

Abstract
Purinergic receptor-7 (P2X7R) signaling controls Th17 and Th1 generation/differentiation, while NOD-like receptor P3 (NLRP3) acts as a Th2 transcriptional factor. Here, we demonstrated the existence of a P2X7R/NLRP3 pathway in T cells that is dysregulated by a P2X7R intracellular region loss-of-function mutation, leading to NLRP3 displacement and to excessive Th17 generation due to abrogation of the NLRP3-mediated Th2 program. This ultimately resulted in poor outcomes in cardiac-transplanted patients carrying the mutant allele, who showed abnormal Th17 generation. Transient NLRP3 silencing in nonmutant T cells or overexpression in mutant T cells normalized the Th profile. Interestingly, IL-17 blockade reduced Th17 skewing of human T cells in vitro and abrogated the severe allograft vasculopathy and abnormal Th17 generation observed in preclinical models in which P2X7R was genetically deleted. This P2X7R intracellular region mutation thus impaired the modulatory effects of P2X7R on NLRP3 expression and function in T cells and led to NLRP3 dysregulation and Th17 skewing, delineating a high-risk group of cardiac-transplanted patients who may benefit from personalized therapy.
Keywords: Immunology, Transplantation
Keywords: Genetic variation, Organ transplantation, T cells

Introduction
Purinergic receptor-7 (P2X7R), primarily expressed on lymphocytes, senses adenosine 5′-triphosphate (ATP) (1) released extracellularly during cell damage (2, 3) and has been shown to regulate T cell activation (4–6). In particular, P2X7R signaling favors T helper 1 (Th1) and Th17 generation and differentiation (7–9), which are involved in alloimmune responses and allograft rejection (10–14). Our group has indeed demonstrated a major role for the extracellular ATP/P2X7R axis in regulating Th1/Th17 differentiation during cardiac allograft rejection (15–17). Interestingly, the opposite T cell immune-polarizing role has been recently described for NOD-like receptor P3 (NLRP3) (18), with NLRP3 acting as a DNA-binding transcriptional factor to promote an IL-4–mediated Th2 program in CD4+ T cells (19). The interaction between NLRP3 and P2X7R has been described in various cells, but not in T lymphocytes (20, 21). We hypothesize that a P2X7R/NLRP3 pathway within T cells controls Th cell fate, which may be dysregulated by mutation of P2X7R, thus driving excessive Th17 generation, particularly upon allostimulation due to abrogation of the NLRP3-mediated Th2 program. Interestingly, a single-nucleotide polymorphism (SNP), rs3751143 (p.Glu496Ala/c.1513A>C), located in exon 13 of P2X7R, targets the intracellular C-terminal portion of P2X7R. This mutation is relatively frequent — with 25% of the general population heterozygous and 2% homozygous — and results in loss of function of P2X7R (22–29). In this study, we have explored the existence of a P2X7R/NLRP3 pathway within T cells that controls Th cell fate and the effect of a P2X7R loss-of-function mutation on this pathway, on Th cell fate, and on clinical outcomes in a large cohort of cardiac-transplanted patients.
Results
A P2X7R/NLRP3 pathway exists in CD4+ T cells.
Given that an interaction between P2X7R and NLRP3 has been documented in other cell types (i.e., microglial cells, leukemic cells) (20, 21), we initially explored whether a similar association exists in T cells, particularly in human CD4+ T cells. We observed that NLRP3 and P2X7R reciprocally immunoprecipitated in CD4+ T cells isolated from peripheral blood mononuclear cells (PBMCs) obtained from human subjects (Figure 1A and Supplemental Figure 1A; supplemental material and complete unedited blots available online with this article; https://doi.org/10.1172/JCI94524DS1). We then confirmed their colocalization via analysis of immunofluorescence distribution at the single-cell level (Figure 1, B and C).
Figure 1. Existence of a P2X7R/NLRP3 pathway within human CD4+ T cells.

(A) P2X7R and NLRP3 immunoprecipitation (IP) in human CD4+ T cells. Expression of NLRP3 (top blot) and P2X7R (bottom blot) is shown. Lane 1: Total protein. Lane 2: IP with NLRP3 Ab. Lane 3: IP with P2X7R Ab. Lane 4: IP with Ab alone (NLRP3 and P2X7R). Lane 5: IP with control IgG (for NLRP3 Ab in top blot, for P2X7R Ab in bottom blot). The experiment was run in triplicate (representative blot shown). (B and C) Confocal microscopy analysis (B, scale bar: 5 μm, ×100 original magnification; C, scale bars: 20 μm, ×40 original magnification) depicting baseline colocalization of P2X7R (green) and NLRP3 (red) in human CD4+ T cells. Cells were stained with DAPI (blue) and immunolabeled with anti-P2X7R (green) and anti-NLRP3 Abs (red) (n = 3). (D–F) Bar graphs depicting expression of NLRP3 mRNA by qRT-PCR (D), and protein by flow cytometry (E) and ELISA (F), evaluated in human CD4+ T cells activated with benzoyl ATP (BzATP) and treated with CE-224,535, a P2X7R inhibitor. Experiments were run in duplicate (n = 5). (G) Bar graph representing expression of NLRP3 on human CD4+P2X7R+ cells analyzed by flow cytometry upon BzATP stimulation (n = 5). (H) Representative flow dot plots of NLRP3 expression upon gating on human BzATP-stimulated CD4+P2X7R+ cells. (I) Confocal analysis (scale bar: 5 μm; ×100 original magnification) depicting colocalization of P2X7R (green) and NLRP3 (red) in CD4+ T cells upon in vitro stimulation of P2X7R with BzATP (n = 3). (J–M) Bar graphs comparing expression of NLRP3 downstream signaling Th2-related factors IL-4 (J), IRF4 (K), GATA-3 (L), and IL-10 (M) by qRT-PCR using mRNA isolated from human CD4+ T cells activated with BzATP and treated with the P2X7R inhibitor CE-224,535. Experiments were run in triplicate (n = 5). Data are expressed as mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; 1-way ANOVA with Bonferroni’s post hoc test or Student’s t test. mRNA expression was normalized to β-actin (ACTB).
From a functional point of view, we demonstrated an increase in NLRP3 expression upon in vitro P2X7R stimulation with ATP (BzATP) in CD4+ T cells (Figure 1, D–F) and in CD4+P2X7R+ cells (Figure 1, G and H). Conversely, P2X7R blockade with a specific inhibitor (CE-224,535) prevented the upregulation of NLRP3 expression in CD4+ T cells stimulated with benzoyl ATP (BzATP) (Figure 1, D–F). Moreover, we confirmed that in CD4+ T cells, the activation of P2X7R signaling with BzATP is associated with an increase in P2X7R and NLRP3 colocalization (Figure 1I). Further analysis of NLRP3 downstream signaling revealed that the expression of NLRP3/Th2-related factors was upregulated when CD4+ T cells were challenged in vitro with BzATP and downregulated when P2X7R inhibitor (CE-224,535) was added (Figure 1, J–M), thus supporting the existence of a P2X7R/NLRP3 pathway that controls human CD4+ T cell fate. We finally demonstrated the absence of a reciprocal link between P2X7R and NLRP3 within human CD4+ T cells by showing that NLRP3 blockade using siRNA silencing or NLRP3 inhibitor (CP-456773) did not alter P2X7R expression (Supplemental Figure 1, B–D).
The P2X7R rs3751143 loss-of-function mutation affects intracellular P2X7R function.
The availability of a large cohort of cardiac-transplanted patient samples and data sets provided by 3 international independent studies (the CTOT-05, the NIT-Bergamo, and the AIRT-Bologna) and the recently established crucial role of P2X7R signaling in alloimmune response activation led us to explore the hypothesis that a mutation in P2X7R may alter T cell phenotype/function and ultimately affect cardiac transplantation outcomes, by disrupting the regulatory effect of P2X7R on NLRP3 expression and function.
Intriguingly, the already described P2X7R loss-of-function mutation rs3751143 is quite frequent in its homozygous mutated state (nearly 2% in the general population, and 7% in cardiac-transplanted patients), and it mainly targets the C-terminal intracellular domain (24, 25). We thus performed bioinformatic protein sequence analysis of P2X7R and determined that the intracellular region is predicted to be composed of significant secondary structure elements, and that mutation of Glu496 likely disrupts some of the molecular functions of P2X7R (Figure 2A). We then identified carriers of the mutation by genotyping the 413 cardiac-transplanted patients enrolled in the 3 aforementioned studies (Supplemental Table 1). First, we observed that P2X7R and NLRP3 reciprocally immunoprecipitate in CD4+ T cells of carriers, in both the heterozygous and the homozygous state (Supplemental Figure 1A). Next, we demonstrated that total protein expression of P2X7R was higher in carrier as compared with noncarrier CD4+ T cells (Figure 2B), but that surface expression was similar (Supplemental Figure 1, E and F). We further observed that P2X7R surface expression did not differ in noncarrier CD4+ T cells among different subsets — naive (CD4+CD45RO–CCR7+), effector (CD4+CD45RO–CCR7–), and memory (CD4+CD45RO+CCR7–) — while it was higher in naive CD4+ T cells of carriers as compared with effector and memory subsets and compared with naive CD4+ T cells of noncarriers (Supplemental Figure 1G).
Figure 2. A C-terminal P2X7R mutation dysregulates NLRP3 expression and function in human CD4+ T cells.

(A) A 3D representation of the full-length structure of P2X7R, highlighting the putative location of the P2X7R mutation in the C-terminal intracellular portion. (B and C) Quantification of P2X7R total protein (B, ELISA, n = 3) and of P2X7R mRNA (C, qRT-PCR, n = 10) on CD4+ T cells of carrier and noncarrier patients. Samples were run in duplicate (B) or in triplicate (C) and normalized to expression level of β-actin (ACTB). (D) Transcriptome profiling of immune-relevant genes (see also Supplemental Table 3) examined in CD4+ T cells of carrier and noncarrier cardiac-transplanted patients (n = 5). (E–G) Expression of NLRP3 mRNA using qRT-PCR (E) and NLRP3 protein using flow cytometry (F) and ELISA (G) in CD4+ T cells of carrier and noncarrier patients (n = 5). (H and I) Flow cytometric expression of NLRP3 on CD4+P2X7R+ cells of carrier patients stimulated with BzATP (n = 5). (J) Percentage of P2X7R+NLRP3+ cells of carrier and noncarrier patients analyzed by immunofluorescence (Figure 1C and Supplemental Figure 2G) (n = 3). (K) Confocal microscopy analysis (×100 original magnification) of P2X7R (green) and NLRP3 (red) coexpression in CD4+ T cells of carrier patients (n = 3). Scale bar: 5 μm. (L) Subcellular localization of NLRP3 in CD4+ T cells of carrier and of noncarrier patients (n = 3). (M and N) IL-4 (M) and IRF4 (N) gene expression detected after ChIP with NLRP3 antibody in CD4+ T cells. (n = 3). (O) Quantification of NLRP3 protein measured in CD4+ T cells treated with the ubiquitin/protease inhibitor MG132 (n = 3). Bars represent mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; Student’s t test or 2-way ANOVA with Bonferroni’s post hoc test.
To further explore any functional alterations in P2X7R, CD4+ T cells were isolated from collected blood samples (Supplemental Table 2), and a significant reduction in the BzATP-stimulated intracellular calcium peak (Supplemental Figure 1H) was observed in carriers of the mutation as compared with noncarriers, while influx of larger cations such as ethidium+ and YO-PRO-1++ was only slightly affected (Supplemental Figure 1, I and J). To further characterize the effects of the P2X7R mutation on other cell functions, we analyzed the intracellular bacterial killing capability of macrophages at different time points (24 and 48 hours) and demonstrated that this function was not altered in carriers as compared with noncarriers (Supplemental Figure 1K). A reduction in mRNA expression of P2X7R was also observed in CD4+ T cells of carriers as compared with noncarriers (Figure 2C), but this was paralleled by a relative increase in mRNA expression of other P2X receptors, such as P2X1R and P2X4R (Supplemental Figure 2, A and B). In line with this observation, delayed ubiquitination/degradation of the P2X7R protein was evident in CD4+ carrier T cells as compared with noncarriers and resulted in protein accumulation within the cells (Supplemental Figure 2C). This likely leads to reduced P2X7R mRNA levels in CD4+ T cells of carriers. Other potential mechanisms such as methylation of the P2X7R promoter or altered P2X7R mRNA half-life were shown not to be associated with a decrease in P2X7R mRNA levels (Supplemental Figure 2, D and E).
Dysregulation of the P2X7R/NLRP3 pathway in CD4+ T cells of patients with the P2X7R mutation.
In order to better understand the effect of the P2X7R mutation, we first conducted transcriptome profiling of immune-relevant genes in CD4+ T cells (Supplemental Table 3). Interestingly, this profiling revealed a reduction in NLRP3 and P2X7R mRNA expression in CD4+ T cells obtained from patients carrying the mutant P2X7R allele as compared with noncarrier patients (Figure 2D), suggesting that the interaction between P2X7R and NLRP3 is altered in the presence of the P2X7R mutation. Quantitative reverse transcriptase PCR (qRT-PCR), flow cytometric, and protein expression analysis confirmed the downregulation of NLRP3 mRNA (Figure 2E) and protein (Figure 2, F and G) in CD4+ T cells, particularly in naive but not in effector or memory T cell subsets (Supplemental Figure 2F), and in CD4+P2X7R+ cells, obtained from patients carrying the mutant allele (Figure 2, H and I). The near absence of NLRP3 and its reduced colocalization with P2X7R in CD4+ carrier T cells were also confirmed with immunofluorescence studies (Figure 2, J and K, and Supplemental Figure 2G). Confocal imaging and subcellular fractionation studies demonstrated that while NLRP3 is present in the cytoplasm in CD4+ T cells of noncarrier patients, it appears confined to the cell membrane of CD4+ T cells in patients carrying the mutant allele (Figure 2, K and L), suggesting that the P2X7R mutation may prevent NLRP3 translocation to the nucleus. Indeed, chromatin immunoprecipitation (ChIP) experiments revealed that DNA encoding IL-4 and IRF4, the transcription factor that in conjunction with NLRP3 transactivates the IL-4 promoter, was clearly bound to NLRP3 precipitated from CD4+ T cells of noncarrier patients, while it was slightly detectably bound to NLRP3 precipitated from CD4+ T cells of carrier patients (Figure 2, M and N), confirming NLRP3 displacement and absence of its binding to DNA in the promoter regions of Th2-related genes in mutant CD4+ T cells. However, NLRP3 translocation could not entirely explain the reduced expression of NLRP3 observed in CD4+ T cells of carrier patients. We thus studied the ubiquitin-mediated degradation of NLRP3 and demonstrated that it was accelerated in CD4+ carrier T cells as compared with noncarrier cells, and that the addition of the proteasome and ubiquitination inhibitor MG132 to cultured CD4+ T cells appears to restore NLRP3 expression (Figure 2O). NLRP3 is thus not only displaced but also is prone to degradation in mutant CD4+ T cells. This may also apply to NLRP3 mRNA, which is decreased in CD4+ T cells of carriers, as a result of reduced mRNA half-life and accelerated mRNA degradation (Supplemental Figure 2H). We further observed reduced levels of the phosphorylated intracellular factor STAT5, a transcriptional regulator of NLRP3, in CD4+ T cells of carriers as compared with noncarriers (Supplemental Figure 2I), which most likely plays a role in reducing NLRP3 mRNA transcription. Finally, we confirmed that epigenetic regulation of NLRP3 DNA via NLRP3 promoter methylation was not altered in carriers as compared with noncarriers (Supplemental Figure 2J), and that P2X7R did not directly bind to DNA of NLRP3 or NLRP3-related genes (i.e., STAT5, NF-κB, MyD88, IL-10) in carrier/noncarrier CD4+ T cells (data not shown) to modulate NLRP3 mRNA transcription.
The dysregulated P2X7R/NLRP3 pathway is associated with altered Th cell fate.
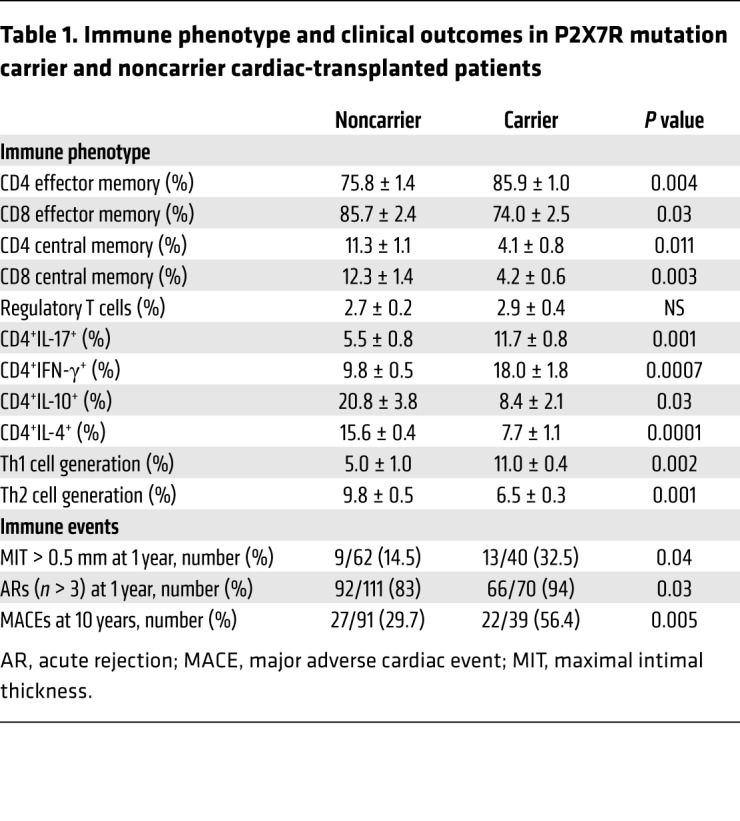
We next confirmed a role for P2X7R/NLRP3 dysregulation in driving Th cell fate during immune activation. In an ex vivo T cell polarization assay in which CD4+ T cells were differentiated by anti-CD3-Ig/anti-CD28-Ig stimulation in the appropriate cytokine milieu for Th1, Th2, or Th17 cells, a skewing to the Th17 — and to a lesser extent Th1 — lineage was evident in patients carrying the mutant allele as compared with noncarriers (Figure 3A and Table 1), and the generation of Th2 cells was significantly reduced in carrier patients as compared with noncarriers (Table 1). This skewing was paralleled by an increase in the percentage of Th1/Th17 cells in the circulation (Figure 3, B and C, and Table 1) as well as a decrease in Th2 cells in carrier patients (Table 1). We further proved the shifted balance toward Th17 by detecting increased peripheral IL-17 levels in the plasma and in the supernatants of unstimulated CD4+ T cells of carrier as compared with noncarrier patients matched for clinical characteristics (Figure 3, D and E). In order to better characterize the Th17 shift in carrier cells, we demonstrated higher expression of the Th17-related markers CCR6, CCR10, and IL-22 — the latter to a lesser degree — in differentiated Th17 cells of carriers as compared with noncarriers by flow cytometric analysis (Figure 3F and Supplemental Figure 2K), which was confirmed by increased production of the Th17 cytokines IL-17, IL-6, and IL-22 in the culture supernatants of these cells (Figure 3F). Finally, Th17-polarized cells of carriers also showed higher production of the proinflammatory cytokines IFN-γ and TNF-α, and reduced release of the antiinflammatory cytokine IL-1ra (Figure 3F and Supplemental Figure 2L), while no difference was found with regard to IL-10 (Figure 3F). Moreover, given the association of Th17 skewing in immune events occurring after transplantation, including cardiac allograft vasculopathy, with the development of an autoimmune response against vimentin (30, 31), we demonstrated that, while anti-vimentin autoimmunity was detectable, no differences were observed in the anti-vimentin immune response in vitro or in peripheral anti-vimentin autoantibodies in carriers as compared with noncarriers (Supplemental Figure 2, M and N). Fewer peripheral central memory CD4+ and CD8+ T cells (CD45RO+CCR7+) paired with a relative increase in effector memory CD4+ and CD8+ T cells (CD45RO+CCR7–) were evident as well in carrier as compared with noncarrier patients (Table 1), thus suggesting an increased effector-type immune response. The P2X7R intracellular domain mutation may thus directly alter T cell phenotype. Transient genetic overexpression and downregulation of NLRP3 were then used to ultimately confirm the reciprocal link between the dysregulated P2X7R/NLRP3 pathway and altered Th cell fate. Silencing of NLRP3 in CD4+ T cells of noncarrier patients reduced mRNA expression of Th2-related factors (Figure 3, G and H, and Supplemental Figure 3A) and was associated with a concomitant increase in the Th17-related factor RORc upon stimulation (Supplemental Figure 3B). Unlike the silencing of NLRP3 (Figure 3, I–K), inhibition of the inflammasome complex by the inhibitor CP-456773 upon ATP triggering in CD4+ T cells of noncarrier patients did not alter either the Th2 profile by flow cytometric analysis (Supplemental Figure 3, C and D) or mRNA expression of Th2-related factors (Supplemental Figure 3, E–G). Expression of the inflammasome-related factors CASP1 and ASC were also unchanged upon NLRP3 silencing but were reduced in inflammasome inhibitor–treated cells (Supplemental Figure 3, H–K). Overall, this suggests that ATP-triggered NLRP3 signaling regulating the Th2 program is inflammasome-independent. In addition, NLRP3 overexpression in CD4+ T cells of carrier patients exerted an opposite effect and restored the Th2 profile (Figure 3, L and M, and Supplemental Figure 3, L and M).
Figure 3. The dysregulated P2X7R/NLRP3 pathway is associated with an abnormal human Th cell immunophenotype.

(A) Percentage of in vitro–generated Th17 cells obtained from CD4+ T cells of carrier and noncarrier patients (n = 8). (B and C) Representative flow zebra plots (B) and quantitative histogram (C) depicting the percentage of peripheral CD4+IL-17+ cells (n = 8). (D) IL-17 plasma levels of carrier and noncarrier patients (n = 10). (E) IL-17 levels (Luminex) measured in the supernatants of unstimulated 24-hour-cultured CD4+ T cells of carrier and noncarrier patients (n = 5). (F) Table summarizing the secretome profile (Luminex, n = 5) and primary phenotypic characteristics (flow cytometry, n = 4) of carrier and noncarrier polarized Th17 cells. (G and H) Normalized mRNA expression of Th2-related factors IL-4 (G) and GATA-3 (H) measured in noncarrier CD4+ T cells exposed to transient knockdown of NLRP3 using silencing RNA (siRNA), before and after anti-CD3-Ig/anti-CD28-Ig stimulation (n = 3). (I–K) Normalized mRNA expression of the Th2-related factors IL-4 (I), IL-10 (J), and GATA-3 (K) measured in noncarrier CD4+ T cells exposed to transient knockdown of NLRP3 (siRNA), upon BzATP exposure (n = 4). (L and M) Normalized mRNA expression of the Th2-related factors IL-4 (L) and GATA-3 (M) measured in carrier CD4+ T cells, in which NLRP3 was overexpressed, before and after anti-CD3-Ig/anti-CD28-Ig stimulation (n = 3). (N) Effects of various treatments (anti–IL-17 antibody, RMT1-10, cyclosporin A [CsA] and rapamycin [Rapa]) on in vitro–generated Th17 cells (n = 5). Experiments were run in triplicate (D, G, H, and N) or in duplicate (F and I–M). mRNA expression was normalized to ACTB. Bars represent mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; Student’s t test or 1-way ANOVA with Bonferroni’s post hoc test.
Table 1. Immune phenotype and clinical outcomes in P2X7R mutation carrier and noncarrier cardiac-transplanted patients.
To finally further establish whether our findings regarding P2X7R/NLRP3 dysregulation in T cells are limited to transplant patients or are also relevant to the general population, we performed the aforementioned experiments in nontransplanted patients (Supplemental Table 4). Indeed, reduced expression of NLRP3 in CD4+ T cells (Supplemental Figure 4A), P2X7R/NLRP3 displacement (Supplemental Figure 4B), reduction in Th2 cells, and Th17 skewing in the periphery (Supplemental Figure 4, C and D) and during Th2/Th17 generation in vitro (Supplemental Figure 4, E and F) were all observed in nontransplanted carriers compared with noncarriers as well.
Anti–IL-17 neutralizing antibody abrogates the deleterious effects of P2X7R loss of function.
With the goal of developing a personalized therapy for carriers of the mutation, we tested whether treatment with an anti–IL-17 neutralizing antibody would be effective in abrogating the altered CD4+ T cell phenotype in carriers of the P2X7R mutation. Reduced Th17 skewing was observed when anti–IL-17 neutralizing antibody, but not immunosuppressive drugs currently used in transplantation (i.e., rapamycin and cyclosporine) or a Th1 inhibitor, was added to an in vitro Th17 generation assay in which CD4+ T cells obtained from carriers were used (Figure 3N). We then tested the effect of an anti–IL-17 neutralizing antibody in a preclinical model of cardiac transplantation. Surprisingly, P2X7R–/– C57BL/6 recipients of bm12 hearts (an established MHC class II–mismatched model of chronic allograft vasculopathy) experienced increased allograft loss as compared with wild-type C57BL/6 recipients (Figure 4A), with higher levels of cell infiltrate, vasculopathy, and myocyte necrosis (Figure 4, B–E). Interestingly, treatment with anti–IL-17 neutralizing antibody significantly reduced graft loss in P2X7R–/– recipients (Figure 4A) and reduced Th17 skewing and immune activation (Figure 4, F–N), as assessed by functional studies (Figure 4, F and G), flow cytometric immunophenotyping (Figure 4, H–M), and cytokine profiling (Figure 4, H–J and N). We further confirmed the relevance of the P2X7R/NLRP3 pathway in the P2X7R–/– mouse model by demonstrating that NLRP3 expression is lower within CD4+ T cells than in C57BL/6 mice (Figure 4O), and we observed a decreased Th2 response, particularly with respect to IL-4, upon in vitro and in vivo allostimulation (Figure 4, G, J, P, and Q). We acknowledge that the murine model is different from cardiac-transplanted patients, who are heavily immunosuppressed and receive a fully MHC-mismatched cardiac allograft. However, these murine data largely parallel our observations in carrier patients (Supplemental Table 5), thus suggesting that an anti–IL-17–based treatment may help to prevent or delay the onset of immune events in cardiac-transplanted patients carrying the P2X7R mutation.
Figure 4. P2X7R loss of function in a preclinical model of heart transplantation reduces allograft survival, with increased vasculopathy and Th1/Th17–mediated responses.

(A) P2X7R–/– mice receiving bm12 heart transplantation demonstrated reduced graft survival as compared with B6 recipients (**P < 0.01), which was significantly prolonged by anti–IL-17 treatment (murine IL-17–depleting antibody) (*P < 0.05 vs. P2X7R–/–) (n = 10 mice per group). (B–D) Semiquantification of graft infiltration (B), coronary vasculopathy (C), and myocyte necrosis (D) confirmed accelerated allograft rejection in P2X7R–/– mice (n = 3). (E) Representative H&E staining (x20 original magnification) showing graft cell infiltration (top panels), vasculopathy (middle panels), and myocyte necrosis (bottom panels) in B6 and P2X7R–/– mice. Scale bars: 200 μm (middle panels), 300 μm (top and bottom panels). (F and G) Numbers of IFN-γ–producing (F) and IL-4–producing (G) cells (ELISPOT) measured in cardiac-transplanted mice (n = 3). (H–M) Percentage of CD4+IL-17+ (H), CD4+IFN-γ+ (I), CD4+IL-10+ (J), CD4+CD44hiCD62Llo (K), CD8+CD44hiCD62Llo (L), and CD4+CD25+Foxp3+ (M) cells detected by flow cytometry in B6 and P2X7R–/– cardiac-transplanted mice and in P2X7R–/– anti–IL-17–treated mice (n = 5). (N) Serum IL-17 level (Luminex) measured in B6 and P2X7R–/– cardiac-transplanted mice and in P2X7R–/– anti–IL-17–treated mice (n = 5). (O) Percentage of CD4+NLRP3+ cells analyzed by flow cytometry in P2X7R–/– and B6 mice (n = 3). (P) Number of IL-4–producing cells (ELISPOT) in P2X7R–/– and B6 mice upon allostimulation (n = 3). (Q) Serum IL-4 level (Luminex), measured in B6 and P2X7R–/– cardiac-transplanted mice (n = 5). Samples were run in duplicate (Luminex) and in triplicate (ELISPOT). Bars represent mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.001; log-rank (Mantel-Cox) test (A), Wilcoxon’s and Student’s t test (2 groups), 1-way ANOVA with Bonferroni’s post hoc test (3 groups).
The dysregulated P2X7R/NLRP3 pathway is associated with poor outcomes in cardiac-transplanted patients.
To finally assess the clinical impact of the dysregulated P2X7R/NLRP3 pathway in cardiac-transplanted patients bearing the P2X7R mutant allele, we studied clinical and immunological outcomes of 413 cardiac-transplanted patients enrolled in the CTOT-05, NIT-Bergamo, and AIRT-Bologna international studies. We first confirmed that the frequency of the P2X7R mutation in the heterozygous condition was similar in the 3 cohorts and in the National Heart, Lung, and Blood Institute (NHLBI) Grand Opportunity (GO) Exome Sequencing Project (ESP) cohort, while the percentage of patients in the homozygous mutant condition was lower in the ESP cohort as compared with what was observed in cardiac-transplanted patients (nearly 7.5%) (Supplemental Table 6).
We next analyzed an early outcome in the CTOT-05 cohort by examining what proportion showed an incremental change greater than 5 mm in intracoronary ultrasonography–measured coronary artery maximal intimal thickness (MIT), which defines rapidly progressive cardiac allograft vasculopathy following heart transplantation. A significant increase in MIT was observed in 13 of 40 carriers (32.5%), and in 9 of 62 noncarrier patients (14.5%; P = 0.04; Figure 5A). Carrying the mutant allele was thus associated with an increased odds ratio (OR) for developing an incremental change in coronary artery MIT within the first year after transplant (OR = 2.83, CI 1.1–7.4, P = 0.04; Table 1), and this association was confirmed after controlling for the most relevant modifying factors (age, sex, pretransplant cardiomyopathy, and immunosuppressive therapy) as shown in Supplemental Table 7. We then studied in the NIT-Bergamo cohort the number of clinically assessed and/or biopsy-proven acute rejection episodes, observing an increased frequency of events in carrier as compared with noncarrier patients (10.4 ± 0.5 vs. 8.7 ± 0.4, P = 0.01; Figure 5B) within the first year after transplantation, with a higher proportion of patients experiencing more than 3 acute rejection events per year (66/70 [94%] in carriers vs. 92/111 [83%] in noncarrier patients, P = 0.03; Table 1). Carrying the mutant allele was thus associated with an increased OR for developing a higher number of acute rejection episodes within the first year after transplant (OR = 3.40, CI 1.1–10.5, P = 0.04; Table 1), which was confirmed after controlling for other factors potentially involved (age, sex, pretransplant cardiomyopathy, and immunosuppressive therapy; Supplemental Table 8). Finally, we analyzed the long-term impact of the mutation and observed in the AIRT-Bologna cohort an increased frequency of major adverse cardiac events (MACEs) within 10 years after transplantation in 22 of 39 carriers (56.4%) compared with 27 of 91 noncarrier patients (29.7%; P = 0.005; Figure 5C). Carrying the mutant allele was thus associated with an increased OR for developing MACEs after transplantation also in the long term (OR = 3.07, CI 1.4–6.6, P = 0.006; Table 1), which was confirmed after adjustment for clinically relevant factors (age, sex, pretransplant cardiomyopathy, and immunosuppressive therapy), as reported in Supplemental Table 9. Despite the heterogeneity in practice among study centers (e.g., suboptimal standardization of clinically relevant outcomes, decision algorithm for immunosuppressive and/or other treatments), our findings collectively demonstrate that the dysregulated P2X7R/NLRP3 pathway in cardiac-transplanted patients bearing the P2X7R mutant allele is associated with an increased risk of developing poor outcomes such as rapidly progressive cardiac allograft vasculopathy and higher frequency of acute rejection episodes and long-term MACEs following transplantation (Figure 5D).
Figure 5. A C-terminal P2X7R mutation is associated with poor outcomes in cardiac-transplanted patients.

(A) Bar graph depicting the percentage of cardiac-transplanted patients who carry the WT or mutant P2X7R allele (n = 102) with an MIT change greater than 0.5 mm (early cardiac allograft vasculopathy, in black) at 1 year after transplantation in the CTOT-05 cohort. (B) Bar graph depicting the number of acute rejection episodes in cardiac-transplanted patients who carry the WT (black) or mutant (white) P2X7R allele (n = 181) within the first year after transplant in the NIT-Bergamo cohort. (C) Bar graph depicting the percentage of cardiac-transplanted patients who carry the WT or mutant P2X7R allele (n = 130) with major adverse cardiac events (MACEs, in black) at 10 years of follow-up in the AIRT-Bologna cohort. In A and C: black, percentage of patients who experienced the event; white, percentage who were free from events. (D) Line graph depicting the estimated odds ratio (OR) for clinical outcomes recorded in the 3 cohorts of cardiac-transplanted patients who carry the WT or mutant P2X7R allele. In the NIT-Bergamo cohort, the OR was calculated based on the requirement of medical intervention for acute rejection episodes with a frequency of greater or less than 3 episodes. *P < 0.05; **P < 0.01. Supplemental Tables 7–9 report detailed analyses. Fisher’s exact and Student’s t tests. (E and F) A stable connection between P2X7R and NLRP3 is necessary to establish a physiological NLRP3-mediated Th2 program (E), while alteration in the P2X7R intracellular domain induces NLRP3 displacement and retains NLRP3 in the cell membrane, thus preventing its nuclear activity and accelerating ubiquitination of NLRP3 (F). This shifts the balance of the immune response toward Th17 cells and favors the development of immune-related events, such as allograft rejection and vasculopathy. Ub, ubiquitin; eATP, extracellular ATP.
Discussion
This study offers a novel understanding of the mechanisms that may control Th cell fate by suggesting that P2X7R exerts a regulatory effect on NLRP3 within CD4+ T cells, which is required to maintain NLRP3 in the proper intracytoplasmic/nuclear position and in an operative mode, thus facilitating its nuclear action (i.e., interaction with the IL-4 promoter and IRF4) and Th2 commitment. Moreover, in our study a group of patients at high risk for immune events related to dysregulation of the P2X7R/NLRP3 pathway and who may require personalized therapy has been identified. We demonstrated that mutation of the P2X7R intracellular region affects Th cell fate, favoring skewing toward Th17 generation, leading to a more pronounced effector/inflammatory phenotype and thus exposing patients to the risk of allograft loss. Given the importance of the Th17 response in allograft rejection (32), understanding the mechanism whereby the mutation of P2X7R favors Th17 skewing is highly clinically relevant. Interestingly, we observed that this mutation in the P2X7R intracellular region directly favors NLRP3 displacement and redirects NLRP3 to the cell membrane compartment. This prevents the nuclear transcriptional and DNA binding activity of NLRP3, as well as results in its accelerated ubiquitination and rapid proteolysis, thus abrogating NLRP3-mediated Th2 differentiation (19) and inducing Th17 skewing. While ubiquitination is a key mechanism in regulating NLRP3 activation within cells that helps to maintain its transcriptional activity (33, 34), we discovered that proper function of the P2X7R/NLRP3 pathway appears to be crucial in rescuing NLRP3 from ubiquitin-mediated degradation and in preserving its function within T cells. A genetic add-back experiment confirmed the link between the dysregulated P2X7R/NLRP3 pathway and altered Th skewing. Transient transgenic overexpression of NLRP3 rescues the Th2 program in CD4+ T cells carrying the mutant allele, while siRNA downregulation of NLRP3 abrogates it, thus promoting Th17 skewing in CD4+ T cells obtained from noncarriers. Interestingly, IL-17 blockade abrogates the abnormal Th17 skewing in human T cells in vitro and prolongs cardiac allograft survival in preclinical models. This immunophenotype has a clear impact on clinical outcomes as demonstrated by our 3 independent international studies (the CTOT-05, the NIT-Bergamo, and the AIRT-Bologna cohorts), which demonstrated an increased frequency of clinical events (i.e., rapidly progressive cardiac allograft vasculopathy, number of acute rejection episodes, and number of MACEs, respectively), even when adjusted for other variables involved.
Our results were also confirmed in a group of nontransplanted patients, among whom reduced expression of NLRP3 in CD4+ T cells, displacement of P2X7R/NLRP3, reduction in Th2 cells, and Th17 skewing (both in the periphery and during Th2/Th17 generation in vitro) were observed in carriers as compared with noncarriers. Further studies are certainly needed to confirm this observation in a larger population, but these data suggest that carriers of the mutant P2X7R allele may represent a population of patients with a fragile immune system at risk of developing a hyper-Th17 syndrome in response to various immune stimulations (e.g., infections, inflammation, or tissue damage).
Some limitations of our study should be noted. Firstly, the bm12 into C57BL/6 heart transplantation murine model does not faithfully replicate the condition of cardiac transplantation in humans, given minor HLA mismatching, a reduced degree of transplant atherosclerosis, and the lack of a concomitant immunosuppression therapy in this model. Secondly, with respect to human studies, a larger cohort would have certainly provided greater statistical power to the association between P2X7R mutation and immune disorders. Thirdly and finally, the complexity of the clinical assessment in our study, which included multiple outcomes evaluated at different time points, as well as the heterogeneity in clinical practice among study centers, may reduce the strength of this analysis. Future studies would benefit from improvement upon these limitations, with the rate of immune events in patients with the P2X7R mutation as a primary endpoint.
In summary, the P2X7R/NLRP3 complex maintains a physiological NLRP3-mediated Th2 program, while intracellular mutation of P2X7R induces NLRP3 displacement in T cells, causing Th17 skewing and resulting in poor allograft outcome (Figure 5, E and F). Our study demonstrates, for the first time to our knowledge, the existence of a novel mechanism controlling T cell fate and delineates a group of cardiac-transplanted patients with Th17 skewing and with worse clinical and immunological outcomes, as well as identifies a population who may benefit from selective targeted therapy.
Methods
A detailed description of the methods used in this study is provided in the Supplemental Methods.
Genotyping of P2X7R rs3751143
DNA was extracted from anticoagulated peripheral blood samples obtained from each patient using the Qiagen QIAamp DNA Blood Mini Kit (51104, Qiagen). Genotypes were determined using an Applied Biosystems TaqMan Human SNP Genotyping Assay (4351376, Thermo Fisher Scientific) for rs3751143 in accordance with the manufacturer’s protocol.
Study cohorts and clinical endpoints
In order to study the effects of rs3751143, we have established a large international cohort of 747 cardiac-transplanted patients, which includes the US-based Clinical Trials in Organ Transplantation-05 (CTOT-05) cohort, the Northern Italian Transplant (NIT, Bergamo, Italy) group, and the Inter-Regional Transplant Association (AIRT, Bologna, Italy). Genotype and immunological and clinical outcomes were evaluated in all patients (Supplemental Table 1).
The CTOT-05, an observational study designed to understand the natural history of cardiac allograft vasculopathy, comprises 200 cardiac-transplanted patients enrolled in the United States from June 2007 to July 2011. Coronary artery vasculopathy, as defined by a change in MIT of ≥0.5 mm through intracoronary ultrasonography (IVUS) (35) between study entry and 12 months, was assessed. Of the 200 patients enrolled in the CTOT-05, 148 were genotyped, and IVUS was performed in 102 patients.
The NIT cohort consists of 187 patients who underwent cardiac transplantation at Divisione Cardiovascolare, Ospedali Riuniti in Bergamo, Italy, from 1985 to 2012. Acute rejection episodes, either clinically assessed and/or proven by biopsy, were recorded over a period of 12 months after transplant. Patients were grouped according to number of episodes (either higher or lower than 3) that required medical intervention and/or hospitalization within the first year. Of the 187 patients enrolled, 187 were genotyped, and acute rejection episodes were observed in 181 patients.
The AIRT cohort consists of 360 cardiac-transplanted patients followed within the Heart Failure and Heart Transplant Program at the Department of Experimental Diagnostic and Specialty Medicine, S. Orsola Hospital in Bologna, Italy, between 1985 and 2013. Clinical outcome was defined by the occurrence of major adverse cardiac events (MACEs), which included nonfatal myocardial infarction, coronary revascularization, and hospitalization for graft failure, over a long-term follow-up (10 years) from the time of transplantation (36). Of the 360 patients enrolled, 359 were genotyped, and data on MACEs for follow-up of at least 10 years from the time of transplantation were available in 130 patients.
Patients carrying the mutant allele for P2X7R in either the homozygous or the heterozygous state were included in the analysis of clinical outcome. Blood was collected according to the following timeline: 1.1 ± 0.4 years of follow-up in the CTOT-05 cohort; 5 ± 1.2 years of follow-up in the NIT-Bergamo cohort; and 15 ± 0.8 years of follow-up in the AIRT-Bologna cohort. Fifty healthy volunteers matched for age and sex were chosen as controls (Supplemental Table 4). The study was approved by the ethics committee of each participating institution.
Immunological studies
PBMC isolation and CD4+ T cell purification.
PBMCs were isolated from 10-ml blood samples collected from cardiac-transplanted patients (Supplemental Table 2) and from nontransplanted patients using Lymphoprep (07801, Stem Cell Technologies) according to the manufacturer’s instructions. Cells were then frozen in vials containing 5 × 106 cells/ml and stored at –80°C. After thawing, PBMCs were recovered in culturing medium consisting of RPMI 1640 (31870017, Life Technologies) supplemented with 10% FBS (12103C-500ML, Sigma-Aldrich), 2 mM l-glutamine (25030081, Life Technologies), 100 U/ml penicillin (15140122, Life Technologies), for 48 hours, and CD4+ T cells were then isolated using a CD4 Positive Isolation Kit (130-090-877, Miltenyi Biotec) according to the manufacturer’s instructions. The purity of the isolated CD4+ T cell population routinely exceeded 95%.
Flow cytometry and intracellular cytokine staining of human cells.
Primary antibodies included anti–human CD45 (560178), CD4 (561030), CD8 (560774), CD25 (560503), CCR7 (557648), CD127 (557938), IFN-γ (554702), IL-4 (554484), IL-10 (554498) (all BD Biosciences); IL-17 (12-7178-41), CD45RO (MHD45R005) (both Thermo Fisher Scientific); and CD3 (300330, BioLegend). FITC-conjugated anti–mouse P2X7R (APR-008-F) was purchased from Alomone Labs. Alexa Fluor 700–conjugated anti–human NLRP3 (IC7578N) was purchased from R&D Systems. Purified anti–human NLRP3 (IC7578G) and Alexa Fluor 488–conjugated anti–rabbit NLRP3 (IC7578G) were purchased from R&D Systems and BD Biosciences, respectively. Resting cells were activated with PMA (5 ng/ml; 13-144, Sigma-Aldrich) and ionomycin (500 ng/ml; 13909, Sigma-Aldrich) for 3 hours; then brefeldin A (2 μM; B7651, Sigma-Aldrich) was added. Cells were harvested in FACS tubes and placed on ice. Cells were then washed with FACSFlow (342003, BD Biosciences) and processed for intracellular cytokine staining with saponin 0.5% (47036, Sigma-Aldrich) and formaldehyde 2% (F8775) for fixation and permeabilization. Anti–phospho-STAT5–PE (pY694, clone 47, 562077, BD Biosciences) was used to assess intracellular phospho-STAT5 expression per the manufacturer’s instructions.
Human Th cell differentiation.
CD4+ T cells isolated from recovered PBMCs were stimulated with anti-CD3-Ig (1 μg/ml; HMCD0300, Life Technologies), anti-CD28-Ig (2 μg/ml; CD2800, Life Technologies), and rIL-2 (50 U/ml; 202-IL, R&D Systems). For Th1 differentiation, rIL-12 (2.5 ng/ml; 219-IL, R&D Systems) and anti–IL-4 (5 μg/ml; 204-IL, R&D Systems) were added. For Th2 differentiation, rIL-4 (12.5 ng/ml; 504-RL, R&D Systems), anti–IFN-γ (5 μg/ml; AHC4032, Invitrogen), and anti–IL-10 (5 μg/ml; 501505, BioLegend) were added. For Th17 differentiation, rIL-1β (1290-IL, R&D Systems), rIL-6 (206-IL, R&D Systems), and rTGF-β (240-B, R&D Systems) (final concentration 10 ng/ml) were added. After 4 days, cells were harvested and stained for FACS analysis. Th1 cells were defined as producing IFN-γ, Th2 cells were defined as producing either IL-4 or IL-10, and Th17 cells were defined as producing IL-17. For detailed Th17 characterization, the expression of CCR6 (563922, BD Biosciences), CCR10 (56769, BD Biosciences), and IL-22 (12-7229-41, Thermo Fisher Scientific) was also assessed.
Human peripheral IL-17 levels.
Levels of IL-17 were measured using an IL-17 Human Quantikine ELISA kit (D1700, R&D Systems) per the manufacturer’s instructions. Plasma samples from cardiac-transplanted patients (Supplemental Table 2) and from nontransplanted patients were diluted per kit protocol and analyzed by an ELISA plate reader according to the manufacturer’s instructions.
Luminex cytokine measurement.
Levels of cytokines were assessed in culture supernatants of CD4+ T cells using the Bio-Plex Pro human cytokine 17-plex panel (M5000031YV, Bio-Rad) according to the manufacturer’s protocol. The Hu Th17 IL-22 set (171BA008M, Bio-Rad) was used to detect IL-22.
Intracellular calcium response of human CD4+ T cells.
Isolated PBMCs were thawed, and recovered CD4+ T cells were purified as described above. CD4+ T cells were then cultured in RPMI 1640 (Life Technologies) supplemented with 10% FBS, 2 mM l-glutamine, and 100 U/ml penicillin (Life Technologies). After 48 hours, cells were resuspended at 1 × 107 cells/ml and incubated in 2.5 μM fluo-3AM (73881, Sigma-Aldrich) for 45 or 60 minutes at 25°C and then diluted and used at 2 × 106 or 4 × 106 cells per time point. Calcium peak response and area under the curve were measured upon stimulation with 500 μM benzoyl ATP (BzATP; B6396, Sigma-Aldrich).
Large cation dye uptake of human CD4+ T cells.
Isolated CD4+ T cells obtained from PBMCs of carrier and noncarrier patients were resuspended in NaCl medium (145 mM NaCl, 5 mM KCl, 5 mM glucose, 0.1% BSA, 10 mM HEPES, pH 7.5), equilibrated at 37°C for 5 minutes, and incubated with 25 μM ethidium+ or 1 μM YO-PRO-1++ in the absence or presence of BzATP (2 mM) for 5 minutes. Incubation with nucleotides was halted by the addition of an equal volume of ice-cold NaCl medium (20 mM MgCl2) followed by centrifugation (300 g for 5 minutes). Cells were washed once with NaCl medium, and events were collected using a BD FACS Celesta (BD Biosciences) (excitation 488 nm, emission collected with 575/26 PE and 515/20 FITC band-pass filters for ethidium+ and YO-PRO-1++, respectively) (37). Membrane integrity was assessed using the BD Horizon Fixable Viability Stain 510 kit (564406, BD Biosciences) to exclude dead cells. The mean fluorescence intensity of relative cation uptake was determined using FlowJo software (version 8, Tree Star).
Interventional in vitro studies
Anti–IL-17 (10 μg/ml; MAB3171-100, R&D Systems), CE-224,535 (50 μM; provided by Pfizer), RMT1-10 (100 μg/ml; BE0133, BioXCell), rapamycin (6 ng/ml; R8781, Sigma-Aldrich), and cyclosporine (100 ng/ml; PHR1092, Sigma-Aldrich) were used in vitro in a Th17 assay using concentrations already tested and described or according to blood levels used in drug monitoring (17, 38–42). Cells were stimulated with BzATP (50 μM) for 24 hours to activate P2X7R in vitro as previously described (20). The effect of inflammasome activation was tested by addition of the inflammasome inhibitor CP-456773 (200 μM; PZ0280, Sigma-Aldrich) to short-term BzATP (5 mM) stimulation for 1 hour in vitro as already described (43).
Transcriptome profiling
Total RNA was extracted from CD4+ T cells isolated from PBMCs obtained from each group of patients, using the RNeasy Mini Kit (74104, Qiagen) with on-column DNase I digestion. Next, 3 μg total RNA from each sample was reverse-transcribed using the RT2 First Strand kit (330401, SABiosciences). The Human Inflammasome RT2 Profiler PCR Array (PAHS-097Z, Qiagen) platform was used to quantitatively measure the expression of a panel of genes using SYBR Green–based real-time PCR (Supplemental Table 3).
qRT-PCR analysis
RNA from isolated CD4+ T cells was extracted using TRIzol Reagent (15596026, Ambion), and quantitative reverse transcriptase PCR (qRT-PCR) analysis was performed using TaqMan assays (Life Technologies) according to the manufacturer’s instructions. qRT-PCR data were determined using the relative quantification method, and ACTB served as a reference housekeeping gene. Statistical analysis compared gene expression across all cell populations for each patient via 1-way ANOVA followed by Bonferroni’s post hoc test for multiple comparisons between the population of interest and all other populations. Statistical analysis was performed also using the software RT2 profiler PCR Array Data Analysis (Qiagen). For 2-group comparisons, Student’s t test was used. Analysis was performed in triplicate after isolation of cells. The main characteristics of primers used are reported in Supplemental Table 10.
Immunoprecipitation
Isolated CD4+ T cells obtained from carrier and noncarrier patients were incubated on ice for 45 minutes in lysis buffer containing 150 mM NaCl, 5 mM EDTA, 20 mM Tris, pH 7.5, 1% Triton X-100, 1 mM benzamidine, 1 mM phenylmethylsulfonyl fluoride, and protease inhibitor cocktail (P8340, Sigma-Aldrich). Lysates were centrifuged at 20,000 g for 5 minutes before precipitation. Total protein content of cell lysates was measured with the Bradford assay. Dynabeads Protein A (10001D, Life Technologies) were used for immunoprecipitation according to the manufacturer’s instructions. Dynabeads were preincubated with polyclonal rabbit anti-NLRP3 (768319, R&D Systems) and polyclonal rabbit anti-P2X7R (P8232-2ML, Sigma-Aldrich) antibodies before addition of lysates (20).
Immunoblotting.
Total proteins of CD4+ T cell samples were extracted in Laemmli buffer (Tris-HCl 62.5 mmol/l, pH 6.8, 20% glycerol, 2% SDS, 5% β-mercaptoethanol), and their concentration was measured. Thirty-five micrograms of total protein was electrophoresed on 7% SDS-PAGE gels and blotted onto nitrocellulose (10401196, Schleicher & Schuell). Blots were then stained with Ponceau S. Membranes were blocked for 1 hour in TBS (Tris 10 mmol/l, NaCl 150 mmol/l), 0.1% Tween-20, 5% nonfat dry milk, pH 7.4 at 25°C, incubated for 12 hours with rat anti-NLRP3 (MAB7578, R&D Systems) diluted 1:250 or with rabbit anti-P2X7R (P8232, Sigma-Aldrich) diluted 1:200 in TBS–5% milk at 4°C, washed 4 times with TBS–0.1% Tween-20, then incubated with goat anti-rat–HRP (sc-2006, Santa Cruz Biotechnology) or goat anti-rabbit–HRP (P0448, Agilent Technologies) diluted 1:500 in TBS–5% milk, and finally washed with TBS–0.1% Tween-20. The resulting bands were visualized using enhanced chemiluminescence (32132, SuperSignal, Pierce Biotechnology Inc.).
Immunofluorescence
Immunofluorescence analysis of CD4+ T cells was performed using the following primary antibodies: rat anti-NLRP3 (MAB7578, R&D Systems) with a 1:100 dilution and rabbit anti-P2X7 (P8232, Sigma-Aldrich) at a 1:50 dilution. Secondary antibodies were as follows: donkey anti-rat–TRITC (712-025-150, Jackson ImmunoResearch) and donkey anti-rabbit–FITC (711-095-152, Jackson ImmunoResearch), at a 1:50 dilution. Analysis of immunofluorescence and collection of images were conducted using a confocal system (TCS SP5 laser scanning confocal, Leica).
Quantification of NLRP3 and P2X7R protein
CD4+ T cells extracted from PBMCs of carrier and noncarrier patients were lysed according to the manufacturer’s instructions. Briefly, cells were pelleted and supernatants removed by centrifugation. Cells were then resuspended in PBS after 3 washing steps and lysed by ultrasonication 4 times. Cellular debris was further removed by centrifugation, and supernatant was collected for use in ELISA (NLRP3 and P2X7R: MBS917009 and MBS9426859, respectively, from LSBio) according to the manufacturer’s instructions. Levels of NLRP3 or P2X7R protein, normalized per milligram or microgram of total protein and/or per cell number when applicable, were assessed and calculated.
NLRP3 small RNA interference and overexpression
CD4+ T cells obtained from noncarrier transplanted patients were cultured in vitro with or without anti-CD3/anti-CD28 stimulation. After 24 hours of culture, 750 ng of small interfering RNA (siRNA; Flexitube siRNA SI02634030, Qiagen) in 100 μl culture medium without serum and with 6 μl HiPerFect Transfection Reagent (301704, Qiagen) was incubated to allow for formation of transfection complexes. Analysis of NLRP3 gene silencing was performed at 24 and 48 hours by evaluation of the mRNA expression of Th2- and Th17-related factors. Control siRNA was used as a negative control to confirm the effect of gene silencing. CD4+ T cells obtained from transplanted carrier patients were cultured in vitro with or without anti-CD3/anti-CD28 stimulation. NLRP3-overexpressing CD4+ T cells were obtained by transfection with Lipofectamine 2000 (11668-019, Life Technologies) per the manufacturer’s instructions. The NLRP3 plasmid (RG220952) was provided by OriGene.
Subcellular localization
Subcellular fractionation of CD4+ T cells was performed using the Qiagen Qproteome Cell Compartment kit (37502, Qiagen), which enables the sequential isolation of proteins associated with the cytosol, membranes, nucleus, and cytoskeleton from cell lysates.
Briefly, cells were isolated as previously described and then pelleted. Cytosolic extraction buffer was added, and the respective fraction was isolated by centrifugation. Addition of membrane extraction buffer was used to obtain the membrane fraction per the manufacturer’s instructions. Expression of NLRP3 and of P2X7R was further assessed in each fraction using an ELISA kit as described above. NLRP3 or P2X7R in each cell fraction was reported as picograms per microgram of total NLRP3 or P2X7R, and data were expressed as percentage of NLRP3 or of P2X7R detected in each compartment.
Assessment of NLRP3 DNA binding using modified ChIP
We performed chromatin immunoprecipitation (ChIP) using an Agarose ChIP Kit (26156, Thermo Fisher Scientific). Briefly, CD4+ T cells were isolated as above, and protein-DNA complexes were stabilized and then extracted. In vivo cross-linking was achieved with formaldehyde. To lyse, extract, and solubilize the cross-linked complexes, the Thermo Fisher Scientific Chromatin Prep Module was used (included in the kit), thus reducing contamination of the chromatin-bound DNA from other cellular compartments and enriching samples for NLRP3. Once chromatin was digested, immunoprecipitation of NLRP3 was performed as described above (see “Immunoprecipitation”). To analyze protein-binding sequences, genomic DNA was sheared into small pieces with enzymatic digestion per the manufacturer’s instructions. After proteinase digestion and cross-link reversal, DNA was purified, and expression of genes of interest (IRF4, IL-4) was assessed using a TaqMan Copy Number Assay (Hs0201197cn, Hs00092447cn, Applied Biosystems). Expression of genes was normalized to that of total non-immunoprecipitated DNA extracted from the same number of cells.
Ubiquitin-mediated culturing experiments
CD4+ T cells isolated from carrier and noncarrier PBMCs were cultured as previously described in the presence of an inhibitor of protein synthesis, cycloheximide (C4859-1ML, Sigma-Aldrich), and NLRP3 and P2X7R protein was quantified at baseline and 30, 60, 90, and 120 minutes and at baseline and 30 and 90 minutes, respectively, in order to evaluate ubiquitin-mediated degradation. The ubiquitin inhibitor MG132 (M8699, Sigma-Aldrich) was added in order to block ubiquitin-mediated degradation, and NLRP3 or P2X7R was quantified at the same time points described above (44, 45).
Bioinformatic analysis and protein modeling
The P2X7R intracellular sequence (from residues 360–595) was submitted to I-TASSER algorithms for 3D protein structure prediction as previously described (46), producing 10 models, the best of which had a normalized Z score of 1.17.
Bacterial killing
CD14+ cells were isolated from PBMCs of carrier and noncarrier patients using CD14 microbeads (130-050-201, Miltenyi), followed by cell culture over 7 days in RPMI 1640 supplemented with 10% FBS, 25 mM HEPES, M-CSF (50 ng/ml), penicillin (100 U/ml), and streptomycin (100 mg/ml). Cells were then coincubated with equal numbers of E. coli for 24 and 48 hours. Lysates obtained were plated onto agar plates, and viable bacteria were counted (CFU) (47, 48).
ELISPOT autoimmune assay
An ELISPOT assay (552138, BD Biosciences) was used to measure IFN-γ production by responder cells when restimulated with vimentin peptide (2105-VI, R&D Systems) (31). Vimentin peptide (20 μg/ml), anti-CD3- and anti-CD28-Ig (1 μg/ml and 2 μg/ml, respectively; CD0300 and CD2800, Life Technologies), tetanus toxoid mixture (1 μl; Diftavax, Sanofi Pasteur), or IA-2 (100 μg/ml; Pierce Biotechnology Inc.) was used to stimulate 1 × 105 responder PBMCs obtained from carrier and noncarrier patients for 48 hours. Spots were counted using an ImmunoSpot analyzer (CTL Europe GmbH) as previously described (15).
Statistics
Continuous variables are presented as means and standard errors, and categorical variables are presented as proportions. We used independent sample t tests to compare continuous variables and χ2 test/Fisher’s exact test to compare categorical variables. For multiple comparisons, 1-way or 2-way ANOVA followed by Bonferroni’s post hoc test between the group of interest and all other groups was used. All multivariable analyses were adjusted for age and sex. Univariable and multivariable logistic regression methods were used to model the relationships between risk factors and outcomes of interest. Two-tailed P values less than 0.05 were considered statistically significant.
Study approval
Human and animal studies were approved by the appropriate institutional review board(s) of Boston Children’s Hospital (Boston, Massachusetts, USA); Ospedale S. Orsola Malpighi (Bologna, Italy); and Ospedali Riuniti di Bergamo (Bergamo, Italy). Written informed consent was received from participants prior to inclusion in the study.
Author contributions
FD designed the study, performed experiments, analyzed data, and wrote the paper. LP collected and analyzed clinical data. AV collected samples and clinical data. AM, MBN, ST, SD, RB, KL, and VU performed experiments and analyzed data. LB, AI, and MI collected data and samples. SDP performed protein structure analysis. DC performed immunostaining and histology analysis. GV, RCS, RA, AC, PSH, BEE, GVZ, FF, MHS, and FG coordinated sample collection, designed research, and edited the paper. PF designed research, and wrote and edited the paper.
Supplementary Material
Acknowledgments
FD is the recipient of a Società Italiana di Diabetologia (SID) Lombardia Grant and of the European Foundation for the Study of Diabetes (EFSD) Rising Star Fellowship Grant. PF is supported by the EFSD/Sanofi European Research Programme and by an American Heart Association Grant-in-Aid. PF and FD are supported by Italian Ministry of Health grant RF-2016-02362512. VU is supported by an FO.DI.RI SID fellowship. We thank the Fondazione Romeo e Enrica Invernizzi for the extraordinary support. We also thank Maddalena Ripamonti for technical assistance in fluorimeter analysis. The work was supported in part by NIH U01 grants AI63594 (to PH) and AI063623 (to AC). ClinicalTrials.gov Identifier: NCT02255123. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Version 1. 07/16/2018
Electronic publication
Version 2. 08/01/2018
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: J Clin Invest. 2018;128(8):3490–3503.https://doi.org/10.1172/JCI94524.
Contributor Information
Andrea Vergani, Email: andrea.vergani80@gmail.com.
Luciano Potena, Email: luciano.potena2@unibo.it.
Anna Maestroni, Email: maestroni.anna26@gmail.com.
Vera Usuelli, Email: vera.usuelli@childrens.harvard.edu.
Moufida Ben Nasr, Email: moufida.benNasr@childrens.harvard.edu.
Roberto Bassi, Email: roberto.bassi@emdserono.com.
Sara Tezza, Email: saratezza@gmail.com.
Sergio Dellepiane, Email: srgdellepiane@gmail.com.
Basset El Essawy, Email: belessawy@gmail.com.
Maria Iascone, Email: miascone@hpg23.it.
Attilio Iacovoni, Email: aiacovoni@hpg23.it.
Laura Borgese, Email: laura.borgese@gmail.com.
Kaifeng Liu, Email: Kaifeng.Liu@childrens.harvard.edu.
Gary Visner, Email: gary.visner@childrens.harvard.edu.
Sirano Dhe-Paganon, Email: dhepag@crystal.harvard.edu.
Domenico Corradi, Email: domenico.corradi@unipr.it.
Reza Abdi, Email: rabdi@rics.bwh.harvard.edu.
Franco Folli, Email: franco.folli@unimi.it.
Gian Vincenzo Zuccotti, Email: gianvincenzo.zuccotti@unimi.it.
Mohamed H. Sayegh, Email: msayegh@rics.bwh.harvard.edu.
Peter S. Heeger, Email: peter.heeger@mssm.edu.
Anil Chandraker, Email: achandraker@partners.org.
Francesco Grigioni, Email: francesco.grigioni@unibo.it.
Paolo Fiorina, Email: paolo.fiorina@childrens.harvard.edu.
References
- 1.Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. N Engl J Med. 2012;367(24):2322–2333. doi: 10.1056/NEJMra1205750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zeiser R, Robson SC, Vaikunthanathan T, Dworak M, Burnstock G. Unlocking the potential of purinergic signaling in transplantation. Am J Transplant. 2016;16(10):2781–2794. doi: 10.1111/ajt.13801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bartlett R, Stokes L, Sluyter R. The P2X7 receptor channel: recent developments and the use of P2X7 antagonists in models of disease. Pharmacol Rev. 2014;66(3):638–675. doi: 10.1124/pr.113.008003. [DOI] [PubMed] [Google Scholar]
- 4.Junger WG. Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol. 2011;11(3):201–212. doi: 10.1038/nri2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schenk U, et al. Purinergic control of T cell activation by ATP released through pannexin-1 hemichannels. Sci Signal. 2008;1(39):ra6. doi: 10.1126/scisignal.1160583. [DOI] [PubMed] [Google Scholar]
- 6.Khakh BS, North RA. P2X receptors as cell-surface ATP sensors in health and disease. Nature. 2006;442(7102):527–532. doi: 10.1038/nature04886. [DOI] [PubMed] [Google Scholar]
- 7.Sullivan JA, et al. Differential requirement for P2X7R function in IL-17 dependent vs. IL-17 independent cellular immune responses. Am J Transplant. 2014;14(7):1512–1522. doi: 10.1111/ajt.12741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Killeen ME, Ferris L, Kupetsky EA, Falo L, Mathers AR. Signaling through purinergic receptors for ATP induces human cutaneous innate and adaptive Th17 responses: implications in the pathogenesis of psoriasis. J Immunol. 2013;190(8):4324–4336. doi: 10.4049/jimmunol.1202045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Atarashi K, et al. ATP drives lamina propria T(H)17 cell differentiation. Nature. 2008;455(7214):808–812. doi: 10.1038/nature07240. [DOI] [PubMed] [Google Scholar]
- 10.Yuan X, et al. Targeting Tim-1 to overcome resistance to transplantation tolerance mediated by CD8 T17 cells. Proc Natl Acad Sci U S A. 2009;106(26):10734–10739. doi: 10.1073/pnas.0812538106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yuan X, et al. A novel role of CD4 Th17 cells in mediating cardiac allograft rejection and vasculopathy. J Exp Med. 2008;205(13):3133–3144. doi: 10.1084/jem.20081937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilhelm K, et al. Graft-versus-host disease is enhanced by extracellular ATP activating P2X7R. Nat Med. 2010;16(12):1434–1438. doi: 10.1038/nm.2242. [DOI] [PubMed] [Google Scholar]
- 13.Chadha R, Heidt S, Jones ND, Wood KJ. Th17: contributors to allograft rejection and a barrier to the induction of transplantation tolerance? Transplantation. 2011;91(9):939–945. doi: 10.1097/TP.0b013e3182126eeb. [DOI] [PubMed] [Google Scholar]
- 14.Sullivan JA, Adams AB, Burlingham WJ. The emerging role of TH17 cells in organ transplantation. Transplantation. 2014;97(5):483–489. doi: 10.1097/TP.0000000000000000. [DOI] [PubMed] [Google Scholar]
- 15.Vergani A, et al. Long-term heart transplant survival by targeting the ionotropic purinergic receptor P2X7. Circulation. 2013;127(4):463–475. doi: 10.1161/CIRCULATIONAHA.112.123653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vergani A, et al. The purinergic system in allotransplantation. Am J Transplant. 2014;14(3):507–514. doi: 10.1111/ajt.12567. [DOI] [PubMed] [Google Scholar]
- 17.Vergani A, et al. Effect of the purinergic inhibitor oxidized ATP in a model of islet allograft rejection. Diabetes. 2013;62(5):1665–1675. doi: 10.2337/db12-0242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baroja-Mazo A, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol. 2014;15(8):738–748. doi: 10.1038/ni.2919. [DOI] [PubMed] [Google Scholar]
- 19.Bruchard M, et al. The receptor NLRP3 is a transcriptional regulator of TH2 differentiation. Nat Immunol. 2015;16(8):859–870. doi: 10.1038/ni.3202. [DOI] [PubMed] [Google Scholar]
- 20.Franceschini A, et al. The P2X7 receptor directly interacts with the NLRP3 inflammasome scaffold protein. FASEB J. 2015;29(6):2450–2461. doi: 10.1096/fj.14-268714. [DOI] [PubMed] [Google Scholar]
- 21.Salaro E, et al. Involvement of the P2X7-NLRP3 axis in leukemic cell proliferation and death. Sci Rep. 2016;6:26280. doi: 10.1038/srep26280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thunberg U, et al. Polymorphism in the P2X7 receptor gene and survival in chronic lymphocytic leukaemia. Lancet. 2002;360(9349):1935–1939. doi: 10.1016/S0140-6736(02)11917-9. [DOI] [PubMed] [Google Scholar]
- 23.Lee KH, et al. P2X7 receptor polymorphism and clinical outcomes in HLA-matched sibling allogeneic hematopoietic stem cell transplantation. Haematologica. 2007;92(5):651–657. doi: 10.3324/haematol.10810. [DOI] [PubMed] [Google Scholar]
- 24.Lees MP, et al. P2X7 receptor-mediated killing of an intracellular parasite, Toxoplasma gondii, by human and murine macrophages. J Immunol. 2010;184(12):7040–7046. doi: 10.4049/jimmunol.1000012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gu BJ, et al. An Arg307 to Gln polymorphism within the ATP-binding site causes loss of function of the human P2X7 receptor. J Biol Chem. 2004;279(30):31287–31295. doi: 10.1074/jbc.M313902200. [DOI] [PubMed] [Google Scholar]
- 26.Shemon AN, et al. A Thr357 to Ser polymorphism in homozygous and compound heterozygous subjects causes absent or reduced P2X7 function and impairs ATP-induced mycobacterial killing by macrophages. J Biol Chem. 2006;281(4):2079–2086. doi: 10.1074/jbc.M507816200. [DOI] [PubMed] [Google Scholar]
- 27.Gidlöf O, et al. A common missense variant in the ATP receptor P2X7 is associated with reduced risk of cardiovascular events. PLoS One. 2012;7(5):e37491. doi: 10.1371/journal.pone.0037491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fuller SJ, Stokes L, Skarratt KK, Gu BJ, Wiley JS. Genetics of the P2X7 receptor and human disease. Purinergic Signal. 2009;5(2):257–262. doi: 10.1007/s11302-009-9136-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gu BJ, et al. A Glu-496 to Ala polymorphism leads to loss of function of the human P2X7 receptor. J Biol Chem. 2001;276(14):11135–11142. doi: 10.1074/jbc.M010353200. [DOI] [PubMed] [Google Scholar]
- 30.Sullivan JA, et al. Th17 responses to collagen type V, kα1-Tubulin, and Vimentin are present early in human development and persist throughout life. Am J Transplant. 2017;17(4):944–956. doi: 10.1111/ajt.14097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mahesh B, Leong HS, Nair KS, McCormack A, Sarathchandra P, Rose ML. Autoimmunity to vimentin potentiates graft vasculopathy in murine cardiac allografts. Transplantation. 2010;90(1):4–13. doi: 10.1097/TP.0b013e3181dfa694. [DOI] [PubMed] [Google Scholar]
- 32.Liu Z, Fan H, Jiang S. CD4(+) T-cell subsets in transplantation. Immunol Rev. 2013;252(1):183–191. doi: 10.1111/imr.12038. [DOI] [PubMed] [Google Scholar]
- 33.Bednash JS, Mallampalli RK. Regulation of inflammasomes by ubiquitination. Cell Mol Immunol. 2016;13(6):722–728. doi: 10.1038/cmi.2016.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song H, et al. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat Commun. 2016;7:13727. doi: 10.1038/ncomms13727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Starling RC, et al. Multicenter analysis of immune biomarkers and heart transplant outcomes: results of the Clinical Trials in Organ Transplantation-05 Study. Am J Transplant. 2016;16(1):121–136. doi: 10.1111/ajt.13422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Potena L, et al. Interplay of coronary angiography and intravascular ultrasound in predicting long-term outcomes after heart transplantation. J Heart Lung Transplant. 2015;34(9):1146–1153. doi: 10.1016/j.healun.2015.01.990. [DOI] [PubMed] [Google Scholar]
- 37.Gadeock S, Pupovac A, Sluyter V, Spildrejorde M, Sluyter R. P2X7 receptor activation mediates organic cation uptake into human myeloid leukaemic KG-1 cells. Purinergic Signal. 2012;8(4):669–676. doi: 10.1007/s11302-012-9320-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miroux C, et al. In vitro effects of cyclosporine A and tacrolimus on regulatory T-cell proliferation and function. Transplantation. 2012;94(2):123–131. doi: 10.1097/TP.0b013e3182590d8f. [DOI] [PubMed] [Google Scholar]
- 39.Ueno T, et al. The emerging role of T cell Ig mucin 1 in alloimmune responses in an experimental mouse transplant model. J Clin Invest. 2008;118(2):742–751. doi: 10.1172/JCI32451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xiao S, et al. Differential engagement of Tim-1 during activation can positively or negatively costimulate T cell expansion and effector function. J Exp Med. 2007;204(7):1691–1702. doi: 10.1084/jem.20062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991;253(5022):905–909. doi: 10.1126/science.1715094. [DOI] [PubMed] [Google Scholar]
- 42.D’Addio F, et al. The link between the PDL1 costimulatory pathway and Th17 in fetomaternal tolerance. J Immunol. 2011;187(9):4530–4541. doi: 10.4049/jimmunol.1002031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coll RC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21(3):248–255. doi: 10.1038/nm.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Azzu V, Brand MD. Degradation of an intramitochondrial protein by the cytosolic proteasome. J Cell Sci. 2010;123(pt 4):578–585. doi: 10.1242/jcs.060004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chun J, et al. NLRP3 localizes to the tubular epithelium in human kidney and correlates with outcome in IgA nephropathy. Sci Rep. 2016;6:24667. doi: 10.1038/srep24667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat Methods. 2015;12(1):7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tang X, Basavarajappa D, Haeggström JZ, Wan M. P2X7 receptor regulates internalization of antimicrobial peptide LL-37 by human macrophages that promotes intracellular pathogen clearance. J Immunol. 2015;195(3):1191–1201. doi: 10.4049/jimmunol.1402845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Placido R, et al. P2X(7) purinergic receptors and extracellular ATP mediate apoptosis of human monocytes/macrophages infected with Mycobacterium tuberculosis reducing the intracellular bacterial viability. Cell Immunol. 2006;244(1):10–18. doi: 10.1016/j.cellimm.2007.02.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.