


Abstract
In metazoan cells, only a limited number of mini chromosome maintenance (MCM) complexes are fired during S phase, while the majority remain dormant. Several methods have been used to map replication origins, but such methods cannot identify dormant origins. Herein, we determined MCM7-binding sites in human cells using ChIP-Seq, classified them into firing and dormant origins using origin data and analysed their association with various chromatin signatures. Firing origins, but not dormant origins, were well correlated with open chromatin regions and were enriched upstream of transcription start sites (TSSs) of transcribed genes. Aggregation plots of MCM7 signals revealed minimal difference in the efficacy of MCM loading between firing and dormant origins. We also analysed common fragile sites (CFSs) and found a low density of origins at these sites. Nevertheless, firing origins were enriched upstream of the TSSs. Based on the results, we propose a model in which excessive MCMs are actively loaded in a genome-wide manner, irrespective of chromatin status, but only a fraction are passively fired in chromatin areas with an accessible open structure, such as regions upstream of TSSs of transcribed genes. This plasticity in the specification of replication origins may minimize collisions between replication and transcription.
INTRODUCTION
DNA replication is a highly orchestrated process that duplicates the entire genome only once per cell cycle, and DNA replication is initiated at multiple chromosomal sites known as replication origins. During the G1 phase of the cell cycle, the stepwise recruitment of origin recognition complex (ORC), CDC6, Cdt1 and mini chromosome maintenance (MCM)2−7 results in the formation of the pre-replication complex (pre-RC). It is now widely believed that, in metazoan cells, MCM2–7 complexes are chromatin-loaded at levels that far exceed the number of ORCs through an unknown mechanism(s) (1). The MCM2−7 complex is the catalytic core of the eukaryotic replicative DNA helicase that remains inactive during the G1 phase. At the G1/S boundary, a fraction of the MCM2−7 complexes become active CDC45-MCM-GINS (CMG) helicases upon binding of CDC45 and GINS. In Saccharomyces cerevisiae, autonomously replicating sequences (ARSs) have been identified as consensus sequences of replication origins (2–4), while in Schizosaccharomyces pombe, although no consensus sequence has been identified, AT-rich regions serve as potential origins (5–8). In mammalian cells, DNA replication origins do not have such consensus sequences, and it remains to be determined how origins are selected (9,10).
Next-generation sequencing-based methods are useful for determining the locations of DNA replication initiation sites in the genome. Such methods have proved successful for the isolation and sequencing of RNA-primed short nascent DNA strand (SNS) (11–13), replication bubbles (14,15) and Okazaki fragments (16). Replication origins often correlate with transcription start sites (TSSs), CpG islands (CGIs), G-quadruplex (G4) sequence motifs and several open histone markers. Other genome-wide studies identified ORC-binding sites as candidate replication origins in human cells (17,18). However, ORCs and MCMs are not necessarily bound to identical chromatin sites in mammalian cells (19–21). In addition, as described above, excess MCM2−7 complexes that outnumber ORCs are loaded onto chromatin (1,22) where they mostly remain inactive and constitute dormant origins as a backup system to maintain genome integrity during replication stress (23,24). Therefore, sites where MCMs are loaded should be identified to better understand the spatiotemporal regulation of both firing and dormant origins.
Herein, we precisely determined MCM7-binding sites by chromatin immunoprecipitation and parallel sequencing (ChIP-Seq), classified them into potential firing and dormant origins using SNS data, and analysed their association with various chromatin signatures.
MATERIALS AND METHODS
Cell culture
HeLa cells were grown in Dulbecco's modified Eagle's medium containing 8% foetal calf serum.
ChIP-Seq
ChIP-Seq was carried out as described below. Asynchronous HeLa cells (5 × 107) were fixed with 1% formaldehyde in culture medium for 15 min, further treated with 125 mM glycine to quench the cross-linking, and then harvested by scraping in ice-cold phosphate-buffered saline containing PMSF and protease inhibitor cocktail (Nacalai). After centrifugation, cells were resuspended in 400 μl ChIP lysis buffer (10 mM Tris–HCl [pH 8.0], 150 mM NaCl, 1 mM CaCl2, 1.5 mM MgCl2, 300 mM sucrose, 0.5% NP-40) containing PMSF and protease inhibitor cocktail and treated with 1000 U micrococcal nuclease (New England Biolabs) at 37°C for 30 min. Then, EDTA (pH 8.0) and SDS was added to a final concentration of 10 mM and 1%, respectively, and chromatin was further sonicated to yield an average fragment size of ∼200 bp. After centrifugation, the lysates were 10-fold diluted with ChIP dilution buffer (16.7 mM Tris–HCl [pH 8.0], 167 mM NaCl, 1.1% Triton X-100, 0.01% SDS, 1.2 mM EDTA [pH 8.0]) containing PMSF and protease inhibitor cocktail.
After removing the input sample (200 μl), each aliquot (950 μl) was pre-cleared with 60 μl Dynabeads (Invitrogen) at 4°C for 2 h and then incubated for 17 h at 4°C with 5 μg of the anti-MCM7 antibody. Immunocomplexes in each tube were collected using 30 μl Dynabeads at 4°C for 3 h. The beads were washed once with RIPA buffer (20 mM Tris–HCl [pH 8.0], 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% SDS), twice with RIPA buffer containing 500 mM NaCl, twice with LiCl wash buffer (10 mM Tris–HCl [pH 8.0], 250 mM LiCl, 1 mM EDTA, 1% NP-40, 0.5% sodium deoxycholate), and once with TE.
After removing the TE buffer, the immunocomplexes were eluted with ChIP elution buffer (100 mM NaHCO3, 1% SDS) at RT for 15 min. After adding NaCl to a final concentration of 0.2 M, formaldehyde reversal was performed at 65°C for 4 h. Samples were serially treated with DNase-free RNase A (Invitrogen) and Proteinase K (Roche), extracted with phenol–chloroform (Nacalai), and precipitated with ethanol. Finally, the DNA was dissolved in TE buffer.
The ChIP library was prepared with the Illumina protocol and sequencing analysis was performed using the HiSeq1500 (Illumina KK). The sequence reads were aligned to the human genome (hg19) using Bowtie software (version 0.12.8; parameter -v3 -m1). Peak detection and identification of the binding sites were obtained by correcting from Input DNA, using MACS2 broad peak detection.
To estimate the ChIP-Seq signals, we utilized the moving-average method in which the number of mapped reads was calculated every 0.1 kb interval with 1 kb window size. In addition, read numbers of the ChIP samples were normalized using Reads Per Million (RPM) (25), and further divided by the RPM of the input samples.
To validate the several peaks detected by ChIP-Seq experiments, we have done ChIP-qPCR (quantitative PCR) experiment according to the Minimum Information for Publication of Quantitative ChIP-qPCR Experiments (MIQE) guidelines (26). The data were already published in our previous papers (27,28). MIQE checklist was shown in Supplemental Table S6.
Antibody
Preparation and specificity of affinity-purified rabbit anti-MCM7 antibody was detailed previously (29,30). Briefly, in the presence of SDS in the buffer, the antibody specifically immunoprecipitates MCM7 without co-immunoprecipitation of the other MCM subunits (29). On the other hand, under milder condition, the antibody specifically immunoprecipitates both soluble and chromatin-bound MCM hexameric complexes (30).
Bioinformatics analysis
Data sets
Genomic annotations were downloaded using the UCSC Genome Browser tool (http://hgdownload.cse.ucsc.edu/downloads.html; human hg19). Genomic annotations of TSSs and TESs were determined using RefSeq (https://www.ncbi.nlm.nih.gov/refseq). Histone marker, transcription factor, Repli-Seq, DNase-Seq and G4-Seq data on build hg19 human genome were downloaded from GEO (https://www.ncbi.nlm.nih.gov/geo). These accession numbers and experimental conditions are listed in Supplemental Table S1. FPKM values were calculated from ENCODE RNA-Seq data (GSE33480) using a command ‘cufflinks’ (31). A list of CGI was downloaded from UCSC website.
Visualization
Positions of mapped sequencing reads and called peaks were visualized with Integrated Genomics Viewer (IGV) version 2.3 (32,33).
Overlap analyses
For overlap analyses, a command ‘bedtools window’ from BEDTools was used (34). Window size is described in figure legends. To generate shuffled peaks containing the same number and length but randomly located to estimate correlation by chance, a command ‘bedtools shuffle’ from BEDTools was used. Venn diagrams were generated with an online tool at the Google Chart API (https://developers.google.com/chart/). Relative co-localisation frequency (RCF) is calculated as below:
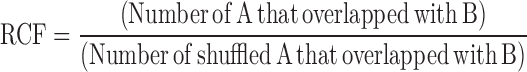 |
Aggregation plot
To investigate the relationships among NGS data, aggregation plots were generated using a command ‘agplus’ or ‘ngsplot’ (35–37). ‘ngsplot’ was used only for generating Supplemental Figure S4C. To calculate the cumulative signals (Figure 2A right, B right, C right, and Supplemental Figure S4A right), the values per 1 bp obtained from aggregation plots were summed up (±1 kb from the centre of each peak). When generating aggregation plots of origins around TSSs, the direction of transcription was also considered.
Figure 2.

Firing but not dormant origins are enriched in open chromatin regions. (A) Aggregation plots showing FAIRE-Seq signals surrounding the indicated origin classes (left). Cumulative FAIRE-Seq signals (the sum of the values per 1 bp obtained from aggregation plots) around firing and dormant origins (±1 kb from the centre of each peak) are also shown (right). (B) Aggregation plots showing DNase-Seq signals surrounding the indicated origin classes (left). Cumulative DNase-Seq signals around firing and dormant origins (±1 kb from the centre of each peak) are also shown (right). (C) Aggregation plots showing the signals of MCM7_1st ChIP-Seq surrounding the indicated origin classes (left). Cumulative MCM7_1st ChIP-Seq signals around firing and dormant origins (±1 kb from the centre of each peak) are also shown (right). (D) Relative co-localisation frequencies (within 0.5 kb) of firing and dormant origins with the indicated histone markers. Values obtained with shuffled peaks are set at 1 (dashed line). (E, F) Relative co-localisation frequencies of firing and dormant origins (E) and the indicated histone markers (F) with the different replication timing regions. Values obtained with shuffled peaks are set at 1. G1b, S1, S2, S3, S4 and G2 mean six different parts of the cell cycle (from early to late replication timing), which are fractionated by flow cytometry according to DNA content of cells pulse-labeled with BrdU.
GC contents
To calculate GC contents of origins, we randomly picked 10 000 peaks by using the RANDBETWEEN and RANK functions of Microsoft Excel software. To convert bed files to fasta files, a command ‘fastaFromBed’ from BEDTools was used (34). GC% of peaks was calculated by using Microsoft Excel software.
Re-analysis of published MCM2 ChIP-Seq data (38)
The sequence reads were aligned to the hg19 using bwa (version 0.7.17-r1188). Peak re-detection and re-identification of the binding sites were obtained by correcting from Input DNA, using MACS2 broad peak detection with P < 0.001 and q < 0.02.
Statistical analysis
Statistical analysis was performed with a Chi-square test or with Wilcoxon rank sum test using R. P-values were used to determine statistical significance as detailed in the figure legends.
RESULTS
Accurate genome-wide identification of MCM7-binding sites as potential origins and classification into firing and dormant origins using SNS data
Previously, we reported genome-wide mapping of MCM7-binding sites in asynchronous HeLa cells by ChIP-Seq (hereafter termed MCM7_1st; ∼340 000 peaks) (28). To identify MCM-binding sites more precisely, we repeated the previous experiment with biological replicates. One reason for why we have used asynchronous HeLa cells is that many of ChIP-Seq and SNS-Seq studies related to ours have been performed with asynchronous HeLa cells (Supplemental Table S1). For specificity of our anti-MCM7 antibody, we had rigorously confirmed it, as reported previously (for detail, see Materials and Methods section). Here, we further confirmed that a remarkably higher amount of chromatin DNA was precipitated with anti-MCM7 antibodies than control IgG and that the amount of the co-precipitated DNA was significantly reduced upon treatment with MCM7 siRNAs (Supplemental Figures S1A–C). Thus, most of the co-precipitated DNAs are specifically derived from the MCM7-bound chromatin DNAs and the obtained ChIP-Seq signals are specific. MACS2 broad peak caller identified ∼520 000 MCM7 peaks in this follow-up experiment (P < 0.001, q < 0.05; Figure 1A and C), hereafter termed MCM7_2nd. We then investigated the concordance between them, and visual inspection of two genomic regions, the lamin B2 and MCM4 loci, revealed both MCM7_1st and MCM7_2nd peaks at both replication origins (Figure 1A). Analysis of MCM7_2nd peak distribution relative to MCM7_1st peak revealed significant enrichment of second peaks and a central sharp peak (Figure 1B). This pattern was lost when using randomized (shuffled) datasets containing the same number and length but randomly located to estimate correlation by chance and indicate the significance. Hereafter, selected MCM7 (sMCM7) is used to represent MCM7_1st peaks that are accompanied by MCM7_2nd peaks within 0.5 kb. This process identified ∼200 000 sMCM7 peaks as MCM2−7-binding sites, including well-known replication origins (Figure 1A and C). The number of sMCM7 peaks was significantly higher than that obtained using randomized MCM7_1st peaks (Figure 1D).
Figure 1.

Genome-wide identification of firing and dormant origin sites. (A) Selected snapshots of the genome browser view around the LMNB2 and MCM4 loci. Visual representations of FAIRE-Seq, DNase-Seq, MCM7 ChIP-Seq (first and second), SNS (13), sMCM7 (MCM7_1st_w0.5_MCM7_2nd), sMCM7_w0.5_SNS (firing origins) and sMCM7_wo0.5_SNS (dormant origins) data from HeLa cells are shown. Green lines indicate known origin regions. (B) Aggregation plots showing the localisation of MCM7_2nd ChIP-Seq peaks and shuffled peaks around MCM7_1st ChIP-Seq peaks. (C) Venn diagram showing the overlap (within 0.5 kb) of MCM7 peaks obtained in two independent experiments. (D) The number of MCM7_1st peaks within 0.5 kb of MCM7_2nd peaks is significantly higher than that obtained with shuffled MCM7_1st peaks. * indicates P < 0.0001 by Chi-square test. (E) Venn diagram showing overlap (within 0.5 kb) of sMCM7 and SNS peaks. (F) The number of sMCM7 peaks within 0.5 kb of SNS peaks is significantly higher than that obtained with shuffled sMCM7 peaks. * indicates P < 0.0001 by Chi-square test. (G) Aggregation plot showing the localization of SNS peaks and shuffled peaks surrounding sMCM7 peaks.
We then classified the MCM2−7-binding sites (sMCM7 sites) into potential firing and dormant origins using previously deposited SNS peak data (∼90 000 sites) (13), which may represent firing replication origins. We defined sMCM7 peaks associated with SNS peaks within 0.5 kb (sMCM7_w0.5_SNS, ∼78 000 sites) as origins that may mainly consist of firing origins, and sMCM7 peaks not associated with SNS peaks within 0.5 kb (sMCM7_wo0.5_SNS, ∼120 000 sites) as origins that may mainly consist of dormant and/or inefficient origins (Figure 1A and E). In the later part of this manuscript, we used the terms ‘firing origins’ and ‘dormant origins’ for simplicity. The association between sMCM7 and SNS was statistically significant when compared with the data obtained with shuffled sMCM7 peaks (Figure 1F). Analysis of SNS peak distribution relative to sMCM7 peaks revealed significant enrichment of SNS peaks but not shuffled SNS (Figure 1G). SNS-Seq data (13) mainly used in this research were obtained by re-analyzing the original SNS-Seq data obtained by Besnard et al. (12). We found that the sMCM7 sites well correlate with the original SNS data (∼250 000 sites; Supplemental Figures S2A–D) as well as the reanalyzed SNS data.
SNS-Seq method is widely used for determining the locations of DNA replication initiation sites in the genome. However, G4s are known to be resistant to lamda exonuclease digestion and therefore it is possible SNS-Seq generates additional false positive sites (39). Therefore, we also used another origin map, Ini-Seq (∼25 000 sites) (40). In this approach, newly replicated DNA is directly labelled with digoxigenin-dUTP near the sites of its initiation in a cell-free system, immunoprecipitated and sequenced. Interestingly, the sMCM7 significantly correlated with Ini-Seq sites as well (Supplemental Figures S3A–C). All these data strongly support the specificity of sMCM7 sites we determined, at least for firing origins.
On the other hand, it is possible that dormant origins are near-randomly distributed across the genome. To rule this out, we pre-classified MCM7_1st and MCM7_2nd into firing and dormant origins using SNS data (13) and then compared dormant MCM7_1st and dormant MCM7_2nd. As a result, we found significant co-localization between them (Supplemental Figures S1D and E). Thus, most of the sMCM7 sites including dormant origins may be specific.
Firing origins are enriched in open chromatin regions, while dormant origins are more uniformly distributed across the genome
To identify chromatin states surrounding firing or dormant origins, we first used formaldehyde-assisted isolation of regulatory elements (FAIRE)-Seq data (28); FAIRE is a method of isolating genome regions with depleted or coarse nucleosomes (41–43). We examined aggregated FAIRE-Seq signals surrounding the identified origins to determine the average chromatin state. The results suggest that firing origins have a more open chromatin structure than dormant origins (Figure 2A; see also snapshot data in Figure 1A). A positive correlation between chromatin openness and firing origin was also apparent using DNase-Seq signal data (Figure 2B; snapshot data in Figure 1A). We next investigated MCM7 signals at origin sites, and aggregation plots of MCM7_1st ChIP signals indicated minimal difference between firing and dormant origins in terms of the efficiency of MCM loading (Figure 2C), especially compared with the clear difference in chromatin openness (Figure 2A and B). Similar results were obtained with the MCM7_2nd ChIP data (Supplemental Figure S4A). We investigated whether the conclusions still hold with the original SNS-Seq (12) or Ini-Seq (40) datasets. As shown in Supplemental Figures S2E–F and S3D–E, essentially the same results were obtained with these datasets.
To further explore the effect of chromatin openness on origin firing, we assessed the relationship between origins and histone markers, and identified strong correlations between firing origins and two euchromatin markers, H3K9ac and H3K4me3 (Figure 2D). H4K20me2 is derived from PR-Set7-mediated H4K20me1 and is the reported binding site for the bromo-adjacent homology (BAH) domain of ORC1 (44,45). Since ChIP-Seq data of H4K20me2 in HeLa cells are not available, we analysed H4K20me1 data. H4K20me1 was moderately associated with firing origins, whereas H3K27me3 and H3K9me3, which are markers of silent chromatin, were not associated with firing origins (Figure 2D). By contrast, dormant origins were not correlated with any histone markers (Figure 2D). We verified whether these histone modifications actually correlated with chromatin openness using FAIRE-Seq data (28). As expected, chromatin regions with active histone modifications have a more open chromatin structure (Supplemental Figure S4B). Taken together, these results suggest that excessive MCMs might be loaded by the active loading machinery in a genome-wide manner, irrespective of the degree of chromatin openness, and firing of origins might be regulated mainly by chromatin openness. On the other hand, we cannot formally exclude other possibilities; e.g. origin firing might create open chromatin structures.
The Repli-Seq technique allows the identification of newly replicated (BrdU-labelled) DNA in synchronised cells during consecutive phases of the cell cycle (46). We investigated the relationship between origins and replication timing using Repli-Seq data from HeLa cells (Supplemental Table S1) and found that firing origins are remarkably enriched in early replicating regions (Figure 2E). We also compared several histone markers with the Repli-Seq data and found that the density of open chromatin markers such as H3K9ac and H3K4me3 is high in early replicating regions, whereas heterochromatic markers are denser in late replicating regions, as expected (Figure 2F). These results also suggest that a more open chromatin structure might promote the firing of origins, leading to early replication.
Firing but not dormant origins are enriched near active promoters
Human and mouse firing origins detected by SNS-Seq mapping and Drosophila and human ORCs detected by ChIP-Seq often co-localise with TSSs (11,12,17,18,47,48). Therefore, we analysed the distribution of firing or dormant origins with respect to TSSs and found that firing origins but not dormant origins are closely associated with regions upstream of TSSs (Figure 3A). As shown in Supplemental Figures S2G and S3F, essentially the same results were obtained with the original SNS-Seq data and Ini-Seq data. To investigate the effect of gene expression levels on the association, we classified all genes into four arbitrary groups (high, middle, low and no expression; Supplemental Table S2) based on relative fragments per kilobase per million mapped reads (FPKM) using RNA-Seq data, and re-plotted firing origin peaks surrounding TSSs in each group. The results confirmed enrichment of firing origins with higher expression levels (Figure 3B). As expected, TSS regions of highly and moderately expressed genes have a more open chromatin structure than those of genes expressed at lower levels (Figure 3C). In Drosophila cells, a small amount of MCMs are initially loaded close to ORC during early G1, but from late G1 to S phase entry, a 10-fold higher amount of MCMs are loaded throughout the genome and subsequently displaced by active transcription, resulting in a binary pattern of broad MCM-containing regions of non-transcribed DNA and broad MCM-free regions consisting of active genes (49). In HeLa cells, sMCM7 peaks are enriched in the genomic segments upstream of the TSSs and the majority of the peaks are coming from the expressed genes (Supplemental Figure S4C). This difference could be explained by species difference.
Figure 3.

Firing but not dormant origins are enriched near active promoters. (A) Firing and dormant origin sites were aggregated together with TSS sites. (B) Effect of gene expression level on enrichment of firing origins around TSSs. Based on FPKM, TSSs were arbitrarily classified into four classes: high expression (N = 7122), middle expression (N = 7122), low expression (N = 7122) and undetectable expression (N = 8162; Supplemental Table S2). Aggregation plots of firing origins surrounding each of the four TSS classes were then calculated. (C) Aggregation plots of FAIRE-Seq signals surrounding each of the four TSS classes. (D, E) Firing origins are enriched at CGI (D) and TBP-bound (E) sites. (F) Relative co-localisation frequencies (within 0.5 kb) of firing origins with CGI or TBP. Values obtained with shuffled peaks are set at 1 (dashed line). (G) Relative co-localisation frequencies (within 0.5 kb) of firing or dormant origins with the indicated transcription factor-binding sites. Values obtained with shuffled peaks are set at 1 (dashed line). (H) Aggregation plots showing FAIRE-Seq signals surrounding the indicated transcription factor-binding sites.
Because promoters are located upstream of TSSs, we then investigated whether firing origins are enriched near promoter sites. Common mammalian promoters can be separated into two major classes: CpG islands (CGIs) and TATA boxes (50). Whereas the former is associated with constitutive expression, the latter correlates with more tissue-specific expression (51). We found that firing but not dormant origins correlated well with both CGI- and TATA box-binding proteins (TBPs; Figure 3D−F). These results suggest that firing origins are enriched near active promoters with a more accessible, open chromatin structure. Again, no specific correlation between dormant origins and TSSs was observed (Figure 3A, D and E).
Several transcription factors may strongly stimulate origin firing by inducing chromatin openness
We next compared firing and dormant origins with the known binding sites of cell cycle-related transcription factors E2F1, c-myc, ELK1, Nrf1, Nrf2, MAZ, c-Jun, c-Fos and STAT3. An outline of relationships between transcription factors and cell-cycle regulation is shown in Supplemental Table S3. We found a strong correlation between firing origins and E2F1-, c-myc-, ELK1-, Nrf1-, Nrf2- and MAZ-binding sites, but not c-Jun, Fos or STAT3 (Figure 3G). Conversely, the association with dormant origins was negligible. Therefore, E2F1, c-myc, ELK1, Nrf1, Nrf2 and MAZ do not appear to be essential for MCM loading. We examined the degree of chromatin openness at these binding sites, and transcription factor-binding sites that correlated well with firing origins have a relatively open structure, while those with a lower correlation have only moderate chromatin openness (Figure 3H). Therefore, we concluded that firing origins associate with transcription factors that induce an opening of the chromatin structure. In other words, a highly accessible and open chromatin structure near active promoters might promote origin firing (i.e. MCM activation) rather than specific transcription factors alone.
Firing but not dormant origins have a high GC content
Although genetic signatures at origins remain elusive in multicellular organisms, several studies report that genome-wide SNS sites in human, mouse and Drosophila are GC-rich (12,52,53). To investigate possible sequential bias in firing and dormant origins, we calculated the GC content. The mean GC content of firing origins is 52.5%, compared with only 45.8% for dormant origins and 40.9% for shuffled peaks (Figure 4A). Many G-rich motifs are predicted to form G4 structures (54,55) that have been linked to origin functions (12,56). We therefore investigated the relationship between origins and G4-Seq results obtained by combining features of the polymerase stop assay with next-generation sequencing (57). Figure 4B shows profiles for FAIRE-Seq, DNase-Seq and G4-Seq data aligned with MYC and AXIN1 genomic regions containing known replication origins. Aggregation plots of G4-Seq peaks reveal enriched G4 formation at firing but not dormant origin sites (Figure 4C), suggesting that G4 motifs may influence the efficiency of origin firing but not the efficiency of MCM loading. We also performed similar analyses with Ini-Seq data (40), which does not involve lamda exonuclease digestion that may generate a bias toward G4 enrichment. As shown in Supplemental Figures S3G and H, essentially the same results were obtained with the Ini-Seq data. Recent anti-G4 ChIP-Seq experiments using HaCaT and NHEK cells identified endogenous genome-wide G4s (58), suggesting that G4 DNA formation is highly dependent on nucleosome-depleted chromatin and elevated transcription, as previously described (59,60). Consistent with this, we found that G4 sites correlated well with open chromatin (Figure 4D). Therefore, an open chromatin structure might be more important for promoting origin firing than G4 sites alone. In support of this idea, firing origins correlated well with both CGI- and TBP-bound TATA-type promoters, as described above (Figure 3D−F).
Figure 4.

Firing but not dormant origins are GC-rich and associated with G4 structures. (A) GC content of 10 000 randomly selected origins. Grey boxes represent results obtained using shuffled datasets. The Wilcoxon rank sum test was used to calculate P-values. (B) Selected snapshots of the genome browser view around the MYC and AXIN1 loci. Visual representations of FAIRE-Seq, DNase-Seq, G4-Seq, firing origin and dormant origin data from HeLa cells are shown. Green lines indicate known origin regions. (C) Aggregation plots of G4-Seq peaks surrounding firing and dormant origins. (D) Aggregation plots of FAIRE-Seq signals surrounding G4 ChIP-Seq peaks and shuffled peaks.
Although no consensus sequence has yet been associated with mammalian origins (61,62), we attempted to identify enriched motifs in firing or dormant origins using the genetic motif discovery tool MEME-ChIP (63). Although some motifs were identified in firing origins, the associated E-values were low (data not shown), suggesting that firing origin sites are not determined by specific sequence motifs, as previously described.
Firing origins are generally of low density but are enriched upstream of TSSs in common fragile sites
Common fragile sites (CFSs) are specific loci characterised by gaps and breaks in metaphase chromosomes following partial inhibition of DNA synthesis (64). Because the instability of CFSs may depend on expression of the underlying long genes and be caused by collisions between replication and transcription complexes (65,66), we investigated MCM loading and firing in CFS sites. Supplemental Table S4 lists 31 human genes known as CFSs (67). It is known that at least FRA3B and FRA16D are fragile in HeLa cells (68). Most genes were found to be larger than 700 kb in length. For comparison, 31 control long genes of >700 kb were randomly selected (Supplemental Table S5). Whether these randomly selected long genes are fragile in HeLa cells is unknown. We found that the density of MCM-loaded sites (i.e. total origins) in both CFSs and randomly selected long genes was about half that of control groups randomly selected from all genes (Figure 5A). Furthermore, ∼24−31% of loaded MCMs were found to fire at non-CFS long genes, which was somewhat lower than control groups randomly selected from all genes (∼37−40%). However, the rate was further reduced to ∼20% for CFS genes (Figure 5A). These findings are consistent with previous DNA combing results showing that origin density is reduced in CFS sites (69–72). As suggested previously, replication of CFS regions was largely associated with the late S phase (Figure 5B). Similarly, randomly selected long genes were also found to replicate mainly in the late S phase (Figure 5B). By contrast, control genes randomly selected from total genes were replicated earlier (Figure 5B). Meanwhile, similar to all firing origins (Figure 3A), firing origins at CFS genes were also enriched upstream of TSSs (Figure 5C and D). Aggregation plots constructed using FAIRE-Seq signals showed that promoter regions of CFSs or long genes also have an open chromatin window, but this is restricted compared with control genes selected from all genes (Figure 5E). These results reveal some common features of CFS genes: (i) In agreement with previous findings, CFS genes are long genes with relatively tight chromatin structure; (ii) the number of replication origins, especially firing origins, is reduced; (iii) despite this overall reduction, firing origins are preferentially located upstream of TSSs, where the window of open chromatin structure is encountered. Our analysis indicates that randomly selected long genes also share such features, but the relative level of transcription may differ (Supplemental Table S5). Therefore, differences in the frequency of collisions between replication and transcription could account for this variation (see Discussion).
Figure 5.

Firing origins are generally of low density but are enriched upstream of TSSs in common fragile sites. (A) Average number of sMCM7 (total origins), firing origins and dormant origins within the indicated genes (per 100 kb). (B) Relative co-localization frequencies of the different replication timing regions with the indicated genes. Values obtained with shuffled genes are set at 1. (C) Number of firing origins surrounding TSSs of CFSs. (D) Selected snapshots of the genome browser view around the FHIT and DPYD CFS loci. Visual representations of FAIRE-Seq, DNase-Seq, firing origin and dormant origin data from HeLa cells. (E) Aggregation plots of FAIRE-Seq signals surrounding TSSs of the indicated genes.
DISCUSSION
This study reports the first accurate genome-wide analysis of a component of the MCM hexamer in mammals. It is widely believed that the number of MCM2−7 double hexamers loaded onto DNA is much higher than the number of origins that are actually active in any individual S phase in various organisms (1,22,73,74). One possible explanation is that only a proportion of origins are actually used in the S phase, while the majority remain dormant as backup origins. Previously, active replication origins have been mapped using SNS-Seq, DNA bubble structure-Seq (Bubble-Seq), Okazaki fragment-Seq (OK-Seq) and Ini-Seq (11–16,40). These studies indicated that DNase I hypersensitive sites, some open histone markers and G4 structures are linked to origin activity. However, such approaches cannot identify dormant origins. Herein, we determined both firing and dormant origins in a genome-wide manner. Firstly, we identified ∼200 000 MCM-binding sites precisely by combining two independent MCM7 ChIP-Seq datasets (Figure 1A−D). The preciseness of our MCM7-binding site data is strongly supported by the fact that the original MCM7_1st and MCM7_2nd peaks are concordant with SNS-Seq and Ini-Seq sites better than other reported or deposited MCM ChIP-Seq data (Supplemental Figure S5). One reason for this might be due to high specificity of our anti-MCM7 antibody (Supplemental Figure S1 and Materials and Methods). Here, we have repeated MCM7 ChIP-Seq twice and combined the two datasets to precisely determine MCM7-binding sites. If the same experiment(s) will be further repeated and combined with the present data, specificity of the MCM7-binding sites would increase but the number of false negative sites would also increase. On the other hand, we also observed many non-concordant peaks in each experiment. This could simply represent experimental bias, but it could also indicate that some origin sites might not be strictly fixed (in other words, plasticity of origin usage). Actually, among many different genome-wide datasets for replication origins, significant overlap, but not near-complete overlap, is observed. We then classified MCM7 sites into firing origins (∼78 000 sites) and dormant origins (∼120 000 sites) using SNS peaks (Figure 1E−G). It should be noted that the number of firing origins we determined is derived from the population of cells and only a subset of them will be actually activated during a single S phase in a single cell (1). We then analysed association of firing and dormant origins with various chromatin signatures. The results suggest that firing origins are non-randomly distributed throughout the genome, and that origin firing might be sensitive to chromatin openness (Figures 2, 3 and 4). Furthermore, firing origins are enriched at active promoters with a more open chromatin structure (Figure 3). Essentially the same results were obtained with the original SNS-Seq data and Ini-Seq data (Supplemental Figures S2 and S3). These findings are consistent with the previous results discussed above. Our results show that the association between firing origins and TSSs is increased as a function of gene expression level (Figure 3B), although replication initiation was reported to be less frequent in regions that exhibit higher levels of transcription (75), and the reason for the apparent discrepancy is unclear at present. In this regard, SNS-Seq peaks at TSSs in MCF7 cells are more enriched in actively expressed genes than in genes expressed at lower levels (76).
By contrast, we found that dormant origins are not tightly associated with any specific chromatin signatures, suggesting that they are more uniformly distributed across the genome. Since excess dormant origins act as backup origins to protect human cells from replicative stress, such a wide distribution may be important for genome stability. Interestingly, the efficiency of MCM loading at dormant origins with a closed chromatin structure is comparable to that at firing origins with a more open chromatin structure (Figure 2A−C and Supplemental Figures S2E, F, S3D, E and S4A), suggesting that the active MCM loading machinery might load MCMs onto chromatin regions with a closed structure. If so, it is possible that Cdt1 recruits chromatin regulatory factors such as SNF2H, HBO1 and GRWD1 to facilitate MCM loading by altering chromatin accessibility (27,28,77,78). This idea may be further supported by the observation that chromatin decondensation mediated by LacI-Cdt1 tethered to LacO stimulates MCM recruitment (79).
Based on our findings, we propose a model in which a large number of MCMs are uniformly loaded in a genome-wide manner, irrespective of chromatin status, but only a small proportion are passively fired in chromatin regions with an accessible, open structure such as those upstream of TSSs of actively transcribed genes. Indeed, a more open chromatin structure rather than MCM loading stimulates MCM activation (80). In addition, in budding and fission yeast, Xenopus and human cells, CDC45 is considered rate limiting for MCM activation (20,81–83). In budding yeast, efficient MCM loading leads to efficient DNA replication initiation (84). However, in our current analysis, the efficiency of MCM loading at dormant origins with a closed chromatin structure was comparable with that at firing origins. Therefore, chromatin openness might affect origin firing more strongly than MCM loading levels.
As replication and transcription compete for the same DNA template, collisions between the processes are unavoidable, and can result in severe DNA damage (65,66). In eukaryotes, the replication machinery progresses at about the same speed as the transcription machinery (85). Therefore, based on the simplest model with one chromosome and one transcribed gene, placing a replication origin upstream of the TSS would be a straightforward and effective way to avoid collision between transcription and replication (Figure 6A). Even in the case of a more complex model chromosome with two genes, if located head-to-head and tail-to-tail, such a strategy would be still effective (Figure 6B–I and B-II). However, if the two genes are located head-to-tail, collision may occur (model shown in Figure 6B-III, showing transcription from the left gene colliding with the replication fork from the right origin). However, if there is a long interval region (intergenic or untranscribed region) between the two genes, a head-on collision could be avoided (Figure 6B-IV). Human cells undergo critical changes in transcriptional programming during differentiation and in response to environmental transitions. In this model, the position of firing origins would be passively changed and located upstream of transcribed genes in response to changes in transcriptional programming. Such plasticity for the specification of firing replication origins may be advantageous for minimising collisions between replication and transcription, as discussed above. Furthermore, this simple model does not preclude the possibility of specific regulation for determining the origin under certain conditions.
Figure 6.

A simple model showing how the passive and dynamic generation of firing replication origins upstream of active TSSs minimises collisions between replication and transcription. The model is discussed in detail in the main text. (A) Simple model chromosome with one transcribed gene. (B) More complex model chromosomes with two genes in head-to-head (I), tail-to-tail (II) or head-to-tail (III) orientations. Collisions between replication and transcription may occur as shown in B-III. However, if an intergenic or untranscribed region between the two genes is longer than the left gene (IV), collisions may be avoided.
Even for long genes, transcription begins at TSSs, and our present analysis indicates that firing replication origins are preferentially located upstream of TSSs (Figure 5). Therefore, when a gene is too long, a single transcription complex would travel a long distance, and a head-on collision between the converging replication fork and transcription machinery would be unavoidable during the latter stages. This model is supported by the fact that break sites in CFS-related genes such as FHIT, WWOX and DPYD are found in parts of these genes that are transcribed later (86–88).
The first step in the assembly of the pre-RC is the binding of ORC1−6 to chromatin. Previous genome-wide human ORC1 mapping in HeLa cells indicated that ORC1 sites (∼13 000) are strongly associated with TSSs, and that TSS expression levels influence the efficiency of ORC1 recruitment at G1, and hence the probability of firing during the S phase (17). Based on recent genome-wide ORC2 mapping in K562 cells (∼52 000 sites) (18), the number of ORC1 sites appears to be limited. Additionally, ORC2 binding in K562 cells is also more enriched in active promoters than in weaker (less active) promoters (18). Approximately 34% and 17% of ORC1 sites were associated with firing and dormant origins, respectively (data not shown). As mentioned above, in human cells, the number of MCM heterohexamers loaded onto chromatin is much greater than the number of ORCs, although the mechanism(s) remain unknown (1,19,21). Therefore, one possible simple scenario might be that ORCs are preferentially assembled at active TSSs with a more open and accessible chromatin structure, allowing the loading of both ORC-proximal and ORC-distal multiple MCM complexes. If so, ORC-proximal MCMs within open chromatin are more likely to fire efficiently.
DATA AVAILABILITY
ChIP-Seq data have been deposited with accession codes DRA005864 (raw data) and GSE107248 (processed data).
Supplementary Material
ACKNOWLEDGEMENTS
We thank M. Kosugi and C. Sueyoshi for technical and secretarial assistance. We also appreciate the technical support of the Research Support Center, Graduate School of Medical Sciences, Kyushu University. Computation was performed in part using the NIG supercomputer at the ROIS National Institute of Genetics.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Ministry of Education, Culture, Sports, Science and Technology of Japan [21370084, 25291027, 26114713 to M.F., 25870509 to N.S.] (in part). Funding for open access charge: Kyushu University, Institutional Funds.
Conflict of interest statement. None declared.
REFERENCES
- 1. Hyrien O. How MCM loading and spreading specify eukaryotic DNA replication initiation sites [version 1; referees: 4 approved]. F1000Research. 2016; 5:(F1000 Faculty Rev):2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stinchcomb D.T., Struhl K., Davis R.W.. Isolation and characterisation of a yeast chromosomal replicator. Nature. 1979; 282:39–43. [DOI] [PubMed] [Google Scholar]
- 3. Rao H., Marahrens Y., Stillman B.. Functional conservation of multiple elements in yeast chromosomal replicators. Mol. Cell. Biol. 1994; 14:7643–7651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wyrick J.J., Aparicio J.G., Chen T., Barnett J.D., Jennings E.G., Young R.A., Bell S.P., Aparicio O.M.. Genome-wide distribution of ORC and MCM proteins in S. cerevisiae: high-resolution mapping of replication origins. Science. 2001; 294:2357–2360. [DOI] [PubMed] [Google Scholar]
- 5. Segurado M., de Luis A., Antequera F.. Genome-wide distribution of DNA replication origins at A+T-rich islands in Schizosaccharomyces pombe. EMBO Rep. 2003; 4:1048–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yompakdee C., Huberman J.A.. Enforcement of late replication origin firing by clusters of short G-rich DNA sequences. J. Biol. Chem. 2004; 279:42337–42344. [DOI] [PubMed] [Google Scholar]
- 7. Dai J., Chuang R.Y., Kelly T.J.. DNA replication origins in the Schizosaccharomyces pombe genome. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hayashi M., Katou Y., Itoh T., Tazumi A., Yamada Y., Takahashi T., Nakagawa T., Shirahige K., Masukata H.. Genome-wide localization of pre-RC sites and identification of replication origins in fission yeast. EMBO J. 2007; 26:1327–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mechali M. Eukaryotic DNA replication origins: many choices for appropriate answers. Nat. Rev. Cell Biol. 2010; 11:728–738. [DOI] [PubMed] [Google Scholar]
- 10. Aladjem M.I., Redon C.E.. Order from clutter: selective interactions at mammalian replication origins. Nat. Rev. 2017; 18:101–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sequeira-Mendes J., Diaz-Uriarte R., Apedaile A., Huntley D., Brockdorff N., Gomez M.. Transcription initiation activity sets replication origin efficiency in mammalian cells. PLoS Genet. 2009; 5:e1000446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Besnard E., Babled A., Lapasset L., Milhavet O., Parrinello H., Dantec C., Marin J.M., Lemaitre J.M.. Unraveling cell type-specific and reprogrammable human replication origin signatures associated with G-quadruplex consensus motifs. Nat. Struct. Mol. Biol. 2012; 19:837–844. [DOI] [PubMed] [Google Scholar]
- 13. Picard F., Cadoret J.C., Audit B., Arneodo A., Alberti A., Battail C., Duret L., Prioleau M.N.. The spatiotemporal program of DNA replication is associated with specific combinations of chromatin marks in human cells. PLoS Genet. 2014; 10:e1004282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mesner L.D., Valsakumar V., Karnani N., Dutta A., Hamlin J.L., Bekiranov S.. Bubble-chip analysis of human origin distributions demonstrates on a genomic scale significant clustering into zones and significant association with transcription. Genome Res. 2011; 21:377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mesner L.D., Valsakumar V., Cieslik M., Pickin R., Hamlin J.L., Bekiranov S.. Bubble-seq analysis of the human genome reveals distinct chromatin-mediated mechanisms for regulating early- and late-firing origins. Genome Res. 2013; 23:1774–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Petryk N., Kahli M., d’Aubenton-Carafa Y., Jaszczyszyn Y., Shen Y., Silvain M., Thermes C., Chen C.L., Hyrien O.. Replication landscape of the human genome. Nat. Commun. 2016; 7:10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dellino G.I., Cittaro D., Piccioni R., Luzi L., Banfi S., Segalla S., Cesaroni M., Mendoza-Maldonado R., Giacca M., Pelicci P.G.. Genome-wide mapping of human DNA-replication origins: levels of transcription at ORC1 sites regulate origin selection and replication timing. Genome Res. 2013; 23:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Miotto B., Ji Z., Struhl K.. Selectivity of ORC binding sites and the relation to replication timing, fragile sites, and deletions in cancers. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:E4810-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ritzi M., Baack M., Musahl C., Romanowski P., Laskey R.A., Knippers R.. Human minichromosome maintenance proteins and human origin recognition complex 2 protein on chromatin. J. Biol. Chem. 1998; 273:24543–24549. [DOI] [PubMed] [Google Scholar]
- 20. Edwards M.C., Tutter A.V., Cvetic C., Gilbert C.H., Prokhorova T.A., Walter J.C.. MCM2–7 complexes bind chromatin in a distributed pattern surrounding the origin recognition complex in Xenopus egg extracts. J. Biol. Chem. 2002; 277:33049–33057. [DOI] [PubMed] [Google Scholar]
- 21. Fujita M., Ishimi Y., Nakamura H., Kiyono T., Tsurumi T.. Nuclear organization of DNA replication initiation proteins in mammalian cells. J. Biol. Chem. 2002; 277:10354–10361. [DOI] [PubMed] [Google Scholar]
- 22. Hyrien O., Marheineke K., Goldar A.. Paradoxes of eukaryotic DNA replication: MCM proteins and the random completion problem. Bioessays. 2003; 25:116–125. [DOI] [PubMed] [Google Scholar]
- 23. Ge X.Q., Jackson D.A., Blow J.J.. Dormant origins licensed by excess Mcm2–7 are required for human cells to survive replicative stress. Genes Dev. 2007; 21:3331–3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ibarra A., Schwob E., Mendez J.. Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:8956–8961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mortazavi A., Williams B.A., McCue K., Schaeffer L., Wold B.. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008; 5:621–628. [DOI] [PubMed] [Google Scholar]
- 26. Bustin S.A., Benes V., Garson J.A., Hellemans J., Huggett J., Kubista M., Mueller R., Nolan T., Pfaffl M.W., Shipley G.L. et al. . The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009; 55:611–622. [DOI] [PubMed] [Google Scholar]
- 27. Sugimoto N., Yugawa T., Iizuka M., Kiyono T., Fujita M.. Chromatin remodeler sucrose nonfermenting 2 homolog (SNF2H) is recruited onto DNA replication origins through interaction with Cdc10 protein-dependent transcript 1 (Cdt1) and promotes pre-replication complex formation. J. Biol. Chem. 2011; 286:39200–39210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sugimoto N., Maehara K., Yoshida K., Yasukouchi S., Osano S., Watanabe S., Aizawa M., Yugawa T., Kiyono T., Kurumizaka H. et al. . Cdt1-binding protein GRWD1 is a novel histone-binding protein that facilitates MCM loading through its influence on chromatin architecture. Nucleic Acids Res. 2015; 43:5898–5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fujita M., Kiyono T., Hayashi Y., Ishibashi M.. hCDC47, a human member of the MCM family. Dissociation of the nucleus-bound form during S phase. J. Biol. Chem. 1996; 271:4349–4354. [DOI] [PubMed] [Google Scholar]
- 30. Fujita M., Kiyono T., Hayashi Y., Ishibashi M.. In vivo interaction of human MCM heterohexameric complexes with chromatin. Possible involvement of ATP. J. Biol. Chem. 1997; 272:10928–10935. [DOI] [PubMed] [Google Scholar]
- 31. Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L.. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012; 7:562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P.. Integrative genomics viewer. Nat. Biotechnol. 2011; 29:24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Thorvaldsdóttir H., Robinson J.T., Mesirov J.P.. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 2013; 14:178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Maehara K., Odawara J., Harada A., Yoshimi T., Nagao K., Obuse C., Akashi K., Tachibana T., Sakata T., Ohkawa Y.. A co-localization model of paired ChIP-seq data using a large ENCODE data set enables comparison of multiple samples. Nucleic Acids Res. 2013; 41:54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shen L., Shao N., Liu X., Nestler E.. ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genomics. 2014; 15:284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Maehara K., Ohkawa Y.. Agplus: a rapid and flexible tool for aggregation plots. Bioinformatics. 2015; 31:3046–3047. [DOI] [PubMed] [Google Scholar]
- 38. Cucco F., Palumbo E., Camerini S., D’Alessio B., Quarantotti V., Casella M.L., Rizzo I.M., Cukrov D., Delia D., Russo A. et al. . Separase prevents genomic instability by controlling replication fork speed. Nucleic Acids Res. 2018; 46:267–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Foulk M.S., Urban J.M., Casella C., Gerbi S.A.. Characterizing and controlling intrinsic biases of lambda exonuclease in nascent strand sequencing reveals phasing between nucleosomes and G-quadruplex motifs around a subset of human replication origins. Genome Res. 2015; 25:725–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Langley A.R., Graf S., Smith J.C., Krude T.. Genome-wide identification and characterisation of human DNA replication origins by initiation site sequencing (ini-seq). Nucleic Acids Res. 2016; 44:10230–10247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Giresi P.G., Kim J., McDaniell R.M., Iyer V.R., Lieb J.D.. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 2007; 17:877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Song L., Zhang Z., Grasfeder L.L., Boyle A.P., Giresi P.G., Lee B.K., Sheffield N.C., Graf S., Huss M., Keefe D. et al. . Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res. 2011; 21:1757–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Simon J.M., Giresi P.G., Davis I.J., Lieb J.D.. Using formaldehyde-assisted isolation of regulatory elements (FAIRE) to isolate active regulatory DNA. Nat. Protoc. 2012; 7:256–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Beck D.B., Burton A., Oda H., Ziegler-Birling C., Torres-Padilla M.E., Reinberg D.. The role of PR-Set7 in replication licensing depends on Suv4-20h. Genes Dev. 2012; 26:2580–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kuo A.J., Song J., Cheung P., Ishibe-Murakami S., Yamazoe S., Chen J.K., Patel D.J., Gozani O.. The BAH domain of ORC1 links H4K20me2 to DNA replication licensing and Meier-Gorlin syndrome. Nature. 2012; 484:115–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hansen R.S., Thomas S., Sandstrom R., Canfield T.K., Thurman R.E., Weaver M., Dorschner M.O., Gartler S.M., Stamatoyannopoulos J.A.. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Karnani N., Taylor C.M., Malhotra A., Dutta A.. Genomic study of replication initiation in human chromosomes reveals the influence of transcription regulation and chromatin structure on origin selection. Mol. Biol. Cell. 2010; 21:393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. MacAlpine H.K., Gordan R., Powell S.K., Hartemink A.J., MacAlpine D.M.. Drosophila ORC localizes to open chromatin and marks sites of cohesin complex loading. Genome Res. 2010; 20:201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Powell S.K., MacAlpine H.K., Prinz J.A., Li Y., Belsky J.A., MacAlpine D.M.. Dynamic loading and redistribution of the Mcm2–7 helicase complex through the cell cycle. EMBO J. 2015; 34:531–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Carninci P., Sandelin A., Lenhard B., Katayama S., Shimokawa K., Ponjavic J., Semple C.A., Taylor M.S., Engstrom P.G., Frith M.C. et al. . Genome-wide analysis of mammalian promoter architecture and evolution. Nat. Genet. 2006; 38:626–635. [DOI] [PubMed] [Google Scholar]
- 51. Schug J., Schuller W.P., Kappen C., Salbaum J.M., Bucan M., Stoeckert C.J. Jr. Promoter features related to tissue specificity as measured by Shannon entropy. Genome Biol. 2005; 6:R33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cayrou C., Coulombe P., Vigneron A., Stanojcic S., Ganier O., Peiffer I., Rivals E., Puy A., Laurent-Chabalier S., Desprat R. et al. . Genome-scale analysis of metazoan replication origins reveals their organization in specific but flexible sites defined by conserved features. Genome Res. 2011; 21:1438–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cayrou C., Ballester B., Peiffer I., Fenouil R., Coulombe P., Andrau J.C., van Helden J., Mechali M.. The chromatin environment shapes DNA replication origin organization and defines origin classes. Genome Res. 2015; 25:1873–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Huppert J.L., Balasubramanian S.. Prevalence of quadruplexes in the human genome. Nucleic Acids Res. 2005; 33:2908–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Burge S., Parkinson G.N., Hazel P., Todd A.K., Neidle S.. Quadruplex DNA: sequence, topology and structure. Nucleic Acids Res. 2006; 34:5402–5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cayrou C., Coulombe P., Puy A., Rialle S., Kaplan N., Segal E., Mechali M.. New insights into replication origin characteristics in metazoans. Cell Cycle. 2012; 11:658–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chambers V.S., Marsico G., Boutell J.M., Di Antonio M., Smith G.P., Balasubramanian S.. High-throughput sequencing of DNA G-quadruplex structures in the human genome. Nat. Biotechnol. 2015; 33:877–881. [DOI] [PubMed] [Google Scholar]
- 58. Hansel-Hertsch R., Beraldi D., Lensing S.V., Marsico G., Zyner K., Parry A., Di Antonio M., Pike J., Kimura H., Narita M. et al. . G-quadruplex structures mark human regulatory chromatin. Nat. Genet. 2016; 48:1267–1272. [DOI] [PubMed] [Google Scholar]
- 59. Halder K., Halder R., Chowdhury S.. Genome-wide analysis predicts DNA structural motifs as nucleosome exclusion signals. Mol. Biosyst. 2009; 5:1703–1712. [DOI] [PubMed] [Google Scholar]
- 60. Halder R., Halder K., Sharma P., Garg G., Sengupta S., Chowdhury S.. Guanine quadruplex DNA structure restricts methylation of CpG dinucleotides genome-wide. Mol. Biosyst. 2010; 6:2439–2447. [DOI] [PubMed] [Google Scholar]
- 61. Vashee S., Cvetic C., Lu W., Simancek P., Kelly T.J., Walter J.C.. Sequence-independent DNA binding and replication initiation by the human origin recognition complex. Genes Dev. 2003; 17:1894–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Schaarschmidt D., Baltin J., Stehle I.M., Lipps H.J., Knippers R.. An episomal mammalian replicon: sequence-independent binding of the origin recognition complex. EMBO J. 2004; 23:191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ma W., Noble W.S., Bailey T.L.. Motif-based analysis of large nucleotide data sets using MEME-ChIP. Nat. Protoc. 2014; 9:1428–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Durkin S.G., Glover T.W.. Chromosome fragile sites. Annu. Rev. Genet. 2007; 41:169–192. [DOI] [PubMed] [Google Scholar]
- 65. Tuduri S., Crabbe L., Conti C., Tourriere H., Holtgreve-Grez H., Jauch A., Pantesco V., De Vos J., Thomas A., Theillet C. et al. . Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol. 2009; 11:1315–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Helmrich A., Ballarino M., Tora L.. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell. 2011; 44:966–977. [DOI] [PubMed] [Google Scholar]
- 67. Smith D.I., Zhu Y., McAvoy S., Kuhn R.. Common fragile sites, extremely large genes, neural development and cancer. Cancer Lett. 2006; 232:48–57. [DOI] [PubMed] [Google Scholar]
- 68. Durkin S.G., Arlt M.F., Howlett N.G., Glover T.W.. Depletion of CHK1, but not CHK2, induces chromosomal instability and breaks at common fragile sites. Oncogene. 2006; 25:4381–4388. [DOI] [PubMed] [Google Scholar]
- 69. Palumbo E., Matricardi L., Tosoni E., Bensimon A., Russo A.. Replication dynamics at common fragile site FRA6E. Chromosoma. 2010; 119:575–587. [DOI] [PubMed] [Google Scholar]
- 70. Le Tallec B., Dutrillaux B., Lachages A.M., Millot G.A., Brison O., Debatisse M.. Molecular profiling of common fragile sites in human fibroblasts. Nat. Struct. Mol. Biol. 2011; 18:1421–1423. [DOI] [PubMed] [Google Scholar]
- 71. Letessier A., Millot G.A., Koundrioukoff S., Lachages A.M., Vogt N., Hansen R.S., Malfoy B., Brison O., Debatisse M.. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature. 2011; 470:120–123. [DOI] [PubMed] [Google Scholar]
- 72. Ozeri-Galai E., Lebofsky R., Rahat A., Bester A.C., Bensimon A., Kerem B.. Failure of origin activation in response to fork stalling leads to chromosomal instability at fragile sites. Mol. Cell. 2011; 43:122–131. [DOI] [PubMed] [Google Scholar]
- 73. Blow J.J., Ge X.Q., Jackson D.A.. How dormant origins promote complete genome replication. Trends Biochem. Sci. 2011; 36:405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wong P.G., Winter S.L., Zaika E., Cao T.V., Oguz U., Koomen J.M., Hamlin J.L., Alexandrow M.G.. Cdc45 limits replicon usage from a low density of preRCs in mammalian cells. PLoS One. 2011; 6:e17533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Martin M.M., Ryan M., Kim R., Zakas A.L., Fu H., Lin C.M., Reinhold W.C., Davis S.R., Bilke S., Liu H. et al. . Genome-wide depletion of replication initiation events in highly transcribed regions. Genome Res. 2011; 21:1822–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Valenzuela M.S., Chen Y., Davis S., Yang F., Walker R.L., Bilke S., Lueders J., Martin M.M., Aladjem M.I., Massion P.P. et al. . Preferential localization of human origins of DNA replication at the 5′-ends of expressed genes and at evolutionarily conserved DNA sequences. PLoS One. 2011; 6:e17308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Miotto B., Struhl K.. HBO1 histone acetylase activity is essential for DNA replication licensing and inhibited by Geminin. Mol. Cell. 2010; 37:57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Aizawa M., Sugimoto N., Watanabe S., Yoshida K., Fujita M.. Nucleosome assembly and disassembly activity of GRWD1, a novel Cdt1-binding protein that promotes pre-replication complex formation. Biochim. Biophys. Acta. 2016; 1863:2739–2748. [DOI] [PubMed] [Google Scholar]
- 79. Wong P.G., Glozak M.A., Cao T.V., Vaziri C., Seto E., Alexandrow M.. Chromatin unfolding by Cdt1 regulates MCM loading via opposing functions of HBO1 and HDAC11-geminin. Cell Cycle. 2010; 9:4351–4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Feng Y., Vlassis A., Roques C., Lalonde M.E., Gonzalez-Aguilera C., Lambert J.P., Lee S.B., Zhao X., Alabert C., Johansen J.V. et al. . BRPF3-HBO1 regulates replication origin activation and histone H3K14 acetylation. EMBO J. 2016; 35:176–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wu P.Y., Nurse P.. Establishing the program of origin firing during S phase in fission Yeast. Cell. 2009; 136:852–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Tanaka S., Nakato R., Katou Y., Shirahige K., Araki H.. Origin association of Sld3, Sld7, and Cdc45 proteins is a key step for determination of origin-firing timing. Curr. Biol. 2011; 21:2055–2063. [DOI] [PubMed] [Google Scholar]
- 83. Kohler C., Koalick D., Fabricius A., Parplys A.C., Borgmann K., Pospiech H., Grosse F.. Cdc45 is limiting for replication initiation in humans. Cell Cycle. 2016; 15:974–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Das S.P., Borrman T., Liu V.W., Yang S.C., Bechhoefer J., Rhind N.. Replication timing is regulated by the number of MCMs loaded at origins. Genome Res. 2015; 25:1886–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Helmrich A., Ballarino M., Nudler E., Tora L.. Transcription-replication encounters, consequences and genomic instability. Nat. Struct. Mol. Biol. 2013; 20:412–418. [DOI] [PubMed] [Google Scholar]
- 86. Wang L., Paradee W., Mullins C., Shridhar R., Rosati R., Wilke C.M., Glover T.W., Smith D.I.. Aphidicolin-induced FRA3B breakpoints cluster in two distinct regions. Genomics. 1997; 41:485–488. [DOI] [PubMed] [Google Scholar]
- 87. Palakodeti A., Han Y., Jiang Y., Le Beau M.M.. The role of late/slow replication of the FRA16D in common fragile site induction. Genes. Chromosomes Cancer. 2004; 39:71–76. [DOI] [PubMed] [Google Scholar]
- 88. Hormozian F., Schmitt J.G., Sagulenko E., Schwab M., Savelyeva L.. FRA1E common fragile site breaks map within a 370kilobase pair region and disrupt the dihydropyrimidine dehydrogenase gene (DPYD). Cancer Lett. 2007; 246:82–91. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ChIP-Seq data have been deposited with accession codes DRA005864 (raw data) and GSE107248 (processed data).