
Table 1.
X-ray diffraction and refinement statistics
| Data collection a | |
| Space group | P43212 |
| Cell constants | |
| a = b, c (Å) | 40.4, 284.6 |
| α = β = γ (°) | 90.0 |
| Resolution (Å) | 38.90–1.80 |
| (1.83–1.80) | |
| R p.i.m. (%)b | 2.6 (45.1) |
| CC1/2 (%)c | 98.7 (69.2) |
| I/σ(I) | 19.9 (1.8) |
| Complete (%) | 99.4 (91.8) |
| Redundancy | 8.8 (7.9) |
| Refinement | |
| Resolution (Å) | 37.2–1.80 |
| No. reflections | 23 297 |
| R work /R free (%) | 18.9/22.1 |
| No. atoms | |
| Protein | 746 |
| RNA | 572 |
| Solvent | 153 |
| B -factors (Å 2 ) | |
| Protein | 39 |
| RNA | 44 |
| Waters | 47 |
| R.M.S. deviations | |
| Bonds (Å) | 0.005 |
| Angles (°) | 0.759 |
| Clash scored | 0.4 |
| Ramachandran (%) | |
| Allowed | 100.0 |
| Outliers | 0.0 |
| Coord. errore (Å) | 0.21 |
a X-ray data collection (λ = 0.9795) was conducted remotely at beamline 12-2 of the Stanford Synchrotron Radiation Lightsource (SSRL, Menlo Park, CA, USA) using Blu-Ice software and the Stanford Auto-Mounter (50).
b
R
precision-indicating merging R-value = 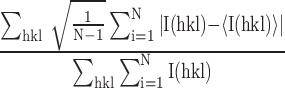 , where N is the redundancy of the data and
, where N is the redundancy of the data and 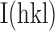 is the average intensity (51). Data were reduced with XDS and AIMLESS (52,53).
is the average intensity (51). Data were reduced with XDS and AIMLESS (52,53).
c The Pearson correlation coefficient calculated for the average intensities resulting from division of the unmerged data into two parts, each containing half of the measurements selected at random for each unique reflection (54).
d Number of unfavorable all-atom steric overlaps ≥0.4 Å per 1000 atoms (55).
e Coordinate error as implemented in PHENIX (29).