

 Our data provide the direct in vivo evidence to indicate the molecular signalling mechanism of endosulfan-induced apoptosis.
Our data provide the direct in vivo evidence to indicate the molecular signalling mechanism of endosulfan-induced apoptosis.
Abstract
Endosulfan as a new member of persistent organic pollutants has been shown to induce apoptosis in various animal models. However, the mechanism underlying endosulfan-induced apoptosis has not been well elucidated thus far. Caenorhabditis elegans N2 wild type and mutant strains were used in the present study to clarify the roles of the mitochondria, the insulin/insulin-like growth factor-1 (IGF-1) signaling pathway, and mitogen-activated protein kinase (MAPK) cascades in α-endosulfan-induced apoptosis. Our results demonstrated a dose- and time-dependent increase of apoptosis in the meiotic zone of the gonad of C. elegans exposed to graded concentrations of endosulfan. The expression levels of sod-3, localized in the mitochondrial matrix, increased greatly after endosulfan exposure. A significant increase in germ cell apoptosis was observed in abnormal methyl viologen sensitivity-1 (mev-1(kn-1)) mutants (with abnormal mitochondrial respiratory chain complex II and higher ROS levels) compared to that in N2 at equal endosulfan concentrations. We found that the insulin/IGF-1 signaling pathway and its downstream Ras/ERK/MAPK did not participate in the endosulfan-induced apoptosis. However, the apoptosis in the loss-of-function strains of JNK and p38 MAPK signaling pathways was completely or mildly suppressed under endosulfan stress. The apoptotic effects of endosulfan were blocked in the mutants of jnk-1/JNK-MAPK, sek-1/MAP2K, and pmk-1/p38-MAPK, suggesting that these downstream genes play an essential role in endosulfan-induced germ cell apoptosis. In contrast, the mkk-4/MAP2K and nsy-1/MAP3K were only partially involved in the apoptosis induction. Our data provide evidence that endosulfan increases germ cell apoptosis, which is regulated by mitochondrial function, JNK and p38 MAPK cascades. These findings contribute to the understanding of the signal transduction pathways involved in endosulfan-induced apoptosis.
Introduction
Endosulfan (6,7,8,9,10,10-hexachloro-1,5,5a,6,9,9a-hexahydro-6,9-methano-2,4,3-benzodioxathiepin-3-oxide), an organochlorine pesticide, binds and inhibits the γ-amino-butyric acid (GABA)-gated chloride channel receptor, thereby inhibiting GABA-induced chloride flux across membranes, resulting in uncontrolled excitation.1 Technical-grade endosulfan is commercially available as a mixture typically containing >95% of two diastereoisomers, α-endosulfan and β-endosulfan, in ratios from 2 : 1 to 7 : 3.2 Endosulfan has been listed as a persistent organic pollutant (POP) by the Stockholm Convention and is being phased out of all manufacturing processes.3 However, endosulfan is still widely used and produced in more than 30 countries because of its insecticidal applications.4 A large number of ecosystem organisms and field workers may potentially be exposed to endosulfan via the food chain and occupational routes.
Recently, there have been increasing concerns about the influence of endosulfan on the function of the reproductive systems of various species.1,5 For example, the exposure of pregnant rats to endosulfan not only increased fetal resorption and induced gross fetal anomalies, but also decreased spermatogenesis in the offspring.6 Short-term exposure to environmentally-relevant concentrations of endosulfan may have long-term effects on reproduction in fish.7
Apoptosis is a programmed event that plays multiple roles and has critical functions in reproductive development by maintaining an appropriate germ cell to Sertoli cell ratio, removing defective germ cells, and controlling sperm production.8 Endosulfan has been reported to induce apoptosis in the spermatogenic cells of mouse, adult rat testes, adult rabbit testes, and Sertoli-germ cells of male rats.5,9–11 Oxidative stress and mitochondrial dysfunction have been shown to play a pivotal role in endosulfan toxicity-induced apoptosis.9,11 Endosulfan induces oxidative stress and apoptosis via possible mechanisms of both mitochondrial and non-mitochondrial pathways.11 Oxidative stress and mitochondrial dysfunction can easily lead to disorders of other intracellular signals. For example, the highly-conserved insulin/insulin-like growth factor (IGF)-1 and mitogen-activated protein kinase (MAPK) cascades are affected by changes in reactive oxygen species (ROS) levels.12–14 Increasing evidence suggests that apoptosis may be directly or indirectly controlled by the extracellular signal-regulated protein kinase (ERK) and the c-Jun N-terminal kinase (JNK).15–17 A study using spermatogonial cells of mice as an assay system demonstrated that endosulfan resulted in a dose-dependent increase in apoptosis by inhibiting the ERK/MAPK signaling pathway.18 However, a recent study demonstrated that in human HaCaT keratinocytes, endosulfan decreases apoptosis under persistent ERK phosphorylation.19 Therefore, contradictory conclusions have been obtained from the studies on endosulfan toxicity. Moreover, we know little about the molecular mechanisms underlying endosulfan-induced apoptosis.
Caenorhabditis elegans (C. elegans) as a model organism was first recognized by Brenner.20 Depending on the particular bioinformatics approach used, C. elegans homologues have been identified for 60–80% of human genes.21 Today, C. elegans is used to study a large variety of biological processes including apoptosis, cell signaling, cell cycle, cell polarity, gene regulation, metabolism, aging, and sex determination.21 Its germ cells keep proliferating and undergo programmed cell death either physiologically or in response to environmental stresses.22–24 The germline cells of C. elegans are sensitive to stress induced by exogenous factors.24 The C. elegans germline apoptosis during oogenesis is a fundamentally important reproductive process and is evolutionarily conserved from nematodes to mammals, including humans. Importantly, C. elegans shares cellular and molecular structures and signaling pathways with higher organisms; thus, biological information learned from C. elegans may be directly applicable to more complex organisms.25 The use of in vivo animal models with mammalian homologous genes is basically needed to better understand the toxic mechanism of endosulfan.
The toxic effects of endosulfan have been investigated in both target and nontarget organisms, which have revealed that endosulfan isomers and their sulfate metabolite exhibit different levels of toxicity.26,27 Evidence suggests that the α-isomer is more toxic to insects and mammals than the β-isomer and sulfate metabolite.26,27 The main objective of the present study was to investigate the roles of conserved signal transduction pathways in α-endosulfan-induced apoptosis in C. elegans. Our results show that the exposure of L4-stage or young adult worms to α-endosulfan significantly enhances germ cell apoptosis. This induction of germ cell apoptosis might be triggered by ROS, JNK, and p38/MAPK signaling pathways.
Materials and methods
Chemicals
α-Endosulfan used in this study was purchased from Sigma-Aldrich (St Louis, MO) and was of the highest purity available. The molecular configuration of endosulfan isomers and sulfate metabolite is shown in Fig. 1. Acridine orange (AO) was a commercial product of Molecular Probes (Eugene, OR). 5-Fluoro-2′-deoxyuridine (FudR) was purchased from Sigma-Aldrich (St Louis, MO) as well.
Fig. 1. Endosulfan isomers and sulfate metabolite. (A) α-Endosulfan. (B) β-Endosulfan and (C) endosulfan sulfate.

Worm strains and culture
Maintenance of C. elegans was carried out according to the standard procedures as described by Brenner.20 All strains were grown at 20 °C in Petri dishes in nematode growth medium (NGM) and fed with the bacterium Escherichia coli strain OP50. The tests on C. elegans were conducted following the institutional criteria for the care and use of laboratory bio-materials approved by the Chinese Academy of Sciences. The wild-type strain used was Bristol N2. The following mutant strains were provided by the Caenorhabditis Genetics Centre (CGC): the strains deficient in the ERK signalling cascade, lin-45(n2520), mek-2 (n1989), ksr-1(ku68) and mpk-1 (ku1); the JNK signalling cascade, mek-1 (ks54), jnk-1 (gk7), jkk-1(km2) and mkk-4 (ju91); the p38 MAPK signalling cascade, nsy-1 (ag3), sek-1(ag1) and pmk-1 (km25); the insulin/IGF-1 signalling pathway, daf-2(e1370), age-1(hx546) and daf-16(mu86); the oxygen sensitive mutant of mev-1(kn-1) and the transgenic strain muIs84 [sod-3p::GFP + rol-6]. The description of all mutants we used in the experiment is listed in Table S1.† To obtain synchronized cultures, gravid hermaphrodites were lysed in an alkaline hypochlorite solution and the gathered eggs were subjected to an overnight hatching at 20 °C. The new hatchers were maintained for 24 h at the L1 stage in the absence of food until inoculated onto NGM.
Worm exposure
Endosulfan was dissolved in dimethyl sulfoxide (DMSO). Before using, the stock solutions were diluted with M9 buffer (3 g KH2PO4, 6 g Na2HPO4, 5 g NaCl, 1 ml 1 M MgSO4, and H2O to 1 L, sterilized by autoclaving) to the working concentrations. Young adult hermaphrodites were transferred into a Costar 24-well tissue plate with M9 buffer or test solutions for 6, 12, and 24 h, respectively. E. coli strain OP50 was added as a food source. The maximum concentration of DMSO was 0.01% in the working solutions of endosulfan, which showed no effects in C. elegans.
Germ cell death/apoptosis assay
Germ cell corpses were measured by AO vital staining. Briefly, the treated worms were stained for 1 h in the dark at 20 °C by transferring worms into a Costar 24-well plate containing 500 μL of 25 μg mL–1 AO and OP50 in M9 buffer and then transferred to NGM for a recovery of 40 min on bacterial lawns also in the dark. AO staining positive cell corpses were scored using an Olympus IX71 fluorescence microscope (Olympus, Tokyo, Japan). The apoptotic cells appeared yellow or yellow-orange after AO vital staining, representing increased DNA fragmentation, while intact cells were uniformly green in color. Only the gonad arm in the posterior part of the body was scored, because the autofluorescence of the intestine shaded the gonad arm near the pharynx.
Life-span assay
All life-span assays were conducted in 96-well plates containing M9 buffer and E. coli OP50 was added as a food source. Briefly, 30 age-synchronized L3 stage hermaphrodites were picked and transferred to a 96-well plate. Each well contained one worm in 200 μL M9 or test solution at 20 °C. 5-FudR (20 μg mL–1), with no significant effect on the lifespan, was added to each well to prevent offspring generation. All worms were continuously exposed to endosulfan until the worm was dead, which was identified by the lack of response to mechanical stimulation.
Sod-3 expression assay
Sod-3 expression was measured as an indicator of oxidative stress in endosulfan-exposed sod-3p::gfp allele worms, which co-express green fluorescent protein with sod-3 precursor in wild type. Worms at L3-larva were exposed to endosulfan for 8 h, and expression of sod-3 was observed by fluorescence microscopy. Nematodes were imaged using an Olympus IX71 fluorescence microscope (Olympus, Tokyo, Japan) and quantified using Image J.
Data analysis
All experiments were performed at least three independent times. All values were expressed as means ± standard error. A significant difference (p < 0.05) level between various concentrations of different strains was tested using ANOVA followed by Tukey's multiple comparison test. For comparisons between different strains, statistical analysis was performed with 2-factor ANOVA with Dunnett's t tests.
Results and discussion
Induction of germ cell apoptosis in C. elegans treated with α-endosulfan
Endosulfan has been reported to exhibit significant genotoxicity and cytotoxicity in several in vitro or in vivo models.9–11 As shown in Fig. 2, α-endosulfan (0.1 to 10 μM) induced germ cell death in a time- and dose-dependent manner. A significant increase in the number of germ cell corpses was observed in C. elegans exposed to 0.1 μM endosulfan for 12 h. These data were consistent with observations in mice spermatogenic cell lines (GC-1 spg) and the human T-cell leukemic line.9,28 Germline apoptosis plays an integral role in oogenesis. An abnormal increase in the level of apoptosis under exogenous stress will inevitably leads to dysfunction of the reproductive system. However, the potential mechanisms underlying endosulfan-induced apoptosis are still largely unknown, and likely involve multiple pathways. C. elegans can serve as a powerful model system for studying the molecular mechanisms underlying endosulfan toxicity in vivo.
Fig. 2. Fluorescence microscopy observations of α-endosulfan exposure-induced germline cell apoptosis in wild type N2 exposed to 0 (A) and 10 μM (B) of endosulfan for 12 h, respectively. Representative pictures of dying cells stained with acridine orange (AO). Apoptotic cells are indicated by white arrows. (C) C. elegans was exposed to graded concentrations of α-endosulfan for 6, 12, and 24 h, and the number of germ cell corpses per gonad arm was determined. About 44 to 50 worms were quantified for each dose. Data were pooled from at least three independent experiments. Error bars indicate ± SD; asterisks indicate statistical significance at p < 0.05.

Contribution of mitochondria to endosulfan-induced apoptosis
ROS, which can cause oxidative damage to proteins, DNA, and lipids are considered to be a key inducer of apoptosis.9–11 Several in vitro and in vivo studies have suggested that exposure to endosulfan induces ROS generation and results in oxidative damage.9,10 The C. elegans sod-3 encodes an iron/manganese superoxide dismutase, localized in the mitochondrial matrix, that might defend against oxidative stress and promote a normal lifespan.29,30 Using CF1553(muls84) strain containing the sod-3p::gfp reporter gene, we found that exposure to endosulfan at 0.1, 1, and 10 μM resulted in an approximately 1.44-, 1.79-, and 2.29-fold increase in fluorescence intensity compared to CF1553 control, respectively (Fig. 3A and B), indicating that endosulfan increased ROS generation in C. elegans. Organisms are equipped with antioxidant defense mechanisms to counter oxidative stress. A significant time-dependent increase of SOD activity was observed in the Sertoli-germ cells of male rats following endosulfan treatment.5 However, the antioxidant enzymes were gradually consumed following the excess production of ROS, which resulted in a decreased antioxidant capacity. An imbalance in the prooxidant/antioxidant homeostasis led to oxidative stress.31,32
Fig. 3. The endosulfan-induced germ cell apoptosis and lifespan decrease were mediated by mitochondria and ROS. (A) Representative fluorescence microscopy images of sod-3 gene expression in L3 larva of C. elegans muIs84 (sod-3p::gfp) exposed to graded concentrations of endosulfan for 8 h. (B) The fluorescence intensity of CF1553 quantified using the Image J software. (C) The average number of endosulfan-induced germline apoptotic cells in N2 worms with or without 1.0% DMSO and in stress-sensitive mev-1(kn-1) mutants. (D) Effect of endosulfan on the lifespan of wild type N2 worms and mev-1(kn-1) mutants. Error bars indicate ±SD; asterisks indicate statistical significance at p < 0.05.

Mev-1 encodes the C. elegans ortholog of the succinate dehydrogenase cytochrome b560 subunit, an integral membrane protein that is a subunit of mitochondrial respiratory chain complex II (ubiquinol-cytochrome c reductase).33Mev-1 is required for oxidative phosphorylation. As shown in Fig. 3C, the spontaneous physiological germ cell apoptosis in stress-sensitive mev-1(kn-1) mutants was higher than that in the wild-type worm. Compared to that in wild-type worms, a greater number of apoptotic cells and a shorter lifespan were observed in mev-1(kn-1) mutants following exposure to equal endosulfan concentrations, indicating that the mev-1(kn-1) mutants were hypersensitive to endosulfan exposure (Fig. 3C and D). However, the enhanced apoptosis induced by endosulfan was effectively rescued upon the addition of DMSO, a ROS quencher (Fig. 3C). These results provide clear evidence that mitochondria and ROS play a pivotal role in endosulfan-induced germ cell death.
Roles of insulin/IGF-1 signaling pathway in endosulfan-induced germ cell apoptosis
The conserved insulin signaling pathway can be positively and negatively regulated by ROS.12 The C. elegans insulin/IGF-1 signaling (IIS) pathway connects nutrient levels to metabolism, growth, development, longevity, and behavior.13 This fundamental pathway is regulated by insulin-like peptide ligands that bind to the insulin/IGF-1 transmembrane receptor (IGFR) ortholog daf-2.34 Daf-2/IGFR controls the activity of the PI3K/Akt kinase cascade, culminating in the regulation of a FoxO transcription factor, daf-16, that governs most of the functions of this pathway.34 The downstream targets of daf-16/FoxO influence germline cell proliferation and p53-dependent apoptosis.34 Therefore, it was expected that the insulin/IGF-1 signaling pathway might play a role in endosulfan-induced germline apoptosis in C. elegans.
As shown in Fig. 4, there were no significant differences in the numbers of germ cell corpses in daf-2(e1370)/IGFR, age-1(hx546)/PI3K, and daf-16(mu86)/FoxO mutants compared to that in N2 worms at all concentrations, suggesting that the insulin/IGF-1 signaling pathway is not involved in regulating endosulfan-induced apoptosis in C. elegans. However, it has been shown that the insulin levels can be significantly altered by endosulfan. Endosulfan has an impact on the B cells of the rat pancreas, which are responsible for insulin secretion.35 Exposure of adult male rats and rabbits to endosulfan inhibited the synthesis and secretion of insulin in pancreatic cells and resulted in a marked decrease in the level of serum insulin.36,37 Activated insulin and IGF-1 receptors phosphorylate a variety of substrates, among which are the insulin receptor substrate (IRS) family of scaffold proteins.38 Tyrosine-phosphorylated IRS proteins promote the recruitment and activation of components of downstream cascades such as the Ras/MAPK pathway (Ras-Raf-MEK-ERK MAPK) described below, as well as the phosphoinositide 3-kinase (PI3K)/Akt and mTOR pathways.13
Fig. 4. Roles of daf-2, age-1 and daf-16 in endosulfan-induced apoptosis. Data were pooled from at least three independent experiments. Error bars indicate ±SD; asterisks indicate statistical significance at p < 0.05.

Contribution of MAPK signaling cascades in endosulfan-induced germ cell death
The Raf/MEK/ERK signaling pathway is one of the most well-known signal transduction pathways that regulates cell proliferation, cell cycle arrest, terminal differentiation, and apoptosis.39 In C. elegans, lin-45(Raf, MAPKKK), mek-2(MEK, MAPKK), and mpk-1(ERK, MAPK) are the components of the ERK signaling pathway, which controls and coordinates multiple biological processes during germline development.40 The C. elegans ksr-1 encodes one of the two C. elegans kinase suppressors of Ras paralogs, and positively regulates Ras signaling.41 As shown in Fig. 5A, germline cells in lin-45(n2520), mek-2 (n1989), ksr-1(ku68), and mpk-1 (ku1) mutants showed a prominent apoptosis response to endosulfan compared to that of the untreated mutants, indicating that the ERK signaling pathway was not indispensable in response to endosulfan-induced apoptosis. However, germ cell apoptosis occurring in the proximal pachytene zone has been shown to be directly or indirectly controlled by ERK.16,17 Endosulfan decreased apoptosis in human HaCaT keratinocytes, and this was partially mediated by a ROS-dependent ERK1/2 mechanism.19 This inconsistency might be due to the differences in the species.
Fig. 5. Induction of germ cell apoptosis by exposure to α-endosulfan in worms deficient in ERK (A), JNK (B), and p38/MAPK (C) signaling pathways. The germline apoptosis of the corresponding mutants was determined after exposure of the worms to 10 μM α-endosulfan for 12 h. Data were pooled from at least three independent experiments. Error bars indicate ±SD; asterisks indicate statistical significance at p < 0.05.

In addition to the ERK signaling pathway, the JNK and p38/MAPK signaling pathways have also been implicated in a variety of biological functions in mammalian cells, such as apoptosis and the responses to ROS related stress.42 The C. elegans nsy-1 encodes a homolog of the human apoptosis signal-regulating kinase (ASK1, MAPKKK), an activator of JNK and p38 kinases.43,44 The mek-1 and jkk-1 genes encode human MKK7-like MAPKKs. The mkk-4 encodes a protein that is homologous to the MKK4 family of MAPKK. Jnk-1 is a member of the JNK homolog.45 As shown in Fig. 5B, germline apoptosis was only slightly altered in jnk-1(gk7) mutants exposed to endosulfan compared to that in the untreated mutants control. Nsy-1(ag3) and mkk-4(ju91) mutants exposed to 10 μM endosulfan showed a significant increase in the apoptotic germ cells (Fig. 5B). These results indicated that jnk-1 was involved in regulating apoptosis induction by endosulfan; however, the function of nsy-1 or mkk-4 was only partially involved in the induction of apoptosis. It has been proved that the C. elegans jkk-1 and mek-1 function in the same pathway as jnk-1 to regulate graphene oxide toxicity,46 however jkk-1 and mek-1 do not seem to be involved in response to endosulfan toxicity in this study.
The p38 MAPK of C. elegans has been associated with germline apoptosis regulation, innate immune responses, and the responses to heavy metals and oxidation.47–50 The C. elegans pmk-1 is a p38 MAPK homolog, and the p38 MAPK pathway of C. elegans is activated by mammalian MKK3/6 homolog sek-1(MAPKK), which is in turn activated by nsy-1.43,44 As shown in Fig. 5C, except for the nsy-1(ag3) mutants treated with 10 μM endosulfan, the number of germ cell corpses was not altered by endosulfan treatment in sek-1(ag1) and pmk-1(km25) mutants, suggesting that sek-1 and pmk-1 play pivotal roles in mediating endosulfan-induced germline apoptosis in C. elegans.
It is known that overexpression of ASK1 induced apoptotic cell death and ASK1 was activated in cells treated with tumor necrosis factor-α (TNF-α). Moreover, TNF-α-induced apoptosis was inhibited by a catalytically inactive form of ASK1.44C. elegans nsy-1/ASK1 may be a key element in endosulfan-induced apoptosis. However, the nsy-1 mutants did not completely block endosulfan-induced germline apoptosis. The elevated levels of apoptosis in nsy-1/ASK1 as well as mkk-4 mutants following treatment with 10 μM endosulfan suggested that a co-regulated pathway may be involved in endosulfan-induced germline apoptosis in C. elegans, as exists for the genotoxic response genes cep-1, egl-1, hus-1, and ced-3/4.51 It has been reported that endosulfan exposure can induce apoptosis in a variety of cells.5,8,11 In this report, we have provided clear evidence that the jnk-1/JNK-MAPK, sek-1/MAP2K, and pmk-1/p38-MAPK genes are critical for regulating endosulfan-induced germ cell apoptosis, which implies that there is a close relationship between the phosphorylation of MAPK and endosulfan-induced apoptosis. Phosphorylation is involved in each MAPK pathway,52 and the genes we found to be important in this study are down-stream targets in this pathway. These key genes are regulated by several up-stream genes, and even if one or more up-stream genes are mutated, the signal can be transduced by other genes.
ROS can be activated by various stresses and are rapidly removed by intracellular antioxidant proteins.53 However, once ROS production exceeds the capacity of the antioxidant proteins, the ROS may induce oxidative modifications of MAPK signaling proteins (e.g., RTKs and MAP3Ks), thereby leading to MAPK activation.53,54 Therefore, we hypothesize that the germ cell apoptosis induced by α-endosulfan in C. elegans occurs via an endosulfan-induced mitochondrial response and ROS generation that activates MAPK signaling pathways. A further investigation is needed to determine how the key signaling molecules, such as jnk-1 and pmk-1, are integrated with the core apoptotic machinery.
Conclusion
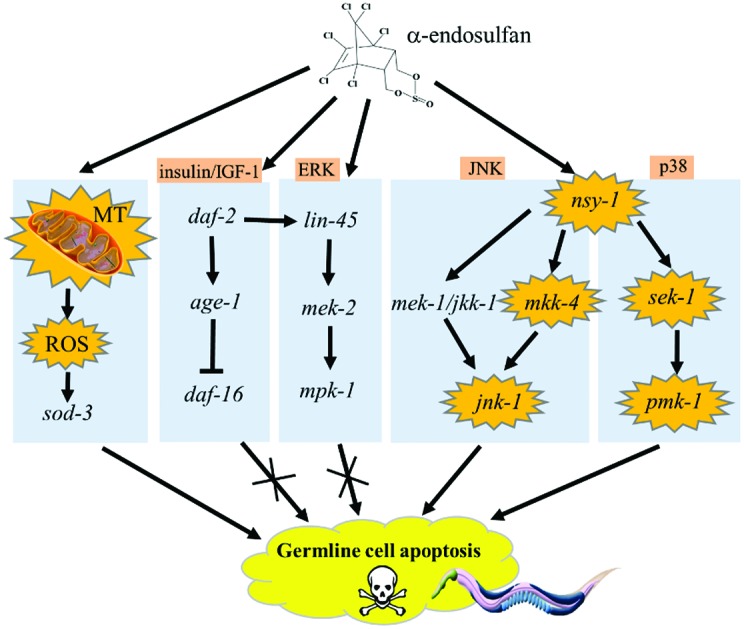
Our data provide direct evidence indicating that endosulfan has toxic effects on nematode germ cells, and suggest potential molecular mechanisms underlying endosulfan-induced germ cell apoptosis. Mitochondria play an essential role in endosulfan-induced apoptosis (Fig. 6). The prooxidant/antioxidant homeostasis was disturbed by endosulfan, making mev-1(kn-1) mutants with increased ROS level in vivo have a higher susceptibility to endosulfan toxicity. We found that the insulin/IGF-1 signaling pathway did not participate in endosulfan-induced apoptosis. Among the MAPK family members, the downstream genes of jnk-1/JNK-MAPK, sek-1/p38-MAP2K, and pmk-1/p38-MAPK appear to play pivotal roles in the induction of apoptosis by endosulfan, whereas mkk-4/JNK-MAP2K and nsy-1/MAP3K were only partially involved (Fig. 6). Our study provides new information that is useful for understanding the molecular basis of endosulfan-induced apoptosis.
Fig. 6. Roles of the mitochondria and MAPK signaling cascades in α-endosulfan-induced germline apoptosis in C. elegans.

Conflict of interest
There is no conflict of interest to declare.
Supplementary Material
Acknowledgments
This work was supported by grants from the Major National Scientific Research Projects, 2014CB932002, the Strategic Leading Science & Technology Program (B), XDB14030502, the National Natural Science Foundation of China, 21677147, 21507136, the China Postdoctoral Science Foundation, 2015 M82023, 2015 M582006, the CPSF-CAS Joint Foundation for Excellent Postdoctoral Fellows, 2015LH0017 and the CASHIPS director's fund, YZJJ201501.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c7tx00046d
References
- Silva M. H., Carr W. C., Beauvais S. CalEPA. 2007;1:4. [Google Scholar]
- Maier-Bode H. Residue Rev. 1968;22:1–44. doi: 10.1007/978-1-4615-8434-6_1. [DOI] [PubMed] [Google Scholar]
- Stockholm Convention, http://chm.pops.int/TheConvention/ConferenceoftheParties/Meetings/COP5/tabid/1267/mctl/ViewDetails/EventModID/870/EventID/109/xmid/4351/Default.aspx.
- Weber J., Halsall C. J., Muir D., Teixeira C., Small J., Solomon K. Sci. Total Environ. 2010;408:2966–2984. doi: 10.1016/j.scitotenv.2009.10.077. [DOI] [PubMed] [Google Scholar]
- Rastogi D., Narayan R., Saxena D. K., Chowdhuri D. K. Chemosphere. 2014;94:104–115. doi: 10.1016/j.chemosphere.2013.09.029. [DOI] [PubMed] [Google Scholar]
- Milesi M. M., Varayoud J., Bosquiazzo V. L., Munoz-De-Toro M., Luque E. H. Reprod. Toxicol. 2012;33:85–93. doi: 10.1016/j.reprotox.2011.12.003. [DOI] [PubMed] [Google Scholar]
- Gormley K. L., Teather K. L. Ecotoxicol. Environ. Saf. 2003;54:330–338. doi: 10.1016/s0147-6513(02)00005-2. [DOI] [PubMed] [Google Scholar]
- Shukla K. K., Mahdi A. A., Rajender S. Front. Biosci. 2012;4:746–754. doi: 10.2741/415. [DOI] [PubMed] [Google Scholar]
- Xu Y., Wang N., Shi Z., Li Y., Zhou X., Sun Z. Toxicol. Ind. Health. 2016;32:1550–1563. doi: 10.1177/0748233714567525. [DOI] [PubMed] [Google Scholar]
- Ozmen O., Mor F. Pestic. Biochem. Physiol. 2012;102:129–133. [Google Scholar]
- Aly H. A. A., Khafagy R. M. Food Chem. Toxicol. 2014;64:1–9. doi: 10.1016/j.fct.2013.11.007. [DOI] [PubMed] [Google Scholar]
- Bashan N., Kovsan J., Kachko I., Ovadia H., Rudich A. Physiol. Rev. 2009;89:27–71. doi: 10.1152/physrev.00014.2008. [DOI] [PubMed] [Google Scholar]
- Taniguchi C. M., Emanuelli B., Kahn C. R. Nat. Rev. Mol. Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- Keshari R. S., Verma A., Barthwal M. K., Dikshit M. J. Cell. Biochem. 2013;114:532–540. doi: 10.1002/jcb.24391. [DOI] [PubMed] [Google Scholar]
- Barateiro A., Vaz A. R., Silva S. L., Fernandes A., Brites D. NeuroMol. Med. 2012;14:285–302. doi: 10.1007/s12017-012-8187-9. [DOI] [PubMed] [Google Scholar]
- Arur S., Ohmachi M., Nayak S., Hayes M., Miranda A., Hay A., Golden A., Schedl T. Proc. Natl. Acad. Sci. U. S. A. 2009;106:4776–4781. doi: 10.1073/pnas.0812285106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. H., Ohmachi M., Arur S., Nayak S., Francis R., Church D., Lambie E., Schedl T. Genetics. 2007;177:2039–2062. doi: 10.1534/genetics.107.081356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F. Z., Zhang L. S., Wei J. L., Li Y. B., Shi Z. X., Yang Y. M., Zhou X. Q., Sun Z. W. Toxicol. Res. 2015;4:508–518. [Google Scholar]
- Antherieu S., Ledirac N., Luzy A. P., Lenormand P., Caron J. C., Rahmani R. J. Cell Physiol. 2007;213:177–186. doi: 10.1002/jcp.21108. [DOI] [PubMed] [Google Scholar]
- Brenner S. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaletta T., Hengartner M. O. Nat. Rev. Drug Discovery. 2006;5:387–398. doi: 10.1038/nrd2031. [DOI] [PubMed] [Google Scholar]
- Gumienny T. L., Lambie E., Hartwieg E., Horvitz H. R., Hengartner M. O. Development. 1999;126:1011–1022. doi: 10.1242/dev.126.5.1011. [DOI] [PubMed] [Google Scholar]
- Gartner A., Milstein S., Ahmed S., Hodgkin J., Hengartner M. O. Mol. Cell. 2000;5:435–443. doi: 10.1016/s1097-2765(00)80438-4. [DOI] [PubMed] [Google Scholar]
- Lant B., Derry W. B. Methods. 2013;61:174–182. doi: 10.1016/j.ymeth.2013.04.022. [DOI] [PubMed] [Google Scholar]
- Leung M. C., Williams P. L., Benedetto A., Au C., Helmcke K. J., Aschner M., Meyer J. N. Toxicol. Sci. 2008;106:5–28. doi: 10.1093/toxsci/kfn121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan M. T., Kuo J. N., Buday C., Schroeder G., Van Aggelen G., Pasternak J. Environ. Toxicol. Chem. 2005;24:1146–1154. doi: 10.1897/04-300r1.1. [DOI] [PubMed] [Google Scholar]
- Du H., Wang M., Dai H., Hong W., Wang M., Wang J., Weng N., Nie Y., Xu A. Environ. Sci. Technol. 2015;49:2460–2468. doi: 10.1021/es504837z. [DOI] [PubMed] [Google Scholar]
- Kannan K., Holcombe R. F., Jain S. K., Alvarez-Hernandez X., Chervenak R., Wolfe R. E., Glass J. Mol. Cell. Biochem. 2000;205:53–66. doi: 10.1023/a:1007080910396. [DOI] [PubMed] [Google Scholar]
- Hunter T., Bannister W. H., Hunter G. J. J. Biol. Chem. 1997;272:28652–28659. doi: 10.1074/jbc.272.45.28652. [DOI] [PubMed] [Google Scholar]
- Giglio M. P., Hunter T., Bannister J. V., Bannister W. H., Hunter G. J. Biochem. Mol. Biol. Int. 1994;33:37–40. [PubMed] [Google Scholar]
- Storz G., Imlay J. A. Curr. Opin. Microbiol. 1999;2:188–194. doi: 10.1016/s1369-5274(99)80033-2. [DOI] [PubMed] [Google Scholar]
- Finkel T., Holbrook N. J. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- Senoo-Matsuda N., Hartman P. S., Akatsuka A., Yoshimura S., Ishii N. J. Biol. Chem. 2003;278:22031–22036. doi: 10.1074/jbc.M211377200. [DOI] [PubMed] [Google Scholar]
- Pinkston-Gosse J., Kenyon C. Nat. Genet. 2007;39:1403–1409. doi: 10.1038/ng.2007.1. [DOI] [PubMed] [Google Scholar]
- Kalender Y., Kalender S., Uzunhisarcikli M., Ogutcu A., Açikgoz F., Durak D. Toxicology. 2004;200:205–211. doi: 10.1016/j.tox.2004.03.017. [DOI] [PubMed] [Google Scholar]
- Kumar S. N., Jain A. K., Singh K. P., Shrivastava N., Telang A. G. Toxicol. Lett. 2008;180:S188–S188. [Google Scholar]
- Ozmen O., Sahinduran S., Mor F. Pancreas. 2010;39:367–370. doi: 10.1097/MPA.0b013e3181bd95d6. [DOI] [PubMed] [Google Scholar]
- White M. F. Mol. Cell. Biochem. 1998;182:3–11. [PubMed] [Google Scholar]
- Peyssonnaux C., Eychene A. Biol. Cell. 2001;93:53–62. doi: 10.1016/s0248-4900(01)01125-x. [DOI] [PubMed] [Google Scholar]
- Arur S., Ohmachi M., Nayak S., Hayes M., Miranda A., Hay A., Golden A., Schedl T. Proc. Natl. Acad. Sci. U. S. A. 2009;106:4776–4781. doi: 10.1073/pnas.0812285106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmachi M., Rocheleau C. E., Church D., Lambie E., Schedl T., Sundaram M. V. Curr. Biol. 2002;12:427–433. doi: 10.1016/s0960-9822(02)00690-5. [DOI] [PubMed] [Google Scholar]
- Tanaka-Hino M., Sagasti A., Hisamoto N., Kawasaki M., Nakano S., Ninomiya-Tsuji J., Bargmann C. I., Matsumoto K. EMBO Rep. 2002;3:56–62. doi: 10.1093/embo-reports/kvf001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagasti A., Hisamoto N., Hyodo J., Tanaka-Hino M., Matsumoto K., Bargmann C. I. Cell. 2001;105:221–232. doi: 10.1016/s0092-8674(01)00313-0. [DOI] [PubMed] [Google Scholar]
- Ichijo H., Nishida E., Irie K., tenDijke P., Saitoh M., Moriguchi T., Takagi M., Matsumoto K., Miyazono K., Gotoh Y. Science. 1997;275:90–94. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- Sakaguchi A., Matsumoto K., Hisamoto N. J. Biochem. 2004;136:7–11. doi: 10.1093/jb/mvh097. [DOI] [PubMed] [Google Scholar]
- Zhao Y., Wu Q., Wang D. RSC Adv. 2015;5:92394–92405. [Google Scholar]
- Cheesman H. K., Feinbaum R. L., Thekkiniath J., Dowen R. H., Conery A. L., Pukkila-Worley R. G3: Genes, Genomes, Genet. 2016;6:541–549. doi: 10.1534/g3.115.025650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y., Zhi L., Wu Q., Yu Y., Sun Q., Wang D. Nanotoxicology. 2016;10:1469–1479. doi: 10.1080/17435390.2016.1235738. [DOI] [PubMed] [Google Scholar]
- Kim D. H., Feinbaum R., Alloing G., Emerson F. E., Garsin D. A., Inoue H., Tanaka-Hino M., Hisamoto N., Matsumoto K., Tan M. W., Ausubel F. M. Science. 2002;297:623–626. doi: 10.1126/science.1073759. [DOI] [PubMed] [Google Scholar]
- Wang S., Tang M., Pei B., Xiao X., Wang J., Hang H., Wu L. Toxicol. Sci. 2008;102:345–351. doi: 10.1093/toxsci/kfm220. [DOI] [PubMed] [Google Scholar]
- Du H., Wang M., Wang L., Dai H., Wang M., Hong W., Nie X., Wu L., Xu A. Toxicol. Sci. 2015;145:118–127. doi: 10.1093/toxsci/kfv035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L., Karin M. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- Son Y., Cheong Y. K., Kim N. H., Chung H. T., Kang D. G., Pae H. O. J. Signal Transduction. 2011;2011:792639. doi: 10.1155/2011/792639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalmi S. K., Sinha A. K. Front. Plant Sci. 2015;6:769. doi: 10.3389/fpls.2015.00769. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.