
Fig. 4. In silico molecular docking calculations for Oxa12.
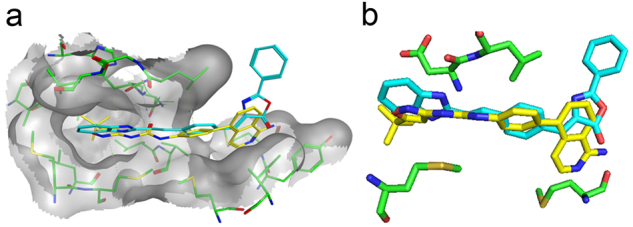
a Optimal poses obtained inside RIP1 active site (grey) for compound Oxa12 (represented in stick model and coloured blue) compared with crystallographic ligand 1-aminoisoquinoline inhibitor (PDBID: 4NEU) (yellow). b Compound Oxa12 and 4NEU co-crystallized inhibitor interacting with Asp156, Leu157, Met67, and Met95. Docking calculations were performed using the X-ray structure obtained for RIP1 complexed with 1-aminoisoquinoline inhibitor at resolution of 2.57 Å, PDBID: 4NEU, by the GOLD 5.2 software