

Abstract
Background and Objective
Verinurad (RDEA3170) is a high-affinity, selective URAT1 transporter inhibitor in development for treating gout and asymptomatic hyperuricemia. This Phase I, single-dose study investigated the pharmacokinetics, pharmacodynamics, and safety of verinurad in adults with renal impairment and controls with normal renal function.
Methods
Males aged 18–85 years were enrolled with serum urate (sUA) 4.5–10 mg/dl and creatinine clearance 60– < 90, 30– < 60, 15– < 30, or ≥ 90 ml/min (mild, moderate, severe renal impairment and controls, respectively; n = 7/8). Verinurad 15 mg was administered orally under fasted conditions. Serial plasma/serum and urine samplings were 30 min pre-dose to 72 h post-dose.
Results
Compared to controls, verinurad maximum observed plasma concentration increased by 53, 73, and 128% and area under the concentration-time curve increased by 24, 148, and 130%, in subjects with mild, moderate, and severe renal impairment, respectively; renal clearance decreased by 5, 42, and 79%. Exposures of major verinurad metabolites also increased with increasing renal impairment. Verinurad decreased sUA in all groups, with greater maximal changes in control and mild renal impairment than moderate and severe impairment groups (− 38.3, − 36.9, − 20.5, − 12.6%, respectively). There were no adverse event-related withdrawals or clinically meaningful changes in laboratory values.
Conclusion
Exposures of verinurad and metabolites increased with decreasing renal function. Consistent with the renal-dependent mechanism of action of verinurad, increasing severity of renal impairment was associated with decreased sUA lowering. Verinurad safety assessments were similar regardless of renal impairment. Continued investigation of verinurad is warranted in patients with gout and renal impairment.
ClinicalTrials.gov identifier
Key Points
| Verinurad is a potent uricosuric agent in clinical development for the treatment of gout and hyperuricemia. |
| In this study of adults with mild, moderate, or severe renal impairment and controls, plasma exposures of verinurad and its major metabolites increased with increasing levels of renal impairment. |
| Verinurad 15 mg lowered serum urate levels and increased renal excretion of uric acid in all groups, with smaller absolute changes in the moderate and severe renal impairment groups, consistent with the renal-dependent mechanism of action of verinurad. |
Introduction
Gout is an inflammatory arthritis associated with elevated serum urate (sUA) levels. Long-term management goals are to maintain sUA concentrations below target levels (e.g., < 6 mg/dl), which leads over time to dissolution of monosodium urate crystal deposits and alleviation of gout symptoms [1–4]. Common management approaches incorporate a urate-lowering therapy (ULT), typically a xanthine oxidase inhibitor (XOI) such as allopurinol or febuxostat, as the first-line therapy.
Many patients fail to attain their target sUA using a single ULT alone [4, 5]. For these patients, management guidelines recommend combining ULTs with complementary mechanisms of action [6, 7]. Typical combination therapies include an XOI to reduce production of urate and a uricosuric—such as probenecid, benzbromarone, or lesinurad—to increase renal excretion of uric acid [8].
Renal impairment is a common co-morbidity in patients with gout that may cause elevation in sUA [9–12]. Hyperuricemia, in turn, can contribute to further renal impairment, creating a vicious cycle. While sUA lowering may slow the deterioration in renal function in these patients, the presence of renal impairment complicates management in terms of dosing, safety, or response to ULTs [8]. In the case of allopurinol, many patients with both gout and renal impairment fail to receive optimized dosing because of perceived safety concerns with uptitration [13]. For these patients, combining two therapies with complementary mechanisms of action represents a promising approach to achieving target sUA without the need for allopurinol dose uptitration.
Lesinurad is a specific inhibitor of the uric acid transporter URAT1, which was recently approved in the USA and Europe in combination with an XOI for treating hyperuricemia associated with gout. In the Phase III clinical development programme, treatment with lesinurad 200 and 400 mg, in combination with febuxostat, resulted in more patients achieving target sUA and experiencing a reduction in overall tophus area compared with febuxostat alone [14]. This treatment combination, however, showed only a trend toward reduction in gout flares at the highest lesinurad dose tested (400 mg), a dose that had increased renal adverse effects (compared with lesinurad 200 mg plus febuxostat or febuxostat alone) and was neither submitted to nor approved by US and European regulatory agencies.
The clinical trial experience with ULTs has demonstrated that greater lowering of sUA below target levels is associated with improved outcomes (e.g., lower gout flare incidence, greater tophus area reduction) [1, 3, 15–17]. Verinurad, a URAT1 inhibitor in clinical development, demonstrates high potency with a half-maximal inhibitory constant of 36 nM in inhibiting URAT1 [18] and in humans provides sUA lowering at doses as low as 2.5 mg [19–21]. The potency of verinurad together with its pharmacokinetic properties may enable it to be used as a low-dose drug, with potentially less drug interaction issues in gout patients receiving concurrent treatments for comorbidities such as hypertension, cardiovascular disease, diabetes, and obesity. The combination of verinurad with an XOI results in a urinary uric acid profile that may offer a low potential for renal adverse effects [22] while also lowering sUA to a greater extent than either agent alone at the same or even higher monotherapy doses [23–25].
Verinurad is predominantly metabolized by glucuronyl transferases to form M1 and by CYP3A4 to form the N-oxide metabolite (M4), which subsequently undergoes secondary metabolism by glucuronyl transferases to form M8. M8 formation can also occur via CYP2C8-mediated metabolism of M1. Metabolites M1 and M8 are the predominant circulating metabolites of verinurad and are devoid of URAT1 activity [26]. Verinurad is predominantly eliminated by hepatic metabolism, with about 2% of parent drug excreted unchanged in urine [21]. The bioavailability of verinurad is unknown, as clinical evaluation has not been conducted, but in animal studies in rats, dogs, and monkeys it ranges from 57% to 80% (unpublished data).
Gout patients frequently have impaired kidney function. Renal failure has been shown to significantly reduce non-renal clearance and alter the bioavailability of drugs predominately metabolized by the liver [27]. The aim of this Phase I study was to investigate the pharmacokinetics, pharmacodynamics, and safety of verinurad and its metabolites in adults with mild, moderate, or severe renal impairment and matched controls (RDEA3170-108, NCT02219516).
Methods
Study Design and Subjects
The single-dose, open-label study included subjects with normal renal function or renal impairment measured by estimated creatinine clearance (eCrCl), calculated by the Cockcroft-Gault formula [28]. The severity of renal impairment was categorized as mild (eCrCl 60– < 90 ml/min), moderate (30– < 60 ml/min), and severe (15– < 30 ml/min). All subjects were male, aged 18–85 years, with sUA levels of 4.5–10 mg/dl.
Screening to determine study eligibility was performed within 21 days before verinurad administration. Subjects in the normal renal function group (eCrCl ≥ 90 ml/min) were enrolled after the other groups to match the baseline characteristics of age and body mass index. Subjects checked into the study centre on Day − 2 and stayed until the last procedures were completed. All subjects had a follow-up visit on Day 7.
Oral verinurad was administered once under fasted conditions at a dose of 15 mg, which has been shown to have significant pharmacodynamic activity [19, 29]. Subjects receiving other medications for gout (including XOIs), strong or moderate CYP3A inhibitors or P-glycoprotein inhibitors, or strong or moderate CYP2C9 inhibitors or organic anion transporter inhibitors (OAT1 or OAT3) within 2 weeks or seven half-lives before the dose of study medication were excluded.
The study was conducted in compliance with the ethical principles of Good Clinical Practice defined by the International Conference on Harmonisation and the Declaration of Helsinki. The protocol and consent form were approved by independent institutional review boards (Crescent City IRB, New Orleans, LA, USA; Aspire IRB, LLC, Santee, CA, USA; Western Institutional Review Board, Puyallup, WA, USA) and all subjects provided informed consent to participate in the study.
Blood and Urine Sampling
Serial blood samples for pharmacokinetic and pharmacodynamic analyses were collected on Day –1 and Day 1 at frequent preset times up to 72 h post-dose. Urine samples (total catch) for pharmacokinetic and pharmacodynamic analyses were collected at 6- to 12-h intervals to 72 h post-dose.
Endpoints and Determinations
Plasma pharmacokinetic parameters were calculated by non-compartmental analysis and included maximum observed concentration (Cmax), time to Cmax (Tmax), area under the concentration-time curve from time 0 to infinity (AUC0–∞), terminal half-life (t1/2), and total body clearance (CL/F) of verinurad and metabolites M1 and M8, where applicable. Urinary pharmacokinetic parameters included renal clearance (CLR), amount excreted in urine as unchanged drug from time 0 up to 72 h post-dose (Ae0–72), and fractional excretion from 0 to 72 h.
Pharmacodynamic parameters included maximal and mean percent change in sUA from baseline (time-matched to Day –1), amount of uric acid recovered in urine (AeUR), rate of excretion of urinary uric acid (ReUR), renal clearance of uric acid (CLUR), and fractional excretion of uric acid (FEUA) from 0 to 24 h.
Safety assessments comprised adverse events (AEs) coded according to the Medical Dictionary for Regulatory Activities (MedDRA; version 17.0), clinical laboratory evaluations, vital signs, electrocardiograms, and physical examinations. Any hematology, chemistry, or urinalysis abnormalities that were considered clinically relevant were to be assigned a severity rating by the investigator based on Rheumatology Common Toxicity Criteria version 2.0 [19, 30].
Analytical Methods
Pharmacokinetic samples were analysed by Ardea Biosciences, Inc. (San Diego, CA, USA). All analytical methods were validated following US Food and Drug Administration bioanalytical method validation guidance [31]. The analytical methodology for the quantitation of verinurad and metabolite M1 in plasma samples was a combination method similar to the methodology described by Kankam et al. [22], although that methodology did not include M1. When M1 was included in the methodology, it (and its internal standard, IS) chromatographed as diastereomers and each peak was integrated and the resulting areas summed for concentration determination. Addition of M1 to the methodology necessitated inclusion of mass spectrometer transitions for M1 and IS (precursor → product ion transitions of m/z were 525 → 263 (M1) and 531 → 263 (IS [D6]RDEA3170-M1) and a dwell time of180 ms for M1 and 30 ms for the IS). In this study, plasma quality control (QC) samples showed % Theoretical [percent coefficient of variation (%CV)] of 97.7–104.0% (3.0–8.5%) for verinurad and 94.0–111.2% (2.8–6.9%) for M1.
Quantitative determination of verinurad metabolite M8 (measured as total glucuronide conjugated M4 and unconjugated M4) in acidified plasma (1% phosphoric acid, v/v) involved alkalizing samples with 2 M potassium hydroxide solution, incubating at room temperature to completely convert M8 (acyl glucuronidated M4) to its aglycone counterpart (M4), then extraction by protein precipitation with 2% formic acid in acetonitrile (v/v) containing internal standard [D6]RDEA3170-M4. Extracted samples were injected onto a Kinetex C18, 4.6 × 50 mm, 2.6 µm column (Phenomenex, Torrance, CA, USA), which was connected to an Agilent 1100 HPLC system (Agilent Technologies, Santa Clara, CA, USA) running a gradient composed of 0.1% formic acid in water (v/v, mobile phase A) with 0.1% formic acid in acetonitrile, (v/v, mobile phase B) at a flow rate of 0.9 ml/min. The column effluent was introduced into an API 5000 triple quadrupole mass spectrometer (AB Sciex, Framingham, MA, USA) operated in positive TurbolonSpray mode. Under unit/low resolution (Q1/Q3), selected reaction monitoring was used to monitor the precursor → product ion transitions of m/z 365 → 279 (M4), 371 → 280 ([D6]M4), and a dummy ion transition of 600 → 200 (to prevent cross-talk), with dwell times of 150 ms for M4, 30 ms for the internal standard, and 20 ms for the dummy ion. The method had a 0.1 ng/ml lower limit of quantitation (LLOQ). M4 QCs showed % Theoretical (%CV) during study conduct of 97.7–101.3% (6.7–7.9%) and M8 QCs (included to show complete conversion) calculated at 100.3–103.7% (5.0–5.5%). Based on limited circulating unconjugated M4, the ng/ml concentration of M8 (acyl glucuronidated RDEA3170-M4) in the clinical sample (CM8, MW = 540.54) was calculated by converting the ng/ml M4 total concentrations (CM4Tot, MW = 364.42) using their respective molecular weights (g/mol), i.e. CM8 = CM4Tot × 540.54/364.42.
Analytical methodology for the quantitation of verinurad in urine samples is described by Shen et al. [21]. Verinurad urine QCs showed % Theoretical (%CV) of 102.3–102.8% (2.9–7.7%) during this study.
For quantitative determination of M1 in urine, samples were diluted with 0.2% formic acid in acetonitrile (v/v) containing [D6]RDEA3170-M1 as an internal standard. An aliquot of the extract was further diluted with 0.2% formic acid in water (v/v) and injected onto a Kinetex C18, 4.6 × 50 mm, 2.6 µm column (Phenomenex), which was connected to an Agilent 1100 HPLC system (Agilent Technologies) running a gradient composed of 0.1% formic acid in water (v/v, mobile phase A) with 0.1% formic acid in acetonitrile (v/v, mobile phase B) at a flow rate of 0.9 ml/min. The column effluent was introduced into an API 4000 triple quadrupole mass spectrometer (AB Sciex) operated in positive TurbolonSpray mode. Under unit/unit resolution (Q1/Q3), selected reaction monitoring was used to monitor the precursor → product ion transitions of m/z 525 → 263 (RDEA3170-M1), 531 → 263 ([D6]RDEA3170-M1), and a dummy ion transition of 600 → 200, with dwell times of 200 ms for M1, 70 ms for the internal standard, and 30 ms for the dummy ion. The method had a 10.0 ng/ml LLOQ and M1 urine QCs showed % Theoretical (%CV) of 98.7–98.9% (3.6–5.5%) during study conduct.
Similar to its analysis in plasma, quantitation of verinurad metabolite M8 in urine was measured as total M4. Urine samples were alkalized with 2 M potassium hydroxide and incubated at room temperature to completely convert M8 to M4. The samples were then fortified with 2% formic acid in acetonitrile containing [D6]RDEA3170-M4 as internal standard and then diluted with acetonitrile:water, 1:2 (v/v). Extracted samples were injected onto a Synergi Polar-RP, 4.6 × 50 mm, 4 µm column (Phenomenex), which was connected to an Agilent 1100 HPLC system (Agilent Technologies) running a gradient composed of 0.1% formic acid in water (v/v, mobile phase A) with 0.1% formic acid in acetonitrile (v/v, mobile phase B) at a flow rate of 0.9 ml/min. The column effluent was introduced into an API 4000 triple quadrupole mass spectrometer (AB Sciex) operated in positive TurbolonSpray mode. Under unit/unit resolution (Q1/Q3), selected reaction monitoring was used to monitor the precursor → product ion transitions of m/z 365 → 279 (M4), 371 → 280 ([D6]M4), and a dummy ion transition of 600 → 200, with dwell times of 150 ms for M4, 30 ms for the internal standard, and 20 ms for the dummy ion. The method had a 10.0 ng/ml LLOQ. M4 QCs showed % Theoretical (%CV) during study conduct of 97.0–100.0% (2.1–3.3%) and M8 QCs calculated at 97.0–103.3% (3.0–3.3%). Based on limited unconjugated M4 in urine, the concentration of M8 (acyl glucuronidated RDEA3170-M4) was calculated using the conversion described above.
Pharmacodynamic parameters were determined by Early Clinical Biometrics at Covance using SAS software (SAS Institute, Inc., Cary, NC, USA). For pharmacodynamic analyses, urate or creatinine in serum and urine samples were analysed by an enzymatic method.
Data Analysis
Descriptive statistics are provided for pharmacokinetic, pharmacodynamic, and safety data in subjects categorized by their severity of renal impairment, using Phoenix WinNonlin v.6.3 (Pharsight Corporation, Mountain View, CA, USA) or SAS v.9.3 or later (SAS Institute, Inc.). To assess the effect of renal impairment on the pharmacokinetics of verinurad, M1, and M8, a fixed-effects model with treatment (renal impairment compared with normal renal function) as a fixed effect was used to analyse natural log-transformed pharmacokinetic parameters. Upon back-transformation, an estimate of the geometric mean ratios of the pharmacokinetic parameters with their corresponding 90% confidence intervals was generated.
All subjects who received at least one dose of verinurad and had evaluable pharmacodynamic or pharmacokinetic data constituted the pharmacodynamic and pharmacokinetic populations, respectively. All subjects who received at least one dose of verinurad made up the safety population.
Results
Subjects
Thirty-one male subjects were enrolled in the study. Pharmacodynamic and pharmacokinetic data were based on seven or eight subjects in each renal function group. One subject with moderate renal impairment was excluded from the pharmacodynamic and pharmacokinetic populations because of concomitant treatment with carvedilol, a P-glycoprotein inhibitor.
Demographic and other baseline characteristics of subjects categorized by severity of renal impairment are summarized in Table 1.
Table 1.
Demographic and other baseline characteristics
| Characteristic | Normal renal function (N = 8) | Mild renal impairment (N = 8) | Moderate renal impairment (N = 8) | Severe renal impairment (N = 7) |
|---|---|---|---|---|
| Age, years, mean (SD) | 56 (4.7) | 63 (6.4) | 67 (14.9) | 58 (11.1) |
| Body weight, kg, mean (SD) | 86.5 (7.50) | 77.9 (14.16) | 89.4 (18.90) | 74.7 (13.07) |
| BMI, kg/m2, mean (SD) | 26.0 (1.20) | 26.2 (3.86) | 29.3 (4.84) | 26.2 (5.12) |
| Race, n (%) | ||||
| Black or African American | 1 (12.5) | 1 (12.5) | 2 (25.0) | 3 (42.9) |
| White | 7 (87.5) | 6 (75.0) | 6 (75.0) | 4 (57.1) |
| sUA (mg/dl), screening, mean (SD) | 5.7 (0.63) | 6.1 (0.84) | 7.3 (1.53)a | 8.6 (1.56) |
| Creatinine clearance (ml/min), screening, mean (SD) | 98.5 (10.67) | 69.6 (8.30) | 41.0 (7.90) | 23.2 (4.74) |
BMI body mass index, SD standard deviation, sUA serum urate
an = 7 subjects
Pharmacokinetics
Plasma pharmacokinetic parameters of verinurad and metabolites M1 and M8 following administration of verinurad 15 mg are summarized in Table 2 and Fig. 1. The median Tmax of verinurad and metabolites M1 and M8 was similar, ranging from 2.00 to 3.50 h in the normal renal function and renal impairment groups, indicating that metabolite formation occurred immediately after verinurad administration. The Cmax of verinurad increased by 53, 73, and 128% and the AUC0–∞ increased by 24, 148, and 130%, respectively, in the mild, moderate, and severe renal impairment groups compared to subjects with normal renal function. For M1, Cmax increased by 29, 112, and 253% and AUC0–∞ increased by 15, 213, and 316%, respectively, in the mild, moderate, and severe renal impairment groups; for M8, Cmax increased by 58, 56, and 270% and AUC0–∞ increased by 36, 141, and 527%, respectively. The t1/2 of verinurad, M1, and M8 increased with decreasing renal function, while the CL/F of verinurad decreased. Plasma protein binding of verinurad ranged from 97.6 to 98.7% and was not influenced by severity of renal function impairment.
Table 2.
Plasma pharmacokinetic parameters of verinurad, metabolite M1, and metabolite M8 following verinurad 15 mg administration in subjects with normal renal function and mild, moderate, and severe renal impairment
| Parameter | Renal function | |||||
|---|---|---|---|---|---|---|
| Normal (N = 8) | Mild impairment (N = 8) | Moderate impairment (N = 7) | Severe impairment (N = 7) | |||
| Verinurad | Cmax (ng/ml) | GM (95% CI) | 16.7 (12.6–22.3) | 25.6 (20.0–32.9) | 29.0 (22.8–37.0) | 38.2 (24.0–60.8) |
| GLSMR (90% CI) | – | 153% (115–203) | 173% (131–230) | 228% (155–336) | ||
| AUC0–∞ (ng·h/ml) | GM (95% CI) | 162 (120–218) | 201 (144–279) | 402 (255–634) | 372 (209–663) | |
| GLSMR (90% CI) | – | 124% (88.8–173) | 248% (168–366) | 230% (146–363) | ||
| Tmax (h), median (range) | GM (95% CI) | 2.50 (0.500–4.00) | 2.50 (1.00–4.00) | 3.00 (1.50–5.00) | 2.00 (1.50–4.00) | |
| GLSMR (90% CI) | – | NA | NA | NA | ||
| t½ (h) | GM (95% CI) | 13.0 (7.91–21.5) | 9.53 (7.42–12.2) | 20.6 (9.96–42.4) | 22.0 (14.2–34.1) | |
| GLSMR (90% CI) | – | NA | NA | NA | ||
| CL/F (L/h) | GM (95% CI) | 92.6 (68.7–125) | 74.8 (53.7–104) | 37.3 (23.7–58.9) | 40.3 (22.6–71.7) | |
| GLSMR (90% CI) | – | 80.8% (57.9–113) | 40.3% (27.3–59.5) | 43.5% (27.5–68.7) | ||
| M1 | Cmax (ng/ml) | GM (95% CI) | 25.7 (19.9–33.3) | 33.3 (24.0–46.3) | 54.6 (44.3–67.3) | 91.0 (65.8–126) |
| GLSMR (90% CI) | – | 129% (94.9–177) | 212% (165–273) | 353% (262–478) | ||
| AUC0–∞ (ng·h/ml) | GM (95% CI) | 233 (175–310) | 266 (172–412) | 727 (495–1070) | 969 (533–1760) | |
| GLSMR (90% CI) | – | 115% (77.6–169) | 313% (221–442) | 416% (262–662) | ||
| Tmax (h), median (range) | GM (95% CI) | 2.50 (1.50–4.00) | 3.00 (1.00–4.00) | 2.00 (1.50–6.00) | 2.00 (1.50–2.00) | |
| GLSMR (90% CI) | – | NA | NA | NA | ||
| t½ (h) | GM (95% CI) | 11.9 (6.61–21.3) | 10.6 (6.72–16.8) | 20.8 (10.9–39.8) | 21.2 (11.8–38.0) | |
| GLSMR (90% CI) | – | NA | NA | NA | ||
| CL/F (L/h) | GM (95% CI) | NA | NA | NA | NA | |
| GLSMR (90% CI) | – | NA | NA | NA | ||
| M8 | Cmax (ng/ml) | GM (95% CI) | 23.6 (19.5–28.5) | 37.2 (28.1–49.1) | 36.9 (22.8–59.7) | 87.2 (70.0–109) |
| GLSMR (90% CI) | – | 158% (123–203) | 156% (109–224) | 370% (299–458) | ||
| AUC0–∞ (ng·h/ml) | GM (95% CI) | 250 (195–320) | 339 (242–476) | 601 (324–1110) | 1570 (902–2720) | |
| GLSMR (90% CI) | – | 136% (99.3–186) | 241% (152–382) | 627% (411–956) | ||
| Tmax (h), median (range) | GM (95% CI) | 3.50 (2.00–5.00) | 3.00 (1.50–4.00) | 3.00 (2.00–6.00) | 3.00 (2.00–5.00) | |
| GLSMR (90% CI) | – | 136% (99.3–186) | 241% (152–382) | 627% (411–956) | ||
| t½ (h) | GM (95% CI) | 14.4 (8.62–24.0) | 10.7 (8.47–13.4) | 23.2 (14.0–38.4) | 22.8 (13.7–37.9) | |
| GLSMR (90% CI) | – | NA | NA | NA | ||
| CL/F (L/h) | GM (95% CI) | NA | NA | NA | NA | |
| GLSMR (90% CI) | – | NA | NA | NA | ||
AUC0–∞ area under the concentration–time curve from time zero to infinity, CI confidence interval, CL/F total body clearance, Cmax maximum observed concentration, GLSMR geometric least squares mean ratio versus normal renal function group, GM geometric mean, NA not applicable, t½ terminal half-life, Tmax time of maximum observed concentration
Fig. 1.
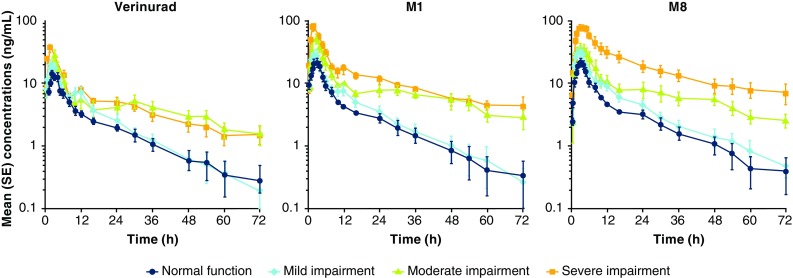
Mean (SE) plasma concentration versus time profile for verinurad, MI, and M8 following verinurad 15 mg administration in subjects with normal renal function and mild, moderate, and severe renal impairment. SE standard error
Urinary pharmacokinetic parameters for verinurad, M1, and M8 are summarized in Table 3. For verinurad, M1, and M8 renal clearance in urine from time zero to 72 h (CLR0–72) and fraction of drug excreted in urine unchanged from time zero to 72 h (fe0–72) tended to decrease with increasing severity of renal impairment. For example, CLR0-72 decreased in the mild, moderate, and severe renal impairment groups by 4.6, 42, and 79% for verinurad; 19, 60, and 90% for M1; and 26, 61, and 88% for M8, respectively.
Table 3.
Urinary pharmacokinetic parameters of verinurad, metabolite M1, and metabolite M8 following verinurad 15 mg administration in subjects with normal renal function and mild, moderate, and severe renal impairment
| Parameter | Renal function | |||||
|---|---|---|---|---|---|---|
| Normal (N = 8) | Mild impairment (N = 8) | Moderate impairment (N = 7) | Severe impairment (N = 7) | |||
| Verinurad | CLR0–72 (ml/min) | GM (95% CI) | 13.5 (9.03–20.2) | 12.9 (8.16–20.4) | 7.87 (3.36–18.4) | 2.86 (1.84–4.45) |
| GLSMR (90% CI) | – | 95.4% (60.6–150) | 58.3% (30.2–113) | 21.2% (13.6–32.8) | ||
| fe0–72 (%) | GM (95% CI) | 0.814 (0.508–1.31) | 1.02 (0.563–1.84 | 1.05 (0.561–1.96) | 0.362 (0.202–0.647) | |
| GLSMR (90% CI) | – | NA | NA | NA | ||
| M1 | CLR0–72 (ml/min) | GM (95% CI) | 201 (168–240) | 163 (108–245) | 80.4 (51.0–127) | 19.6 (14.4–26.7) |
| GLSMR (90% CI) | – | 81.0% (58.2–113) | 40.0% (28.5–56.1) | 9.78% (7.60–12.6) | ||
| fe0–72 (%) | GM (95% CI) | 11.7 (8.83–15.4) | 11.2 (7.87–16.0) | 13.1 (9.08–18.8) | 4.32 (2.71–6.88) | |
| GLSMR (90% CI) | – | NA | NA | NA | ||
| M8 | CLR0–72 (ml/min) | GM (95% CI) | 298 (255–347) | 220 (158–307) | 117 (82.4–167) | 35.0 (23.6–52.0) |
| GLSMR (90% CI) | – | 73.9% (56.3–97.0) | 39.5% (30.2–51.6) | 11.8% (8.77–15.8) | ||
| fe0–72 (%) | GM (95% CI) | 18.0 (13.8–23.4) | 18.8 (14.6–24.3) | 15.0 (8.32–26.9) | 11.9 (7.73–18.4) | |
| GLSMR (90% CI) | – | NA | NA | NA | ||
CI confidence interval, CLR0–72 renal clearance in urine from time zero to 72 h, fe0–72 fraction of drug excreted in urine unchanged from time zero to 72 h; GLSMR geometric least squares mean ratio versus normal renal function group, GM geometric mean, NA not applicable
Pharmacodynamics
Pre-dose mean sUA levels were higher in subjects with moderate and severe renal impairment (7.3 and 8.6 mg/dl, respectively) compared with the normal renal function or mild impairment groups (5.7 and 6.1 mg/dl, respectively).
Verinurad decreased mean sUA in all groups, with greatest decreases in the normal function and mild renal impairment groups. Mean (standard error [SE]) maximal percent change in sUA from baseline (Emax%) was − 38.3% (5.24), − 36.9% (4.79), − 20.5% (2.51), and − 12.6% (2.62), respectively, in the normal function and mild, moderate, and severe renal impairment groups, which occurred approximately 12 h post-dose. By 24 h post-dose, mean (SE) percent change in sUA from baseline was − 31.6% (2.9), − 31.7% (6.0), − 15.2% (3.1), and − 8.6% (2.9), respectively. The absolute changes in mean (SE) sUA concentration in each group over time are shown in Fig. 2.
Fig. 2.

Mean (SE) sUA concentration (time-matched to Day − 1) following verinurad 15 mg administration in subjects with normal renal function and mild, moderate, and severe renal impairment. SE standard error, sUA serum urate
Mean ReUR following verinurad administration is depicted in Fig. 3. ReUR was highest in the 0–6-h collection interval and returned to baseline by 12 h post-dose. Urinary pharmacodynamic parameters for uric acid are shown in Table 4. Following verinurad administration, AeUR, ReUR, and CLUR increased in all groups, with smaller absolute increases in the renal impairment groups compared with controls. Unlike pre-dose, where FEUA tended to increase with increasing renal insufficiency, administration of verinurad resulted in no difference in FEUA between the groups.
Fig. 3.

Mean (SE) urinary uric acid excretion rate (ReUR) following verinurad 15 mg administration in subjects with normal renal function and mild, moderate, and severe renal impairment. SE standard error, ReUR rate of excretion of urinary uric acid
Table 4.
Mean (SD) uric acid parameters at baseline and following verinurad 15 mg administration in subjects with normal renal function and mild, moderate, and severe renal impairment
| Parameter | Normal renal function (N = 8) | Mild renal impairment (N = 8) | Moderate renal impairment (N = 7) | Severe renal impairment (N = 7) | ||||
|---|---|---|---|---|---|---|---|---|
| Baseline | Verinurad | Baseline | Verinurad | Baseline | Verinurad | Baseline | Verinurad | |
| AeUR,0–24 h (mg) | 525 (100) | 969 (299) | 439 (153) | 880 (225) | 440 (188) | 707 (334) | 246 (111) | 393 (144) |
| ReUR,0–24 h (mg/h) | 21.9 (4.18) | 40.1 (12.1) | 18.4 (6.37) | 36.6 (9.39) | 18.4 (7.85) | 29.3 (13.8) | 10.3 (4.62) | 16.3 (6.02) |
| CLUR,0–24 h (ml/min) | 6.41 (1.53) | 17.1 (8.15) | 5.35 (1.40) | 16.5 (7.25) | 4.52 (2.11) | 8.62 (4.66) | 2.05 (0.99) | 3.49 (1.48) |
| FEUA,0–24 h (%) | 5.24 (1.22) | 13.8 (6.24) | 5.43 (0.76) | 16.2 (5.68) | 6.86 (2.90) | 13.0 (7.48) | 9.45 (7.19) | 12.7 (6.43) |
AeUR amount of urinary uric acid recovered in urine, CLUR renal clearance of uric acid, FEUA fractional excretion of uric acid, ReUR rate of urinary uric acid excretion, SD standard deviation
Safety
Verinurad 15 mg was well tolerated, with few AEs, no serious AEs, and no AE-related withdrawals during treatment. Treatment-emergent AEs (TEAEs) occurred at similar frequencies across the renal impairment and control groups (Table 5). One subject in each renal impairment group had one TEAE possibly related to verinurad, which were each categorized as gastrointestinal: dyspepsia in the mild, diarrhoea and nausea in the moderate, and diarrhea in the severe renal impairment group. One subject in the control group had oropharyngeal pain and rhinorrhea possibly related to verinurad.
Table 5.
Summary of treatment-emergent adverse events
| Normal renal function (N = 8) | Mild renal impairment (N = 8) | Moderate renal impairment (N = 8) | Severe renal impairment (N = 7) | |
|---|---|---|---|---|
| Subjects with adverse events | 1 (12.5%) | 3 (37.5%) | 1 (12.5%) | 2 (28.6%) |
| Number of adverse events | 5 | 3 | 2 | 2 |
| Severity (all adverse events) | ||||
| Mild | 1 (12.5%) [4] | 2 (25.0%) [2] | 1 (12.5%) [1] | 1 (14.3%) [1] |
| Moderate | 1 (12.5%) [1] | 1 (12.5%) [1] | 1 (12.5%) [1] | 1 (14.3%) [1] |
| Severe/life-threatening | 0 | 0 | 0 | 0 |
N number of subjects studied, () percentage of subjects with adverse events, [ ] number of adverse events
No clinically meaningful changes in laboratory values or vital signs were observed. One subject in the severe renal impairment group had serum creatinine elevation ≥ 1.5× baseline during treatment, which resolved (to ≤ 1.2× baseline) before study end and was not considered clinically significant.
Discussion
This Phase I, open-label study investigated the pharmacokinetics, pharmacodynamics, and safety of a single 15-mg dose of oral verinurad in adult subjects with mild, moderate, or severe renal impairment and matched normal controls.
Verinurad is predominantly eliminated by hepatic metabolism via glucuronyl transferase and CYP3A4 metabolism, with about 2% of the parent drug excreted unchanged in urine [26]. In this study, plasma exposure of verinurad and its acyl glucuronides increased with the extent of decreased renal function. Changes in metabolites were a magnitude greater than the increase in verinurad exposure. As M1 and M8 are exclusively renally eliminated, it was anticipated that a decrease in renal function would increase their plasma exposures and significantly decrease their urinary elimination. The CLR of M1 and M8 decreased similarly, from approximately 18% in mild to 90% in severe renal impairment. Consequently, the fe of M1 and M8 were not affected by renal function except in severe renal impairment, where the fe of M1 and M8 were approximately 63% and 33%, respectively, of values for normal renal function. This observation was also made with dapagliflozin, in which kidney metabolism decreased with increased renal insufficiency, attributed to loss of functional nephrons which manifested in decreased uridine glucuronyl transferase (UGT) activity [32].
Plasma exposures (Cmax and AUC) of verinurad increased modestly, as noted above, with decreasing levels of renal function, which is an unexpected finding considering that renal elimination is a minor pathway. The results of the study emphasize the importance of studying the impact of renal impairment for an agent that is eliminated mainly via the liver, as previously observed [27, 33, 34]. There are several possible explanations for the increased exposures of verinurad in subjects with renal impairment, including: (1) hydrolysis of acyl glucuronide metabolites to verinurad in the blood and small intestine (via biliary excretion), which is then reabsorbed into the circulation, and (2) reduced kidney metabolism via UGT. The verinurad acyl glucuronide metabolites are primarily eliminated by the kidney [26]. With impaired kidney function, renal elimination of M1is reduced, resulting in higher circulating blood levels which can either undergo hydrolysis-releasing verinurad and/or are taken up by the liver and undergo enterohepatic cycling, resulting in reabsorption of verinurad and contributing to the increase in systemic exposure. This has been demonstrated with mycophenolate mofetil, in which the acyl glucuronide metabolite undergoes enterohepatic recirculation, releasing aglycone for reabsorption into systemic circulation [35]. M8 may undergo some enterohepatic recycling as there are low levels of M4 excreted in the urine as it is not eliminated in feces [26]. Both M1 and M8 are shown to be substrates for multidrug resistance protein 2 (MRP2; unpublished data), supporting the potential for enterohepatic recycling, as both metabolites were not detected in feces [26]. Therefore, increased renal insufficiency is expected to increase the systemic exposure of metabolites which can increase exposure of verinurad via intestinal reabsorption via several mechanisms.
Subjects with greater renal impairment had increased sUA and decreased uric acid excretion at baseline compared with normal controls, which can be explained by decreased filtration of uric acid. Verinurad lowered sUA in all renal impairment groups. The sUA-lowering effect of verinurad in subjects with mild renal impairment was similar to normal controls (Emax% − 36.9 and − 38.3%, respectively). Consistent with the renal-dependent mechanism of action of verinurad, decreases in sUA were less in subjects with moderate or severe renal impairment (Emax% − 20.5 and − 12.6%, respectively), as there is likely less functional URAT1 activity in these two groups compared with normal controls owing to a lower number of functional nephrons [36]. Another explanation is that renal insufficiency may not allow verinurad to reach concentrations to achieve maximal inhibition of URAT1 on the apical membrane of the kidney proximal tubule, resulting in partial inhibition. Overall, the complexity of renal impairment alters uric acid directly by decreased filtration as well as decreased URAT1 activity by loss of nephrons or partial inhibition of URAT1.
Verinurad increased the amount and rate of uric acid excretion in all groups, with smaller absolute changes in the moderate and severe renal impairment groups compared with normal controls. Consistent with this observation, the effects of lesinurad (another URAT1 inhibitor) on urinary pharmacodynamics were also related to renal impairment status [37]. Pre-dose FEUA was greater in subjects with more severe renal impairment, which may reflect a decline in URAT1 activity due to decreased numbers of functional nephrons. FEUA did not differ between groups following verinurad administration, which may be explained by the net effect of multiple mechanisms: less uric acid reabsorption occurred, while less verinurad reached URAT1 due to a decreased glomerular filtration rate and less active uptake. Although verinurad dose adjustment may not be needed based on the degree of renal insufficiency, we would recommend that verinurad is not administered in patients with severe renal impairment, similar to the dosing recommendations for lesinurad. The benefit/risk of verinurad dosing in patients with severe renal impairment will need to be assessed in future studies.
The safety and tolerability of verinurad were similar to normal controls across all stages of renal impairment in this single-administration study. Laboratory parameters were consistent with impaired renal function and no clinically significant changes were associated with verinurad administration.
Potential limitations of the study include the inclusion of exclusively male and predominantly white subjects and the administration of a single dose of study drug. Further, the sUA response to verinurad may differ between subjects with or (as in this study) without gout, potentially as a consequence of differences in urate handling [38]. Another potential issue in the study was the proper handling of blood samples for accurate measurement of the acyl glucuronides (M1 and M8) of verinurad; however, care was taken to acidify plasma samples to ensure the integrity of the M1 and M8 measurements.
In conclusion, following single-dose administration of verinurad 15 mg, plasma exposures of verinurad and its major metabolites increased, and clearance and urinary excretion decreased, with increasing levels of renal impairment. Verinurad lowered sUA and increased renal excretion of uric acid in all groups, with smaller changes in moderate and severe renal impairment groups, consistent with the renal-dependent mechanism of action of verinurad. The safety and tolerability of verinurad were similar across all stages of renal impairment. These data support the continued investigation of oral verinurad in patients with gout who have renal impairment.
Acknowledgements
Editorial support for this manuscript was provided by Bill Wolvey of PAREXEL, which was funded by AstraZeneca.
Funding
Funding for this study was provided by Ardea Biosciences/AstraZeneca.
Conflict of interest
William B. Smith: Full-time employee of Volunteer Research Group, University of TN Medical Center, Knoxville, TN, USA. Jesse Hall, Amy Yamamoto, Susan Walker, Caroline A. Lee, Zancong Shen, David M. Wilson, Dongmei Zhou: Full-time employees of Ardea Biosciences, Inc., a member of the AstraZeneca Group, San Diego, CA, USA at the time of the study. Jolene K. Berg: Full-time employee of DaVita Clinical Research, Minneapolis, MN, USA. Michal Kazimir: Part-time employee of DaVita Clinical Research, Lakewood, CO, USA. Michael Gillen: Full-time employee of AstraZeneca Pharmaceuticals LP, Gaithersburg, MD, USA. Thomas C. Marbury: Full-time employee and equity owner of Orlando Clinical Research Center, Orlando, FL, USA.
Research involving human participants and/or animals
The study was approved by the appropriate institutional board and was performed in accordance with the ethical standards as laid down in the 1964 Declaration of Helsinki and its later amendments.
Informed consent
Study subjects provided written, informed consent to participate in the study and had the right to withdraw from the study at any time.
Data-sharing
De-identified patient data from this study is made available for scientific research on a case by case basis through AstraZeneca’s Data Request Portal: https://astrazenecagroup-dt.pharmacm.com//DT/Home/Index/
References
- 1.Pascual E, Andres M, Vela P. Gout treatment: should we aim for rapid crystal dissolution? Ann Rheum Dis. 2013;72(5):635–637. doi: 10.1136/annrheumdis-2012-202594. [DOI] [PubMed] [Google Scholar]
- 2.Perez-Ruiz F, Dalbeth N, Bardin T. A review of uric acid, crystal deposition disease, and gout. Adv Ther. 2015;32(1):31–41. doi: 10.1007/s12325-014-0175-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shiozawa A, Buysman EK, Korrer S. Serum uric acid levels and the risk of flares among gout patients in a US managed care setting. Curr Med Res Opin. 2017;33(1):117–124. doi: 10.1080/03007995.2016.1239193. [DOI] [PubMed] [Google Scholar]
- 4.Singh JA, Akhras KS, Shiozawa A. Comparative effectiveness of urate lowering with febuxostat versus allopurinol in gout: analyses from large U.S. managed care cohort. Arthritis Res Ther. 2015;17:120. doi: 10.1186/s13075-015-0624-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Becker MA, Fitz-Patrick D, Choi HK, Dalbeth N, Storgard C, Cravets M, et al. An open-label, 6-month study of allopurinol safety in gout: The LASSO study. Semin Arthritis Rheum. 2015;45(2):174–183. doi: 10.1016/j.semarthrit.2015.05.005. [DOI] [PubMed] [Google Scholar]
- 6.Khanna D, Fitzgerald JD, Khanna PP, Bae S, Singh MK, Neogi T, et al. American College of Rheumatology guidelines for management of gout. Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res (Hoboken). 2012;64(10):1431–1446. doi: 10.1002/acr.21772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Richette P, Doherty M, Pascual E, Barskova V, Becce F, Castaneda-Sanabria J, et al. 2016 updated EULAR evidence-based recommendations for the management of gout. Ann Rheum Dis. 2017;76(1):29–42. doi: 10.1136/annrheumdis-2016-209707. [DOI] [PubMed] [Google Scholar]
- 8.Rees F, Hui M, Doherty M. Optimizing current treatment of gout. Nat Rev Rheumatol. 2014;10(5):271–283. doi: 10.1038/nrrheum.2014.32. [DOI] [PubMed] [Google Scholar]
- 9.Bardin T, Richette P. Definition of hyperuricemia and gouty conditions. Curr Opin Rheumatol. 2014;26(2):186–191. doi: 10.1097/BOR.0000000000000028. [DOI] [PubMed] [Google Scholar]
- 10.Johnson RJ, Nakagawa T, Jalal D, Sanchez-Lozada LG, Kang DH, Ritz E. Uric acid and chronic kidney disease: which is chasing which? Nephrol Dial Transplant. 2013;28(9):2221–2228. doi: 10.1093/ndt/gft029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mende C. Management of chronic kidney disease: the relationship between serum uric acid and development of nephropathy. Adv Ther. 2015;32(12):1177–1191. doi: 10.1007/s12325-015-0272-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stamp LK, Chapman PT. Gout and its comorbidities: implications for therapy. Rheumatology (Oxford). 2013;52(1):34–44. doi: 10.1093/rheumatology/kes211. [DOI] [PubMed] [Google Scholar]
- 13.El-Zawawy H, Mandell BF. Managing gout: how is it different in patients with chronic kidney disease? Cleve Clin J Med. 2010;77(12):919–928. doi: 10.3949/ccjm.77a.09080. [DOI] [PubMed] [Google Scholar]
- 14.Dalbeth N, Jones G, Terkeltaub R, Khanna D, Kopicko J, Bhakta N, et al. Lesinurad, a selective uric acid reabsorption inhibitor, in combination with febuxostat in patients with tophaceous gout: a phase III clinical trial. Arthritis Rheumatol. 2017;69(9):1903–1913. doi: 10.1002/art.40159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perez-Ruiz F, Calabozo M, Pijoan JI, Herrero-Beites AM, Ruibal A. Effect of urate-lowering therapy on the velocity of size reduction of tophi in chronic gout. Arthritis Rheum. 2002;47(4):356–360. doi: 10.1002/art.10511. [DOI] [PubMed] [Google Scholar]
- 16.Perez-Ruiz F. Treating to target: a strategy to cure gout. Rheumatology (Oxford). 2009;48(Suppl 2):ii9–ii14. doi: 10.1093/rheumatology/kep087. [DOI] [PubMed] [Google Scholar]
- 17.Terkeltaub R, Perez-Ruiz F, Kopicko J, Fung M, Bhakta N, Adler S, et al. The safety and efficacy of lower serum urate levels: a pooled analysis of gout subjects receiving lesinurad and xanthine oxidase inhibitors. Arthritis Rheumatol. 2015;67(Suppl 10):Abstract 2354. http://acrabstracts.org/abstract/the-safety-and-efficacy-of-lower-serum-urate-levels-a-pooled-analysis-of-gout-subjects-receiving-lesinurad-and-xanthine-oxidase-inhibitors/. Accessed 27 Feb 2018.
- 18.Tan PK, Liu S, Gunic E, Miner JN. Discovery and characterization of verinurad, a potent and specific inhibitor of URAT1 for the treatment of hyperuricemia and gout. Sci Rep. 2017;7(1):665. doi: 10.1038/s41598-017-00706-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gillen M, Miner JN, Valdez S. Pharmacokinetics, pharmacodynamics, and tolerability of verinurad, a selective uric acid reabsorption inhibitor, in healthy Japanese male subjects. Arthritis Rheumatol. 2016;68(Suppl 10):Abstract 193. http://acrabstracts.org/abstract/pharmacokinetics-pharmacodynamics-and-tolerability-of-verinurad-a-selective-uric-acid-reabsorption-inhibitor-in-healthy-japanese-male-subjects/. Accessed 27 Feb 2018. [DOI] [PMC free article] [PubMed]
- 20.Hall J, Gillen M, Yang X, Shen Z. Pharmacokinetics, pharmacodyamics, and tolerability of concomitant administration of verinurad and febuxostat in healthy male volunteers. Clin Pharmacol Drug Dev. 2018 doi: 10.1002/cpdd.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen Z, Gillen M, Miner JN, Bucci G, Wilson DM, Hall JW. Pharmacokinetics, pharmacodynamics, and tolerability of verinurad, a selective uric acid reabsorption inhibitor, in healthy adult male subjects. Drug Des Devel Ther. 2017;11:2077–2086. doi: 10.2147/DDDT.S140658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kankam M, Hall J, Gillen M, Yang X, Shen Z, Lee C, et al. Pharmacokinetics, pharmacodynamics, and tolerability of concomitant multiple dose administration of verinurad (RDEA3170) and allopurinol in adult male subjects with gout. J Clin Pharmacol. 2018 doi: 10.1002/jcph.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fleischmann R, Winkle P, Hall J, Valdez S, Liu S, Yan X, et al. Pharmacodynamic and pharmacokinetic effects and safety of verinurad in combination with febuxostat in adults with gout: a phase IIa, open-label study. RMD Open. 2018;4(1):e000647. doi: 10.1136/rmdopen-2017-000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fleischmann R, Winkle P, Miner JN, Yan X, Hicks L, Valdez S, et al. Pharmacodynamic and pharmacokinetic effects and safety of verinurad in combination with allopurinol in adults with gout: a phase IIa, open-label study. RMD Open. 2018;4(1):e000584. doi: 10.1136/rmdopen-2017-000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shiramoto M, Liu S, Shen Z, Yan X, Yamamoto A, Gillen M, et al. Verinurad combined with febuxostat in Japanese adults with gout or asymptomatic hyperuricaemia: a phase 2a, open-label study. Rheumatology. 2018. (in press). [DOI] [PMC free article] [PubMed]
- 26.Lee C, Yang C, Shah V. Metabolism and disposition of verinurad, a uric acid reabsorption inhibitor, in humans. Drug Metab Dispos. 2018;46(5):532–541. doi: 10.1124/dmd.117.078220. [DOI] [PubMed] [Google Scholar]
- 27.Reidenberg MM, Kostenbauder H, Adams WP. Rate of drug metabolism in obese volunteers before and during starvation and in azotemic patients. Metabolism. 1969;18(3):209–213. doi: 10.1016/0026-0495(69)90040-7. [DOI] [PubMed] [Google Scholar]
- 28.Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31–41. doi: 10.1159/000180580. [DOI] [PubMed] [Google Scholar]
- 29.Shen Z, Rowlings C, Kerr B, Hingorani V, Manhard K, Quart B, et al. Pharmacokinetics, pharmacodynamics, and safety of lesinurad, a selective uric acid reabsorption inhibitor, in healthy adult males. Drug Des Devel Ther. 2015;9:3423–3434. doi: 10.2147/DDDT.S85193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Woodworth T, Furst DE, Alten R, Bingham CO, III, Yocum D, Sloan V, et al. Standardizing assessment and reporting of adverse effects in rheumatology clinical trials II: the Rheumatology Common Toxicity Criteria v. 2.0. J Rheumatol. 2007;34(6):1401–1414. [PubMed] [Google Scholar]
- 31.U.S.Food and Drug Administration. Guidance for industry: Bioanalytical method validation. U S Department of Heath and Human Services Food and Drug Administration. http://www.fda.gov/downloads/Drugs/Guidance/ucm070107.pdf Accessed 27 Feb 2018.
- 32.Kasichayanula S, Liu X, Pe BM, Yao M, Pfister M, LaCreta FP, et al. The influence of kidney function on dapagliflozin exposure, metabolism and pharmacodynamics in healthy subjects and in patients with type 2 diabetes mellitus. Br J Clin Pharmacol. 2013;76(3):432–444. doi: 10.1111/bcp.12056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lalande L, Charpiat B, Leboucher G, Tod M. Consequences of renal failure on non-renal clearance of drugs. Clin Pharmacokinet. 2014;53(6):521–532. doi: 10.1007/s40262-014-0146-1. [DOI] [PubMed] [Google Scholar]
- 34.U.S.Food and Drug Administration. Guidance for Industry: Pharmacokinetics in patients with impaired renal function–study design, data analysis, and impact on dosing and labeling. 2010. FDA Website. https://www.fda.gov/downloads/drugs/guidances/ucm204959.pdf. Accessed 27 Feb 2018.
- 35.Bullingham RE, Nicholls AJ, Kamm BR. Clinical pharmacokinetics of mycophenolate mofetil. Clin Pharmacokinet. 1998;34(6):429–455. doi: 10.2165/00003088-199834060-00002. [DOI] [PubMed] [Google Scholar]
- 36.Kriz W, LeHir M. Pathways to nephron loss starting from glomerular diseases-insights from animal models. Kidney Int. 2005;67(2):404–419. doi: 10.1111/j.1523-1755.2005.67097.x. [DOI] [PubMed] [Google Scholar]
- 37.Gillen M, Valdez S, Zhou D, Kerr B, Lee CA, Shen Z. Effects of renal function on pharmacokinetics and pharmacodynamics of lesinurad in adult volunteers. Drug Des Devel Ther. 2016;10:3555–3562. doi: 10.2147/DDDT.S119944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu S, Perez-Ruiz F, Miner JN. Patients with gout differ from healthy subjects in renal response to changes in serum uric acid. Joint Bone Spine. 2017;84(2):183–188. doi: 10.1016/j.jbspin.2016.04.007. [DOI] [PubMed] [Google Scholar]