranking of genetic and laboratory environment factors influencing a behavioral trait, thermal nociception, via computational analysis of a large data archive. Neurosci. Biobehav. Rev. 26, 907–923 (2002). [DOI] [PubMed] [Google Scholar]
- 70.Gao V., Vitaterna M. H., Turek F. W., Validation of video motion-detection scoring of forced swim test in mice. J. Neurosci. Methods 235, 59–64 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Laposky A. D., Shelton J., Bass J., Dugovic C., Perrino N., Turek F. W., Altered sleep regulation in leptin-deficient mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 290, R894–R903 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/7/eaat1294/DC1
Fig. S1. Distributions of phenotypic measurements.
Fig. S2. Regulatory relationships among gene coexpression network modules.
Data file S1. Phenotypic categories and descriptions.
Data file S2. eQTL and phenotypic associations.
Data file S3. Coexpression network modules.
Data file S4. SD signatures.
Data file S5. MDD signatures.
Data file S6. Bayesian network and key drivers.
Supplementary Methods