

Abstract
Mutations in the Nebulin gene (NEB) may cause core-rod myopathy. The large size of the gene so far prevented inclusion of its routine analysis by didesoxy resequencing methodology in the diagnostic regime for muscular dystrophy cases. Here we report a 54-year-old female with a rare histological myopathy presentation of co-occurring cores and rods. The patient reported early childhood onset weakness. Muscle-MRI showed mainly proximal muscle involvement. We identified two compound heterozygous non-sense mutations in NEB (c.19653G > A, p.W6551* exon 127 and c.25441C > T, p.R8481* exon 182) using a comprehensive next generation sequencing (NGS)-based approach named Mendeliome Sequencing. The p.W6551* mutation has not been reported elsewhere. Early diagnosis by NGS shall be chased since even a scoliosis surgery at the age of 18 years had failed to initiate a neurological workup. Rather, cosmetic surgery for facial weakness had been performed recently, albeit with an unsatisfactory outcome.
Key words: core-rod myopathy, Nebulin, Mendeliome, muscle magnetic resonance imaging, cosmetic surgery
Introduction
Mutations in giant genes like Titin (TTN) and Nebulin (NEB) have been described to cause congenital myopathy. Till recently, the sheer size of these large genes made it difficult to sequence larger cohorts of patients in order to decipher genotype-phenotype correlations. However, next-generation sequencing now enables us to uncover the underlying mutations. Congenital myopathies are primary muscle diseases of broad phenotypic and genetic heterogeneity. Two of the more common congenital myopathies, Central Core Disease (CCD) (1) and Nemaline Myopathy (NM) (2), are associated with unique structural characteristics in muscle fibers. Histology of biopsies from affected individuals revealed oxidative enzyme activity lacking central cores in CCD or rod shaped Nemaline bodies in NM. CCD typically manifests as an early-onset but non-progressive disease most frequently caused by pathogenic ryanodine receptor 1 (RYR1) mutations (3, 4). Limb girdle and axial muscles are predominantly involved in core myopathies. Orthopedic complications such as hip dislocation, scoliosis, and foot deformities are also often found in CCD. NM is clinically and genetically more heterogeneous, ranging from severe fetal akinesia to adult-onset mild weakness, with causative mutations described in 10 genes involved in thin filament structure (MYPN, NEB, ACTA1, TPM2, TPM3, TNNT1, CFL2, LMOD3) (5) or ubiquitin pathway (KBTBD13, KLHL40, KLHL41). Nebulin (NEB) mutations, the most frequent cause of NM, account for ~50 % of cases (6). This large, 183-exon-spanning gene encodes a critical component of sarcomere thin filament assembly, regulating minimum filament length (7) and contractile force (8, 9). Large parts of the protein are comprised of repetitive 35-mer modules, containing an actin binding site, and super repeats of seven modules, containing a motif with putative tropomyosin binding ability (10). Patients with NEB mutations present with facial weakness, nasal speech, and dysarthria frequently. Sanger sequencing of Nebulin (11) is complicated by the large size and highly repetitive sequence of the gene, thus the screening of large cohorts was not feasible in the past. Therefore, many patients with NM were not genetically characterized in the past, which in turn resulted in an under-evaluation of this genotype-phenotype correlation in myopathy patients. We here applied a comprehensive screening panel for clinically significant genes, called the Mendeliome (12, 13), in a patient suffering from a myopathy featuring both cores and rods in muscle fibres for over 50 years, revealing compound heterozygous mutations in the Nebulin gene. We present the long-term disease course of a Nebulin-related Nemaline myopathy.
Material and methods
Patient
This is a case report of a patient in clinical follow-up at the Department of Neurology, University Hospital Cologne. The patient consented for publication of the clinical information. The genetic study was approved by the local institutional review board of the University Hospital Cologne and informed consent for genetic work-up was obtained from the patient and family members.
Genomic analysis
We used targeted gene sequencing on the Illumina Trusight One panel, (Illumina, San Diego, CA, USA) providing comprehensive coverage of 4.813 clinically relevant genes, a technique also called Mendeliome Sequencing (MS) (further details see supplementary material page 127) (13, 14).
Neuropathology
The biopsied left deltoid muscle tissue was snap-frozen in isopentane (Fluka, Neu-Ulm, Germany), pre-cooled in liquid N2 immediately after biopsy, and stored at – 80°C (15). Muscle biopsy work-up methods are presented in the supplementary material (page 127).
Muscle magnetic resonance imaging (MRI)
Muscle MRI of the lower limb was performed using conventional T1-, T2-, and STIR-weighted spin echo. Non-contrast images were obtained from pelvis, thighs, and legs.
Results
Case presentation
This 54-year-old female was referred because of general weakness, difficulty in climbing stairs, and stress dyspnea. Symptoms had been present since early childhood, and she reported that she had been slower in walking and running than her peers during childhood. She developed scoliosis in early adolescence and underwent stab osteosynthesis surgery at the age of 18 years. Furthermore, the patient had cosmetic surgery a few years before being referred to us due to bilateral ptosis. The family history was unremarkable for neuromuscular diseases. On clinical examination at referral, she had a myopathic face, high-arch palate, nasal speech, and right-sided tongue atrophy with deviation to the right. Weakness was prominent at distal muscles of upper limbs and proximal muscles of lower limbs. Substantial weakness was also noted at neck flexor muscles and axial muscles, which resulted in an inability to rise from supine position with positive Gowers’ sign. However, the patient was able to walk without any help. Deep tendon reflexes were equal at both sides, and sensory system was intact. Serum studies demonstrated a slightly elevated creatine kinase (CK) level of 428 U/L. Serum lactate levels were normal. Electromyography (EMG) showed sparse pathological spontaneous activity, small polyphasic potentials, early density interference, and occasional pseudomyotonic discharges. Subsequently, a muscle biopsy was performed from the left deltoid muscle.
Muscle magnetic resonance imaging
MRI of the lower limbs revealed predominant proximal muscle involvement (Fig. 1B). Within the thigh, the ischiocrural and medial muscles showed fatty atrophic changes with the semimembranosus muscles being mostly affected, whereas the anterolateral and the lower leg muscles revealed only minor changes in signal intensity without any selective involvement.
Figure 1.
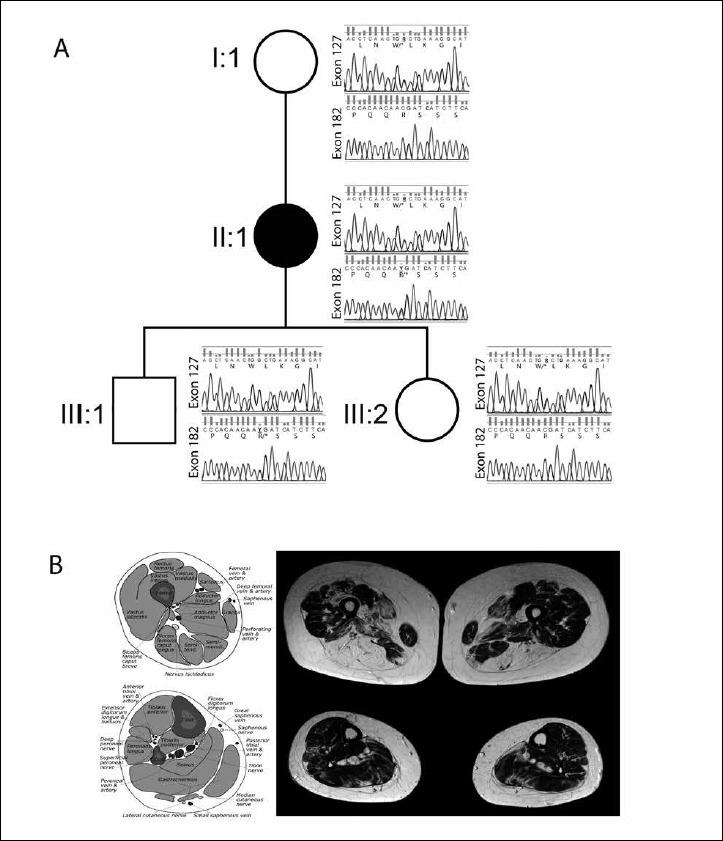
Pedigree of the patient (II:1) shows two heterozygous frameshift mutations in the nebulin (NEB) gene c.19653 G > A in exon 127 causing p.W6551* and c.25441 C > T in exon 182 causing p.R8481*. The bioinformatics analysis showed that the aberrant amino acid changes are located on the super repeats (S15-S21) and serine-rich domains of the protein. The variants were confirmed by Sanger sequencing, which revealed that the mother (I:1) and the daughter (III:2) have a recessive allele in exon127 (p.W6551*). Furthermore, the son (III:1) has a recessive allele in exon 182 (p.R8481*). The mother and the children were clinically unaffected (A). Muscle magnetic resonance imaging of the lower limbs showing predominant proximal involvement, within the thigh the ischiocrural and medial muscles shows fatty atrophic changes (B).
Histopathology
A deltoid muscle biopsy showed variable fiber sizes and increased internalization of nuclei on H&E staining (Fig. 2A). Modified Gomori trichrome staining revealed fuchsinophilic rods in most of the fibers (Fig. 2B, C). NADH staining showed numerous cores with absent enzyme activity in several fibers (Fig. 2D) while mATPase staining was inconspicuous (Fig. 2E). Toluidine blue staining detected especially rods and single cores on longitudinal sections (Fig. 2F). The presence of rods and cores was confirmed by electron microscopy (Fig. 3A-C). Up to 5 µm long rods could be identified, preferentially around nuclei. Numerous muscle fibers harbored core-like disintegration of myofibrils associated with the breakup of Z bands, enlargement of many muscle fiber mitochondria and contained paracrystalline inclusions; several muscle fiber nuclei showed prominent lobulation (Fig. 3D). The mitochondrial and myonuclear alterations have been considered secondary and unspecific.
Figure 2.
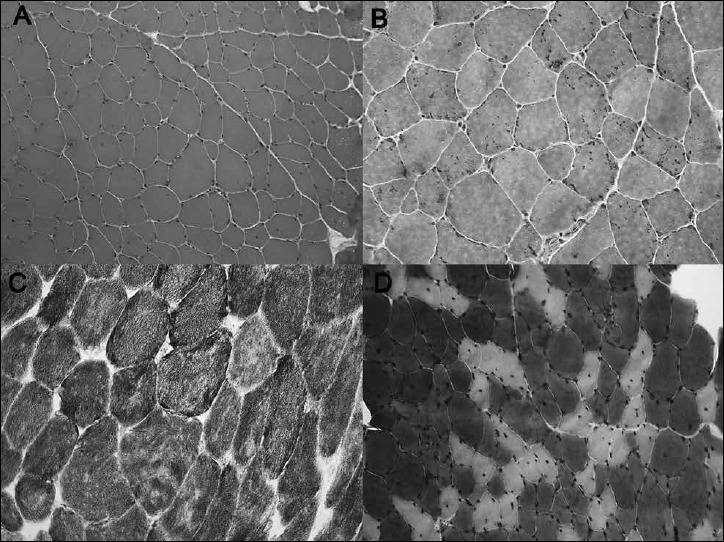
Muscle biopsy showed variation in fiber size and increased internalization of nuclei on H&E staining (A). Modified Gomori trichrome staining revealed fuchsinophilic rods in most of the fibers (B). NADH staining showed numerous cores with absent enzyme activity in several fibers (C), while mATPase staining was inconspicuous (D).
Figure 3.
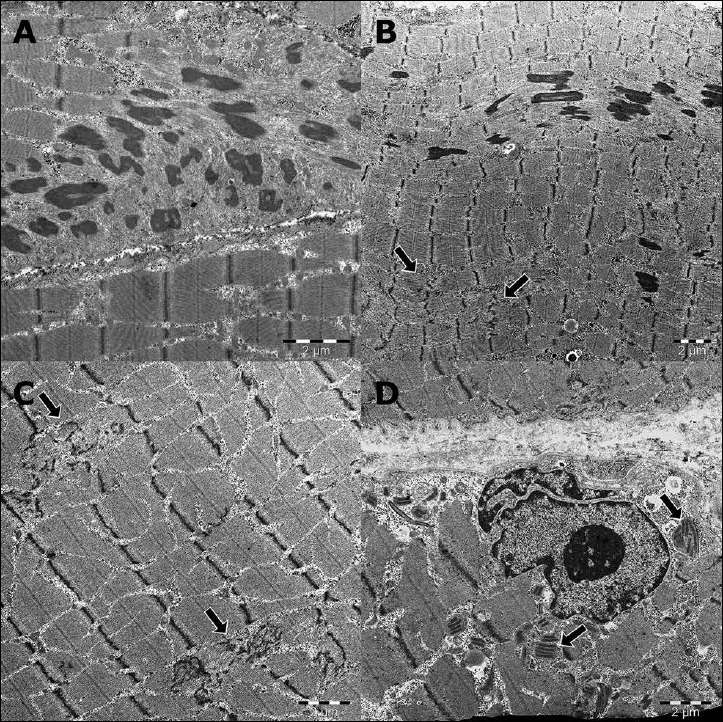
The presence of rods and cores were confirmed by electron microscopy, up to 5 µm long rods could be identified preferentially around nuclei. Numerous muscle fibers harbored core-like disintegration of myofibrils associated with breakup of Z bands (A-C). Many muscle fiber mitochondria were enlarged and contained paracrystalline inclusions, several muscle fiber nuclei showed prominent lobulation (D).
Genetic results
Mendeliome sequencing revealed two heterozygous frameshift mutations in the Nebulin (NEB) gene c.19653G > A in exon 127 causing p.W6551* and c.25441C > T in exon 182 causing p.R8481* according to NM_001271208.1 (Fig. 1A). The p.R8481* allele was detected in 6/120725 population chromosomes by the Exome Aggregation Consortium (http://exac.broadinstitute.org), 10/276544 heterozygous alleles in the gnomAD server (http://gnomad-beta.broadinstitute.org), the ClinVar (https://www.ncbi.nlm.nih.gov/clinvar) accession number is 449500 and was also reported earlier by Lehtokari et al. (6). The clinical significance is reported as pathogenic for this mutation (https://www.ncbi.nlm.nih.gov/clinvar). But the p.W6551* mutation was not reported in the Exome Variant Server, gnomAD, ClinVar and 1000 genomes databases. Arginine at 8481 and Tryptophan at 6551 are conserved in vertebrates (16).
Discussion
This case presents the long-term disease course of core-rod-myopathies revealing compound heterozygous stop-mutations in the Nebulin gene. The p.W6551* is a novel mutation, not described in the literature to our knowledge. The p.R8481* mutation in compound heterozygosity with another more proximal frameshifting mutation p.Ser2820fs was reported earlier in a family with typical Nemaline myopathy features by Lehtokari et al., however lacking further clinical details (6). The combination of cores and rods as revealed by muscle biopsy has been described in a few NEB-related cases with both a severe form (17) and mild forms with normal strength in the leg muscles (6, 18).
The two truncating mutations (p.W6551* in exon 127 and p.R8481* in exon 182) are located in super repeats (S15-S21) and serine rich domains of the protein, respectively (19). The repeat domains have been shown to interact with thin filament components, whereas C-terminal regions have unique sequences for binding to proteins at the Z-line. The serine-rich region at the C-terminal contains several predicted phosphorylation sites adjacent to a Src homology 3 (SH3) domain, which was shown to play a vital role in binding to titin and myopalladin (20).
The muscle biopsy represents a rod-core myopathy. The mitochondrial and myonuclear alterations revealed by electron microscopy were considered to be secondary and not specific. Unfortunately, no immunoblotting or immunohistochemistry was performed earlier for Nebulin and no frozen muscle biopsy was left over to perform these investigations. However, other publications show reduced levels of Nebulin in patients with truncating mutations compared to healthy controls (21). Thus, we may speculate that our further C-terminal stop mutation might lead to a truncated Nebulin protein expression with reduced levels.
The muscle MRI of our case showed predominantly proximal involvement, whereas in previous reports an early and isolated involvement of the tibialis anterior muscle (22) or a predominant distal involvement (12) has been described as the most typical finding in NEB-related NM. A possible explanation for this discrepancy could be that corresponding to the expanding range of phenotypes and the identification of an increasing number of mutations in Nebulin, we are now uncovering the entire clinical spectrum and earlier have been underpowered to claim a specific MRI pattern.
The number of genes associated with congenital myopathies exceeds 20, including giant genes such as TTN with 363 exons, NEB with 183 exons, and RYR1 with 106 exons (23, 24). NGS technologies, which provide a more practical and cost-effective approach compared to conventional gene sequencing techniques, are now being widely implemented in the diagnosis of neuromuscular diseases.
Finally, this case illustrates that despite typical muscle pathology features the diagnosis of a structural myopathy was not made until the patient was 54 years old. Interestingly, even the operation (to correct for scoliosis) at the age of 18 did not trigger further neurological workup. The patient had already shown symptoms during childhood, a congenital form that is not compatible with an adult manifestation. Unfortunately, cosmetic surgery for ptosis and facial weakness was performed with a predictable unsatisfactory outcome.
Acknowledgements
Our patient’s and her family’s cooperation is gratefully acknowledged. We are grateful to the Cologne Center for Genomics for performing the Next Generation Sequencing. We thank Mert Karakaya for technical assistance.
Supplementary Material
Genomic Analysis
We used targeted gene sequencing on the Illumina Trusight One panel (Illumina, San Diego, CA, USA) providing comprehensive coverage of 4.813 clinically relevant genes, a technique also called Mendeliome Sequencing (MS) (1). The coverage was 118-fold i.e., 10x coverage for 98.1% of target sequences and 30x coverage for 93.8% of target sequences. The Cologne Center for Genomics VARBANK pipeline v.2.12 (https://varbank.ccg.uni-koeln.de/) was used for data analysis. Variants were filtered for dominant and compound heterozygous mutations, as well as reading quality and allele frequency among populations. Filtered functional variants in compound heterozygous inheritance were tested based on in silico prediction databases as described elsewhere (2).
Muscle biopsy work–up
The biopsied left deltoid muscle tissue was snap-frozen in isopentane (Fluka, Neu-Ulm, Germany), pre-cooled in liquid N2 immediately after biopsy, and stored at -80°C. For enzyme histochemistry, 9 µm thick serial frozen sections were stained with hematoxylin and eosin (H&E), adenosine triphosphatase (mATPase, pH 9.4, 4.6, and 4.2), nicotinamide adenine dinucleotide dehydrogenase-tetrazolium reductase (NADH TR), modified Gomori trichrome, and cytochrome oxidase (COX). Semithin section histology and preparation for electron microscopy were performed as described previously (3). For electron microscopy, glutaraldehyde-fixed muscle specimen was post-fixed with OsO4 1% in 0.1M cacodylate buffer containing 50 mM K3Fe(CN)6. Muscle specimen were embedded in epoxy resin. Ultra-thin sections were contrasted with uranyl acetate and lead citrate and, finally, examined with a Philips EM 400 T electron microscope.
References
- 1.Fazeli W, Karakaya M, Herkenrath P, et al. Mendeliome sequencing enables differential diagnosis and treatment of neonatal lactic acidosis. Mol Cell Pediatr 2016;3:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Willkomm L, Heredia R, Hoffmann K, et al. Homozygous mutation in Atlastin GTPase 1 causes recessive hereditary spastic paraplegia. J Hum Genet 2016;61:571-3. [DOI] [PubMed] [Google Scholar]
- 3.Weis J, Schroder JM. Adult polyglucosan body myopathy with subclinical peripheral neuropathy: case report and review of diseases associated with polyglucosan body accumulation. Clin Neuropathol 1988;7:271-9. [PubMed] [Google Scholar]
Footnotes
Source of funding
This work was funded by Deutsche Forschungsgemeinschaft, Germany grant CI 218/1–1 to Dr. Sebahattin Cirak.
References
- 1.Magee KR, Shy GM. A new congenital non-progressive myopathy. Brain 1956;79:610-21. [DOI] [PubMed] [Google Scholar]
- 2.Shy GM, Engel WK, Somers JE, et al. Nemaline Myopathy. A new congenital myopathy. Brain 1963;86:793-810. [DOI] [PubMed] [Google Scholar]
- 3.Quane KA, Healy JM, Keating KE, et al. Mutations in the ryanodine receptor gene in central core disease and malignant hyperthermia. Nature genetics 1993;5:51-5. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Y, Chen HS, Khanna VK, et al. A mutation in the human ryanodine receptor gene associated with central core disease. Nature genetics 1993;5:46-50. [DOI] [PubMed] [Google Scholar]
- 5.Lornage X, Malfatti E, Cheraud C, et al. Recessive MYPN mutations cause cap myopathy with occasional nemaline rods. Ann Neurol 2017;81:467-73. [DOI] [PubMed] [Google Scholar]
- 6.Lehtokari VL, Kiiski K, Sandaradura SA, et al. Mutation update: the spectra of nebulin variants and associated myopathies. Hum Mutat 2014;35:1418-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Witt CC, Burkart C, Labeit D, et al. Nebulin regulates thin filament length, contractility, and Z-disk structure in vivo. EMBO J 2006;25:3843-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chandra M, Mamidi R, Ford S, et al. Nebulin alters cross-bridge cycling kinetics and increases thin filament activation: a novel mechanism for increasing tension and reducing tension cost. J Biol Chem 2009;284:30889-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bang M-L, Li X, Littlefield R, et al. Nebulin-deficient mice exhibit shorter thin filament lengths and reduced contractile function in skeletal muscle. J Cell Biol 2006;173:905-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang K, Knipfer M, Huang QQ, et al. Human skeletal muscle nebulin sequence encodes a blueprint for thin filament architecture. Sequence motifs and affinity profiles of tandem repeats and terminal SH3. J Biol Chem 1996;271:4304-14. [DOI] [PubMed] [Google Scholar]
- 11.Pelin K, Hilpela P, Donner K, et al. Mutations in the nebulin gene associated with autosomal recessive nemaline myopathy. Proceedings of the National Academy of Sciences of the United States of America 1999;96:2305-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scoto M, Cullup T, Cirak S, et al. Nebulin (NEB) mutations in a childhood onset distal myopathy with rods and cores uncovered by next generation sequencing. EJHG 2013;21:1249-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fazeli W, Karakaya M, Herkenrath P, et al. Mendeliome sequencing enables differential diagnosis and treatment of neonatal lactic acidosis. Mol Cell Pediatr 2016;3:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Willkomm L, Heredia R, Hoffmann K, et al. Homozygous mutation in Atlastin GTPase 1 causes recessive hereditary spastic paraplegia. J Hum Genet 2016;61:571-3. [DOI] [PubMed] [Google Scholar]
- 15.Weis J, Schroder JM. Adult polyglucosan body myopathy with subclinical peripheral neuropathy: case report and review of diseases associated with polyglucosan body accumulation. Clin Neuropathol 1988;7:271-9. [PubMed] [Google Scholar]
- 16.Schwarz JM, Rodelsperger C, Schuelke M, et al. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 2010;7:575-6. [DOI] [PubMed] [Google Scholar]
- 17.Romero NB, Lehtokari VL, Quijano-Roy S, et al. Core-rod myopathy caused by mutations in the nebulin gene. Neurology 2009;73:1159-61. [DOI] [PubMed] [Google Scholar]
- 18.Park YE, Shin JH, Kang B, et al. NEB-related core-rod myopathy with distinct clinical and pathological features. Muscle Nerve 2016;53:479-84. [DOI] [PubMed] [Google Scholar]
- 19.Donner K, Sandbacka M, Lehtokari VL, et al. Complete genomic structure of the human nebulin gene and identification of alternatively spliced transcripts. EJHG 2004;12:744-51. [DOI] [PubMed] [Google Scholar]
- 20.Yamamoto DL, Vitiello C, Zhang J, et al. The nebulin SH3 domain is dispensable for normal skeletal muscle structure but is required for effective active load bearing in mouse. J Cell Sci 2013;126:5477-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lawlor MW, Ottenheijm CA, Lehtokari VL, et al. Novel mutations in NEB cause abnormal nebulin expression and markedly impaired muscle force generation in severe nemaline myopathy. Skeletal muscle 2011;1:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jungbluth H, Davis MR, Muller C, et al. Magnetic resonance imaging of muscle in congenital myopathies associated with RYR1 mutations. NMD 2004;14:785-90. [DOI] [PubMed] [Google Scholar]
- 23.Wang H, Castiglioni C, Kacar Bayram A, et al. Insights from genotype-phenotype correlations by novel SPEG mutations causing centronuclear myopathy. NMD 2017;27:836-42. [DOI] [PubMed] [Google Scholar]
- 24.Vasli N, Harris E, Karamchandani J, et al. Recessive mutations in the kinase ZAK cause a congenital myopathy with fibre type disproportion. Brain 2017;140:37-48. [DOI] [PMC free article] [PubMed] [Google Scholar]