

Abstract
Indoleamine 2, 3-dioxygenase 1 (IDO1) is a rate-limiting metabolic enzyme that converts the essential amino acid tryptophan (Trp) into downstream catabolites known as kynurenines. Coincidently, numerous studies have demonstrated that IDO1 is highly expressed in multiple types of human cancer. Preclinical studies have further introduced an interesting paradox: while single-agent treatment with IDO1 enzyme inhibitor has a negligible effect on decreasing the established cancer burden, approaches combining select therapies with IDO1 blockade tend to yield a synergistic benefit against tumor growth and/or animal subject survival. Given the high expression of IDO1 among multiple cancer types along with the lack of monotherapeutic efficacy, these data suggest that there is a more complex mechanism of action than previously appreciated. Similar to the dual faces of the astrological Gemini, we highlight the multiple roles of IDO1 and review its canonical association with IDO1-dependent tryptophan metabolism, as well as documented evidence confirming the dispensability of enzyme activity for its immunosuppressive effects. The gene transcript levels for IDO1 highlight its strong association with T-cell infiltration, but the lack of a universal prognostic significance among all cancer subtypes. Finally, ongoing clinical trials are discussed with consideration of IDO1-targeting strategies that enhance the efficacy of immunotherapy for cancer patients.
Keywords: glioblastoma, glioma, IDO, immunosuppression, kynurenine, immunotherapy, melanoma, Treg
Introduction
Within the last decade, there has been incredible success regarding application of immune checkpoint inhibitors, with an emphasis on targeting CTLA-4 and/or PD-(L)1, to improve patient survival for otherwise untreatable melanoma,1 non-small-cell lung2 and renal3 cancers. This progress has motivated medical oncologists and/or tumor immunologists to better understand the regulation, role and functions of co-inhibitory pathways that are expressed by immune and cancer cells.4 Preclinical and clinical studies have demonstrated a trend of immunotherapeutic combination strategies to confer a greater survival benefit over single-agent approaches.5,6 There are, however, notable exceptions to the general belief that more is better, and several recent studies have highlighted the fact that multi-therapy approaches do not universally provide an advantage to the host and/or immune system.7,8 These considerations reflect the complicated regulatory network that governs the human immune system’s response to cancer, its failure to have a ‘one-size-fits-all’ approach, the toxicities induced by certain immunotherapeutic combinations and the critical need to further understand why checkpoint therapies (i) provide benefits to some patients, but not others; (ii) enhance tumoricidal effects against some cancers, but not others; and (iii) beneficially stimulate immune responses with certain combinations, but not others.
In this review, we focus on the novel immune checkpoint target, indoleamine 2, 3-dioxygenase 1 (IDO1), which is characterized as a rate-limiting metabolic enzyme that converts tryptophan (Trp), into downstream kynurenines (Kyn) (Figure 1). IDO1 is interferon-inducible and has been associated with mediating potently immunosuppressive effects in cancer.9,10 While a growing body of data suggest that there is a lack of therapeutic efficacy when targeting IDO1 alone, there is strong support for combination approaches to provide a synergistic benefit.11,12,13,14 Due to the limited effect(s) of single agents, a growing number of active clinical trials utilize IDO1 as an adjuvant alongside other cancer treatment modalities (Vacchelli et al. 15 and Table 1), which raises the question of whether immunological therapies somehow ‘activate’ IDO1 so that it becomes therapeutically targetable. These developments highlight additional questions that have yet to be answered, including the following: (i) Is enzyme metabolism the sole characteristic that endows IDO1 with immunosuppressive activity? (ii) Why is IDO1 enzyme inhibition not effective as a monotherapy16 given that IDO1 is highly expressed in a variety of human cancers?10,17 (iii) Why does coupling radiotherapy and/or chemotherapy tend to synergize with IDO1 inhibition?18 (iv) How do IDO1 pathway inhibitors that have no effect on converting Trp into Kyn,19 such as indoximod (dextrorotatory 1-methyl-Trp; D-1-MT), affect IDO1-mediated immune suppression?20 (v) Do select IDO1 inhibitors confer gain-of-function toxicity under certain therapeutic contexts? (vi) Are there immunosuppressive effects of IDO1 in cancer cells that are independent of enzyme activity and similar to effects that have been reported in innate immune cells?21
Figure 1.
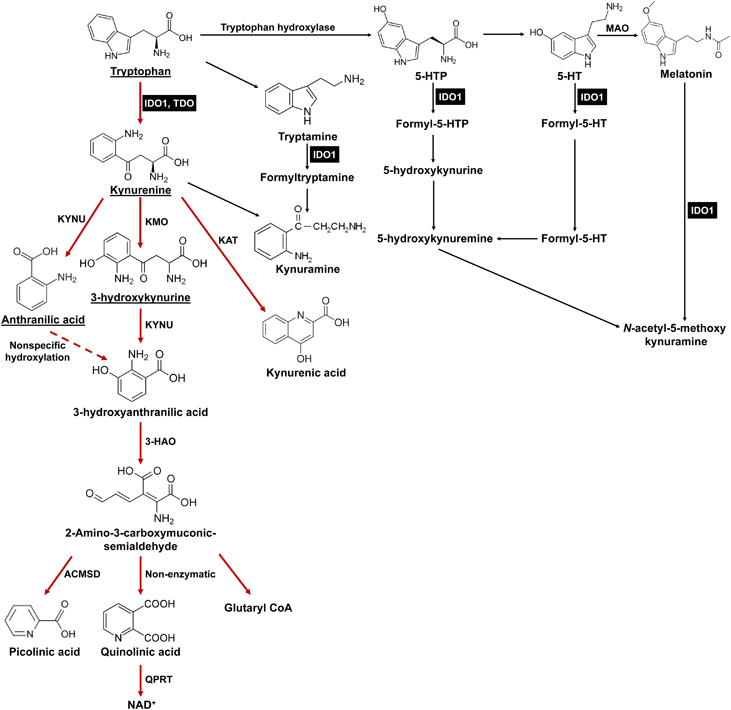
Tryptophan (Trp) catabolic pathways. In addition to being used as a building block for protein synthesis, the majority of dietary Trp (95%) is catabolized via the Trp→Kyn pathway (red arrows). Other minor pathways include conversion to tryptamine or melatonin. Within the Kyn pathway, the underlined metabolites can cross the blood brain barrier (BBB). IDO1 (and TDO) are highlighted in black boxes. ACMSD: 2-amino-3-carboxymuconate semialdehyde carboxylase; 3-HAO, 3-hydroxyanthranilate 3, 4-dioxygenase; IDO1, indoleamine 2, 3-dioxygenase 1; KAT, kynurenine aminotransferase (I, II, III); KMO, kynurenine 3-monooxygenase; KYNU, kynureninase; MAO, monoamine oxidase; QPRT, quinolinic-acid phosphoribosyl transferase; TDO, tryptophan 2,3-dioxygenase.
Table 1.
Ongoing and historical clinical trials that target IDO1 in cancer
| Agent | Indication(s) | Phase | Status | Notes | NCT no. |
|---|---|---|---|---|---|
| Indoximod (D-1-MT) | Metastatic solid tumor | I | Completed | Combined with docetaxel | NCT01191216 |
| Solid tumor | I | Completed | Single agent | NCT00567931 | |
| Metastatic breast cancer | I/II | Active, not recruiting | Combined with vaccine | NCT01042535 | |
| II | Recruiting | Combined with fulvestrant or tamoxifen | NCT02913430 | ||
| II | Active, not recruiting | Combined with docetaxel | NCT01792050 | ||
| Melanoma | I/II | Recruiting | Combined with ipilimumab (CTLA-4 mAb) | NCT02073123 | |
| Metastatic adenoma of pancreas | I/II | Recruiting | Combined with gemcitabine and nab-paclitaxel | NCT02077881 | |
| Acute myeloid leukemia | I/II | Recruiting | Combined with cytarbine, idarubicin | NCT02835729 | |
| GBM, glioma, gliosarcoma | I/II | Recruiting | Combined with temozolomide, bevacizumab (VEGF mAb) and radiation | NCT02052648 | |
| GBM, glioma, ependymoma, medulloblastoma | I | Recruiting | Combined with temozolomide and radiation | NCT02502708 | |
| Prostate carcinoma | II | Active, not recruiting | Combined with sipuleucel-T | NCT01560923 | |
| NSCLC | II | Recruiting | Combined with docetaxel and tergenpumatucel-L | NCT02460367 | |
| INCB024360 | Advanced neoplasms | I | Completed | As single agent | NCT01195311 |
| Myelodysplastic Syndromes | II | Completed | As single agent | NCT01822691 | |
| Melanoma | I/II | Recruiting | Combined with ipilimumab | NCT01604889 | |
| II | Recruiting | Combined with a multipeptide-based vaccine | NCT01961115 | ||
| Reproductive tract tumors | II | Completed | Compared to tamoxifen | NCT01685255 | |
| I | Recruiting | Combined with vaccine and cyclophosphamide | NCT02785250 | ||
| I | Active, not recruiting | As single agent | NCT02042430 | ||
| I | Recruiting | Combined with adoptive transfer of NK cells and IL-2 | NCT02118285 | ||
| I/II | Recruiting | Combined with CRS-207 and pembrolizumab (PD-1 mAb) | NCT02575807 | ||
| I/II | Recruiting | Combined with DC-targeted NY-ESO-1 and poly-ICLC | NCT02166905 | ||
| Solid tumors | I/II | Recruiting | Combined with a PDCD1 mAb | NCT02178722 | |
| I | Recruiting | Alone or combined with pembrolizumab | NCT02862457 | ||
| I | Active, not recruiting | Combined with itacitinib (JAK inhibitor) | NCT02559492 | ||
| I/II | Recruiting | Combined with nivolumab or pembrolizumab and chemotherapy | NCT03085914 | ||
| I/II | Recruiting | Combined with MEDI4736 (PD-L1 mAb) | NCT02318277 | ||
| Meta. colorectal cancer | I/II | Not yet recruiting | Combined with pembrolizumab and azacitidine | NCT03182894 | |
| Gastric cancer | II | Not yet recruiting | As single agent | NCT03196232 | |
| Meta. Pancreatic cancer | II | Not yet recruiting | Combined with CRS-207 and pembrolizumab | NCT03006302 | |
| NSCLC, urothelial carcinoma | I | Active, not recruiting | Combined with atezolizumab (PD-L1 mAb) | NCT02298153 | |
| GDC-0919 (formerly NLG-919) | Solid tumors | I | Completed | Single agent | NCT02048709 |
| Locally advanced or metastatic solid tumors | I | Active, not recruiting | Combined with MPDL3280A (PD-L1 mAb) | NCT02471846 | |
| IDO1 peptide | NSCLC | I | Completed | As single agent | NCT01219348 |
| Melanoma | I | Recruiting | Combined with ipilimumab or vemurafenib (BRAF inhibitor) | NCT02077114 | |
| Melanoma | II | Recruiting | Combined with temozolomide, imiquimod, GM-CSF and survivin peptide | NCT01543464 | |
| PF-06840003 | GBM or grade III anaplastic glioma | I | Recruiting | As single agent | NCT02764151 |
| BMS986205 | Cervical, DLBCL, SCCHN, UC, pancreatic, melanoma, NSCLC | I | Recruiting | As single agent | NCT02658890 |
| Advanced cancer | I | Recruiting | Combined with nivolumab | NCT03192943 |
Abbreviations: DC, dendritic cell; DLBCL, diffuse large B-cell lymphoma; D-1-MT, dextrorotatory 1-methyl-tryptophan; IDO1, indoleamine 2, 3-dioxygenase 1; GM-CSF, granulocyte–macrophage colony-stimulating factor; NK, natural killer cell; GBM, glioblastoma; IL-2, interleukin-2; mAb, monoclonal antibody; NSCLC, non-small-cell lung cancer; SCCHN, squamous cell carcinoma of head/neck; UC, urothelial carcinoma; VEGF, vascular endothelial growth factor.
Trp dioxygenases
L-Trp, which is the least abundant essential amino acid, can be metabolized via four distinct mechanisms: decarboxylation to tryptamine; protein synthesis; the serotonergic pathway; and the Kyn pathway (Figure 1).22 Kyn pathway metabolism accounts for ~95% of all mammalian dietary Trp.23 The first rate-limiting step required for conversion of Trp into Kyn is oxidative cleavage of a 2,3-indole ring double bond that forms N-formylkynurenine, which is almost immediately converted to L-Kyn. Three different Trp dioxygenases have been identified in mammals, including IDO1, tryptophan 2,3-dioxygenase (TDO) and IDO2. Human IDO1 is a monomeric heme-containing protein encoded by chromosome 8p12 and has high enzyme activity for Trp (Km~20 μM).24 TDO is located on chromosome 4 and forms a tetrameric heme-containing complex with lower enzyme activity for Trp (Km~190 μM)25 compared to IDO1. IDO2 is a genetic paralog of IDO1 and is directly adjacent to IDO1 on the same chromosome.26 Although it can allegedly convert Trp into Kyn, it has an almost 1000-fold lower enzyme activity (Km~6.8 mM).27
In addition to the differences in structure and rates of enzyme conversion, the three Trp catabolic enzymes have varying substrate specificities, tissue distribution and regulation of expression. While TDO is only capable of metabolizing the L-Trp isomer,25 IDO1 mediates oxidative cleavage of several indole substrates, including D- and L-Trp, tryptamine, 5-hydroxy-L-tryptophan, serotonin and melatonin (Figure 1).28 Compared to IDO1 and TDO, the exceptionally low enzyme activity of IDO2 raises the question of whether L-Trp is a relevant physiological substrate.24 Under normal conditions, TDO is primarily expressed in the liver, placenta and brain, which likely reflects its professional role of satisfying the energetic needs required by the body. In contrast, IDO1 is detected throughout mammalian tissues at various levels, including the central nervous system, epididymis, intestine, thymus, respiratory tract, spleen, pancreas, placenta, lens, kidney, myeloid cells and endothelial cells,17 and has been shown to be increased in select tissues with age.29 Interestingly, IDO1 is noticeably absent from the liver. Constitutive expression of murine IDO2 mRNA and protein is detected in the liver, epididymis and brain, but little information has been reported for human IDO2 due to the lack of antibody specificity and complexity of human IDO2 transcription.30 In addition to their expression in normal tissues, IDO1, TDO and IDO2 are selectively expressed in different types of human and mouse cancers.31 Notably, although IDO1 and TDO are expressed at a high level in many cancer subtypes, a recent gene expression profiling study evaluating two large RNA-sequencing data sets among 31 cancer subtypes revealed negligible expression of IDO2 in the majority of human cancers (>99%), which possibly indicates that IDO2 has a less important role in supporting tumorigenesis due to is reduced expression and/or activity.30
Previous mechanistic studies demonstrated that expression of TDO is regulated by glucocorticoid hormones and dietary Trp levels,32,33 whereas IDO1 is regulated and expressed in response to a variety of inflammatory stimuli, including interferon (IFN)-α,34 IFN-γ,35 lipopolysaccharide (LPS),36,37 interleukin-1 (IL-1), tumor necrosis factor,38 CpG oligodeoxynucleotides (CpG-ODN)39 and prostaglandin-E2 (PGE2).40 IDO2 is also increased by treating cells with IFN-γ, IL-10, LPS and PGE2, although its expression is less robust than that of IDO1.41 Coincidently, activation of the aryl hydrocarbon receptor (AhR), which is a transcription factor that Kyn has been proposed to serve as a physiologically relevant ligand for,42 has been shown to be upregulated through an IDO2-dependent pathway in dendritic cells (DCs).43,44
The involvement of multiple Trp-catabolizing enzymes contributes to Kyn metabolite generation and/or accumulation, which raises a potential challenge for therapeutic strategies that target this metabolic pathway. A clear question for the field is determining whether inhibiting a single player is sufficient for enhancing immune-mediated antitumor effects or whether simultaneous inhibition of all three enzymes is required. A complete answer to this question will likely depend on the following: (i) the type of cancer under investigation; (ii) expression levels and metabolic activity of IDO1, TDO and IDO2; (iii) intratumoral and serological levels of Trp and Kyn; and (iv) cellular origin of the expressed functional gene products. To date, an IDO1–TDO dual inhibitor has been discovered, although the in vivo relevance of this agent in antitumor therapy has yet to be revealed.45 Furthermore, while addressing whether inhibition of all three Trp dioxygenases is interesting, it is important to note that the more limited anatomical expression of TDO as well as the full role of IDO2 may detract from the undeniably immunosuppressive effects of IDO1, which is a clear therapeutic priority for achieving greater immune-mediated antitumor efficacy.
Kyn pathway and tumor immune escape
The relationship between cancer and elevated Trp catabolism was recognized as early as the 1950s.46 Since Trp is the least abundant amino acid and must be ingested through the diet, IDO1 was originally thought to be part of an ancient, innate mechanism that was designed to slow the growth of neoplastic tissues and/or infectious agents that require Trp stores for continued metabolic activity.27 In support of this hypothesis, Munn et al. demonstrated that female mice pregnant with allogeneic pups and treated with 1-methyl Trp (1-MT) resulted in maternal immune-mediated rejection.47 Later studies of experimental autoimmune encephalomyelitis suggested that Trp catabolites and their derivatives contribute to a shift in primarily Th1-mediated disease to a Th2-associated non-pathological condition.48 Collectively, these studies indicate an important role for IDO1 in general mechanisms that support immune tolerance. The role of IDO1 in immune-mediated evasion of cancer was first introduced in 2002 when Friberg et al. 49 showed that Lewis lung carcinoma (LLC) cells stimulated a more robust allogeneic T-cell response when cultured in the presence of an IDO1 inhibitor, which commensurately delayed LLC tumor growth after systemic treatment in vivo.
Currently, three major hypothetical mechanisms are proposed to explain the role of IDO1 in tumor-associated immunosuppression. First, enzyme activity results in local depletion of Trp, which results in an increase of uncharged transfer RNA in neighboring T cells and activation of the amino-acid-sensitive GCN2 and mTOR stress-kinase pathways. In turn, GCN2 signaling causes cell cycle arrest and induction of anergy in responding T cells. An additional hypothesis is that downstream Kyns, including L-Kyn, 3-hydroxy-L-Kyn, 3-hydroxyanthranilate (3HAA) and quinolinic acid, have an immune modulatory effect that acts by inducing effector T-cell arrest or apoptosis, both in vitro and in vivo. 50 Downstream Kyn accumulation may also contribute to the conversion of naive CD4+ T cells into immunosuppressive FOXP3-expressing regulatory T cells (Treg, CD3+CD4+CD25+FOXP3+) by virtue of the interaction between L-Kyn and Ahr.51 It is important to note, however, that this mechanism is unlikely to represent the majority of Treg in solid tumors, since the infiltrating component is primarily thymus-derived natural Treg (nTreg)52,53 combined with the absence of Ahr expression in nTreg.54 Collectively, these mechanisms may play a role in contributing to the suppression of tumor immunity via IDO1 expression in cancer.
The above hypotheses are supported by several lines of experimental evidence, with the majority of observations derived from an in vitro cell culture-based investigation. The limitation of cell culture analyses for studying the effects of Trp depletion and/or Kyn accumulation is that there is an obvious lack of physiological relevance unless the observations can be mirrored in vivo. This limitation has introduced a potential form of ‘in vitro bias’ that leaves researchers with a series of challenges and questions. For example, previous work has demonstrated that to inhibit T-cell proliferation in vitro, Trp concentrations are required to be below 0.5–1 μM.55 While this observation is interesting and scientifically well-supported, its physiological significance should be considered. Notably, plasma Trp levels range from 50 to 100 μM in humans, and local Trp reservoirs can be rapidly replenished by diffusion and/or active transport across the large amino-acid transporter from surrounding blood vessels. The Trp depletion theory incorporates the premise that non-T cells are more resistant to Trp starvation and is partially explained by the identification of a high-affinity Trp transporter that is selectively expressed on myeloid-derived macrophages, but not in T cells.56 It is not clear whether other types of IDO1-expressing cells, including tumor cells, use the same mechanism to survive Trp depletion. Further questions arise that need to be answered to determine how downstream Kyns suppresses T cells, with hypotheses suggesting that phosphoinositide-dependent protein kinase 1 (PDK1) acts as a direct target for 3HAA and 3HAA-mediated PDK1 inhibition and is responsible for the induction of type 2 T helper cell (TH2 cell) dysfunction and apoptosis.57
Novel aspects of ido1 in cancer immunity
The majority of experimental data supporting a non-enzymatic immunosuppressive role for IDO1 is derived from studies in mouse plasmacytoid DCs, which is a type of professional antigen-presenting cell. In these studies, 1-MT was used as an IDO1 enzyme inhibitor to demonstrate that IDO1-mediated immune suppression was independent of Trp catabolism.58,59,60 The mechanism of action involved two IDO1-intrinsic immunoreceptor tyrosine-based inhibitory motifs (ITIMs). Transforming growth factor-β (TGF-β) signaling caused phosphorylation of the ITIMs, which triggered noncanonical nuclear factor-κB (NF-κB) activation and phosphorylation of inhibitor for NF-κB subunit alpha and led to further autocrine reinforcement of IDO1 and TGF-β expression; ultimately, this signaling led to long-term tolerance of DCs. Although this unique IDO1 signaling pathway was not inhibited by 1-MT, it was abolished in cells lacking IDO1 expression.61
The relevance of non-enzyme IDO1 activity has yet to be addressed in a cancer setting. It is therefore unclear whether IDO1 possesses the same signaling circuitry in tumor cells as has been demonstrated in DCs. Coincidently, our recent work found that the intracranial engraftment of murine glioblastoma cells into syngeneic immunocompetent mice resulted in decreased tumor-infiltrating Treg (P<0.01) and increased animal subject survival (P<0.001) when the brain tumor cells were silenced for IDO1 expression with stably expressing small hairpin RNA.62 Notably, IDO1-silenced tumor cells kill animal subjects similar to IDO1-wild-type control cells when the cells are engrafted intracranially into mice deficient for either CD4+ and/or CD8+ T cells, which suggests that the mechanism regulating survival from tumor cells requires the suppression of IDO1 expression and is immune system-dependent. Interestingly, these outcomes were independent of the IDO1 effects on the Trp and Kyn levels mediated by tumor cells.63 Instead, the majority of IDO1 metabolism was mediated by non-tumor cells of the engrafted intracranial glioblastoma. Also, IDO1 metabolism did not decrease the endogenous Trp levels within the brain tumor compared to a naive mouse brain without tumor cells. Collectively, these data suggest that IDO1 in tumor cells and non-tumor cells possess different functions that are non-overlapping, which infers a potential difference in targetability with the current generation of IDO1 inhibitors primarily focused on enzyme activity.
One recent study revealed a novel role for IDO1 in which it affects tumor repopulating cell survival via induction of the tumor dormancy program.64 Unexpectedly, IFNγ treatment of differentiated tumor cells led to higher rates of apoptosis via STAT1-dependent signaling. By contrast, when IDO1 and Ahr were co-expressed in tumor cells with IFNγ treatment, STAT1 signaling was inhibited, which led to suppression of cell death and activation of the tumor cell dormancy program. Mechanistically, IFNγ induced high IDO1 and AhR expression as well as increased the Trp transporter levels in tumor-repopulating cells, which did not occur in differentiated cells. Mechanistically, IDO1/AhR pathway activation upregulated the cell cycle inhibitor p27, which diverted tumor-repopulating cells from the pro-apoptotic STAT1-dependent pathway toward the survival dormancy program. Therapeutically, L-1-MT-driven IDO1 enzyme inhibition diminished IFNγ-induced dormancy and suppressed tumor growth in vitro and in vivo. This latest finding further highlights the multi-faceted role of IDO1 in tumorigenesis and its complex mechanism of action with respect to cancer immunotherapy.
IDO1 and innate immune modulation in the tumor microenvironment
Over the last decade, interactions between IDO1 and a large range of immune modulators have been discovered. CpG-ODN induces expression of IDO1 through toll-like receptor 9 activation,39 with similar increases of expression by DNA nanoparticle-mediated activation of the stimulator of IFN genes (STING) adaptor pathway coupled with type I IFN (IFN-α/β) signaling.65 STING-mediated IDO1 induction in the setting of tumor immune evasion and tumor progression was demonstrated using a STING knockout mouse with engrafted LLC cells.66 Given the promising results of STING agonists in preclinical tumor immunotherapy67 and the potential commensurate induction of IDO1 via STING activation, it has become critical to evaluate STING-targeted cancer therapies for their potential synergistic potential with IDO1 inhibitors.
Another mechanism resulting in increased IDO1 expression is mediated through cell apoptosis and/or necrosis, which is a pathological hallmark in many cancers. The balance between immunogenic and tolerogenic cell death determines the outcome of the immune response within the tumor microenvironment.68 IDO1 appears to play a significant role in maintaining tolerance of apoptotic cells by the following: (i) altering the phenotype of macrophages and neighboring cells through upregulation of IL-10 and TGF-β as well as inhibition of IL-1; (ii) inducing phenotypic changes of local cross-presenting DCs; and (iii) recruiting Tregs.69,70,71 Furthermore, a subcutaneous injection of apoptotic tumor cells causes activation of IDO1, which induces suppressive phosphatase and tensin homolog (PTEN)-expressing Tregs72 and indicates that there is a potential role for IDO1 in apoptotic tumor cell-induced immune suppression.
Myeloid-derived suppressor cells (MDSCs) have been recognized as an important group of heterogeneous mediators in cancer that convey potent immunosuppressive effects on T cells.73 Recent studies have demonstrated that IDO1 is highly induced in tumor-infiltrating MDSCs and is responsible for MDSC-associated activation and/or recruitment of Tregs in human breast cancer, sarcoma and chronic lymphocytic leukemia.74,75,76 In addition to its role in immunotolerance, studies utilizing a mouse melanoma model also revealed that tumor-expressed IDO1 recruits and activates MDSCs through a Treg-dependent mechanism,77 which demonstrates the functional versatility of IDO1 in MDSC-associated immunoevasion.
IDO1 is also implicated in the inhibition of T-cell-dependent complement system activation, which was initially reported in an early study of allogeneic mouse fetal rejection.78 However, the linkage between IDO1 and the complement system in cancer was not discovered until recently. In a study of intracranial mouse glioblastoma, combination radiation and chemotherapy mediated extensive complement deposition when non-brain tumor cell IDO1 was targeted and/or inhibited through pharmacological and/or genetic methods, respectively. Importantly, complement deposition was mechanistically required for the pro-survival effect of an IDO1 pathway inhibitor.18 Given that we previously demonstrated that non-glioblastoma cells are the predominant mediators of IDO1-dependent Trp catabolism,8 the data collectively suggest that metabolically active IDO1 becomes targetable when other forms of cytotoxic therapy are used synergistically and highlights the IDO1-dependent non-tumor cell mechanisms that contribute to immunosuppression in solid tumors.
IDO1 and other key immune checkpoints
An increasing number of recognized immune checkpoints act to coordinately influence the local tumor-immune environment. To obtain the maximal therapeutic benefit with combination approaches that incorporate multiple forms of immunotherapy, a critical question is how IDO1 inhibition will interact with other key modulators of tumor-induced immune suppression (that is, CTLA-4 and/or PD-1 blockade)? Despite some preclinical cancer studies showing synergy when combining pharmacological IDO1 and CTLA-4/PD-1 blockade,13,14,79 the molecular mechanism of this relationship remains largely unknown. It has been reported that Treg cell-expressed CTLA-4 upregulates IDO1 expression by DCs,80 and there is reciprocal activation of Treg cells. In addition, IDO1 upregulates PD-1 expression on Tregs, which contributes to the maintenance of PTEN activity.72 One of our recent studies also demonstrated that treatment with a dual blockade of CTLA-4 and PD-L1 in glioblastoma-bearing mice resulted in increased tumor IDO1 mRNA expression commensurate with elevated transcripts of CD3ɛ, CD8α and IFNγ,63 possibly suggesting that tumor-infiltrating effector T cells (CD3+CD8+) activated by immune checkpoint inhibition increase IDO1 expression via IFNγ stimulation. Our recent analysis of the cancer genome atlas supports this hypothesis and found a correlation between increasing IDO1 levels with other immune checkpoints, including PD-1, PD-L1, PD-L2, CTLA-4, signal transducer and activator of transcription 3, CD39, B- and T-lymphocyte attenuator, lymphocyte-activation gene 3 and FoxP3 in surgically resected human glioblastoma.81 Taken together, the data suggest that as IDO1 expression increases in tumors, so do other immune checkpoints. It is therefore possible that combination strategies targeting multiple immune checkpoints may lead to greater synergistic effects in cancer immunotherapy, although the enhancement of host toxicity may also increase as well.
IDO1 in different cancer types: functional diversity?
Since the discovery of increased IDO1 levels in human cancer, there have been several reports correlating IDO1 expression with poor patient prognosis, including those diagnosed with acute myeloid leukemia,82 colorectal cancer,83 non-small-cell lung cancer,84,85 prostate cancer,86 ovarian carcinoma,87,88 endometrial cancer89 and esophageal cancer.90 Unexpectedly, high IDO expression levels in renal cell carcinoma and hepatocellular carcinoma patients are correlated with better survival outcomes.91,92,93 The complexity of outcomes associated with utilizing IDO1 expression as a stratifying factor for cancer patient prognosis likely reflects the complexity of IDO1 expression, regulation and functional effects within different types of human cancer. In support of this concept, our analysis of The Cancer Genome Atlas reveals that distinct IDO1 gene expression correlates with overall survival when comparing patients diagnosed with glioblastoma and melanoma.81 As shown in Figure 2, higher IDO1 transcript levels correlate with decreased glioblastoma patient survival, which is diametrically opposed to the correlation between increased IDO1 mRNA levels and its association with increased survival in melanoma patients. These results are somewhat surprising given the preclinical work suggesting that IDO1 inhibition synergizes with CTLA-4 blockade to mediate rejection of mouse melanoma.14 Further analysis demonstrates that IDO1 expression positively correlates with gene expression markers for CD8+ cytolytic T and Tregs, which are both found in glioblastoma and melanoma. Although there is a difference between these cancers regarding their correlation with IDO1 expression and survival, both diagnoses appear to have a strong correlation between the presence of intratumoral T cells and increased IDO1 expression. This led us to ask whether there is also a difference between T cell infiltration and survival outcomes. Whereas higher gene expression for cytolytic T cell markers was associated with decreased glioblastoma patient survival (Figure 2), the opposite trend was true for melanoma patients, which reflects the results of published studies.94,95
Figure 2.
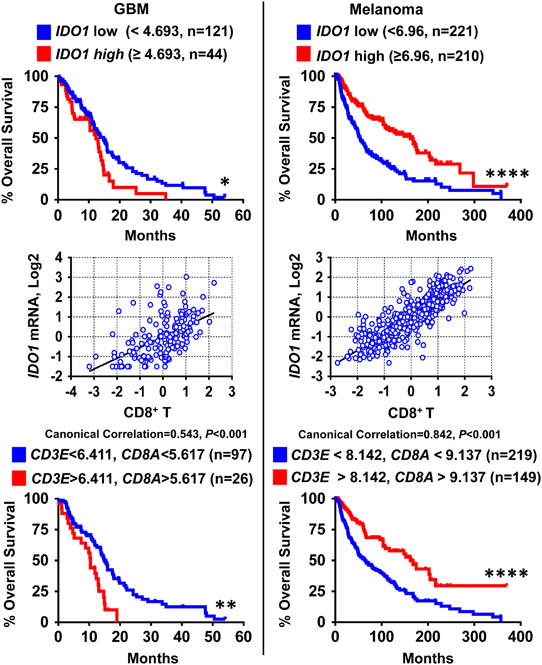
The Cancer Genome Atlas analysis reveals distinct correlations between patient survival, IDO1 transcript levels and markers for tumor-infiltrating lymphocytes between glioblastoma (GBM) and melanoma. Top panel: Kaplan–Meier analysis is based on the mRNA expression level of IDO1 in GBM (left column) and melanoma (right column). Expression of IDO1 is divided into low (blue) and high (red) groups as determined by the indicated cutoff value (calculated by Cutoff Finder, Supplementary Table and Methods). The sample size of each group is listed in the parenthesis. Middle panel: canonical correlation analysis (Supplementary Table and Methods) between IDO1 and tumor-infiltrating CD8+ T lymphocytes within GBM (left column) and melanoma (right column). Bottom panel: Kaplan–Meier analysis based on the mRNA expression level for the CD8+ T cell marker genes CD3E and CD8A in GBM (left column) and melanoma (right column). Expression of CD3E and CD8A are divided into low (blue) and high (red) groups, which were determined by the indicated cutoff value (calculated by Cutoff Finder, Supplementary Table and Methods). The patient sample size for each group is listed in parentheses. *P<0.05; **P<0.01; ****P<0.0001.
While these data are straightforward for patients diagnosed with glioblastoma, in regard to providing a rationale for including IDO1 adjuvant therapy in treatments that enhance T cell-mediated IDO1 expression increases, there are many questions about melanoma, including the following: (i) Does IDO1 play a negative role in the tumors of human patients with malignant skin cancer? (ii) Why does increased IDO1 expression correlate with increased patient survival? (iii) Will the threshold for immunotherapeutic intervention be different between patients diagnosed with glioblastoma and melanoma? (iv) What is the composition of IDO1 expression by tumor and stromal cells among different malignant subtypes? (v) If IDO1 possesses different functions among distinct cell types, do these differences contribute to the differences in outcomes between IDO1 expression and patient survival when glioblastoma and melanoma are compared?
IDO1 inhibitors in cancer immunotherapy
To date, IDO1 inhibitors have been designed, screened, and tested in preclinical models of disease (reviewed in Vacchelli et al. 15; Röhrig et al. 96). Currently, no IDO1-targeting agent is approved by the Food and Drug Administration as a standalone cancer therapeutic. However, the results of recent phase I–II studies suggest that the IDO1 pathway modulator indoximod (D-1-MT), the best-in-class IDO1 enzyme inhibitor INCB024360 (Epacadostat), and the IDO1 vaccine are well tolerated by cancer patients.97,98,99,100,101 With confirmation that targeting IDO1 is safe and well tolerated, the number of trials evaluating IDO1 inhibition in cancer therapy continue to grow (Table 1). Consistent with preclinical evaluation, the objective response rates for the non-enzyme-targeting IDO1 pathway inhibitor indoximod has yielded objective response rates of 10–18%.101,102 Combined with other immunotherapeutics, such as PD-1/PD-L1 or CTLA-4 inhibitors, this value ranges from 10 to 57% among different cancer types.103,104 One potential explanation for this wide range of variable response rates is based on the complexity of IDO1 functions among different cancer types, which suggests that the elucidation of IDO1 in different cancer subtypes is imperative for its efficacy as a therapeutic target.
Precision medicine initiatives that tailor targeted therapy against IDO1 may enhance the effectiveness of treatment, but this ideological concept still requires verification in clinical trials and across cancer diagnoses. This effect may also be a moving target since immunotherapies that enhance T-cell infiltration may also increase immunosuppressive molecule expression and activity. Furthermore, to effectively evaluate IDO1 inhibitors, target validation, pharmacodynamic properties on Trp and Kyn levels, as well as their impact on conformational activity and protein stability, are critical future requirements. While it is easy to measure the Trp and Kyn levels in vitro, quantification of IDO1 metabolism is more challenging in vivo. Recent developments for noninvasive in vivo metabolite imaging may provide a solution for this technical hurdle.105,106 It should be noted, however, that systemic Trp levels can be affected by TDO, which is expressed in the liver constitutively and induced in some types of cancer,42 suggesting that evaluation of this amino acid is not necessarily a sole reflection of IDO1 enzyme activity. Finally, understanding how IDO1 works in cancer cells, versus non-cancer cells, in terms of the enzyme and signal transduction properties, is essential for targeting the full effects of this pleiotropic mediator of immune suppression.
Concluding remarks
Substantial knowledge of IDO1 and its role in cancer has been generated over the past two decades. However, new questions continue to be raised regarding its full spectrum of function(s). Similar to the astrological description of Gemini, the Trp catabolic function of IDO1 appears to be one feature of a multifunctional player. Cell lines that express IDO1, but do not catabolize Trp, have become important tools for recognizing this phenomenon. Similarly, the IDO1 pathway inhibitor indoximod (D-1-MT), which does not convert Trp to Kyn,107,108 but is a potentially important and clinically meaningful treatment for cancer patients, has further highlighted the possibility that the non-enzyme activity of IDO1 is a relevant target in cancer immunotherapy. To address some of the questions raised earlier in this review, our group is currently constructing three novel transgenic mouse models that (i) possess a point mutation that nullifies IDO1 enzyme activity; (ii) have a 2-TA linker connecting the C terminus of endogenous IDO1, with an eGFP reporter; and (iii) contain a floxed STOP codon upstream of FLAG-tagged IDO, for future knock-in experiments under tissue-specific promoters. It is our hope that ongoing work by our team and others will answer some of the unanswered questions surrounding IDO1 in malignant cancers and, perhaps, other diseases as well.
Electronic supplementary material
Acknowledgements
This work was supported by NIH grants R00 NS082381 (DAW) and R01 NS097851-01 (DAW), the Cancer Research Institute—Clinic and Laboratory Integration Program (DAW), the Robert H. Lurie Comprehensive Cancer Center—Zell Scholar Program of the Zell Family Foundation Gift (DAW) and the Northwestern Brain Tumor Institute.
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Supplementary Information for this article can be found on the Cellular & Molecular Immunology website 10.1038/cmi.2017.143
References
- 1.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rizvi NA, Mazieres J, Planchard D, Stinchcombe TE, Dy GK, Antonia SJ, et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): a phase 2, single-arm trial. Lancet Oncol. 2015;16:257–265. doi: 10.1016/S1470-2045(15)70054-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373:1803–1813. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 5.Smyth MJ, Ngiow SF, Ribas A, Teng MWL. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat Rev Clin Oncol. 2015;13:143–158. doi: 10.1038/nrclinonc.2015.209. [DOI] [PubMed] [Google Scholar]
- 6.Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol. 2016;13:273–290. doi: 10.1038/nrclinonc.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shrimali RK, Shamim A, Verma V, Zeng P, Ananth S, Gaur P, et al. Concurrent PD-1 blockade negates the effects of OX40 agonist antibody in combination immunotherapy through inducing T-cell apoptosis. Cancer Immunol Res. 2017;5:755–766. doi: 10.1158/2326-6066.CIR-17-0292. [DOI] [PubMed] [Google Scholar]
- 8.Zhai L, Ladomersky E, Dostal CR, Lauing KL, Swoap K, Billingham LK, et al. Non-tumor cell IDO1 predominantly contributes to enzyme activity and response to CTLA-4/PD-L1 inhibition in mouse glioblastoma. Brain Behav Immun. 2017;62:24–29. doi: 10.1016/j.bbi.2017.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Munn DH, Mellor AL. IDO in the tumor microenvironment: inflammation, counter-regulation, and tolerance. Trends Immunol. 2016;37:193–207. doi: 10.1016/j.it.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9:1269–1274. doi: 10.1038/nm934. [DOI] [PubMed] [Google Scholar]
- 11.Hou DY, Muller AJ, Sharma MD, DuHadaway J, Banerjee T, Johnson M, et al. Inhibition of indoleamine 2,3-dioxygenase in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with antitumor responses. Cancer Res. 2007;67:792–801. doi: 10.1158/0008-5472.CAN-06-2925. [DOI] [PubMed] [Google Scholar]
- 12.Koblish HK, Hansbury MJ, Bowman KJ, Yang G, Neilan CL, Haley PJ, et al. Hydroxyamidine inhibitors of indoleamine-2,3-dioxygenase potently suppress systemic tryptophan catabolism and the growth of IDO-expressing tumors. Mol Cancer Ther. 2010;9:489–498. doi: 10.1158/1535-7163.MCT-09-0628. [DOI] [PubMed] [Google Scholar]
- 13.Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim C, Tobias AL, et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4 and PD-L1 in mice with brain tumors. Clini Cancer Res. 2014;20:5290–5301. doi: 10.1158/1078-0432.CCR-14-0514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med. 2013;210:1389–1402. doi: 10.1084/jem.20130066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vacchelli E, Aranda F, Eggermont A, Sautes-Fridman C, Tartour E, Kennedy EP, et al. Trial watch: IDO inhibitors in cancer therapy. Oncoimmunology. 2014;3:e957994. doi: 10.4161/21624011.2014.957994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beatty GL, O'Dwyer PJ, Clark J, Shi JG, Bowman KJ, Scherle PA, et al. First-in-human phase I study of the oral inhibitor of indoleamine 2,3-dioxygenase-1 epacadostat (INCB024360) in patients with advanced solid malignancies. Clin Cancer Res. 2017;23:3269–3276. doi: 10.1158/1078-0432.CCR-16-2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Theate I, van Baren N, Pilotte L, Moulin P, Larrieu P, Renauld JC, et al. Extensive profiling of the expression of the indoleamine 2,3-dioxygenase 1 protein in normal and tumoral human tissues. Cancer Immunol Res. 2015;3:161–172. doi: 10.1158/2326-6066.CIR-14-0137. [DOI] [PubMed] [Google Scholar]
- 18.Li M, Bolduc AR, Hoda MN, Gamble DN, Dolisca SB, Bolduc AK, et al. The indoleamine 2,3-dioxygenase pathway controls complement-dependent enhancement of chemo-radiation therapy against murine glioblastoma. J Immunother Cancer. 2014;2:21. doi: 10.1186/2051-1426-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu X, Shin N, Koblish HK, Yang G, Wang Q, Wang K, et al. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood. 2010;115:3520–3530. doi: 10.1182/blood-2009-09-246124. [DOI] [PubMed] [Google Scholar]
- 20.Opitz CA, Litzenburger UM, Opitz U, Sahm F, Ochs K, Lutz C, et al. The indoleamine-2,3-dioxygenase (IDO) inhibitor 1-methyl-D-tryptophan upregulates IDO1 in human cancer cells. PLoS ONE. 2011;6:e19823. doi: 10.1371/journal.pone.0019823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pallotta MT, Orabona C, Volpi C, Vacca C, Belladonna ML, Bianchi R, et al. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat Immunol. 2011;12:870–878. doi: 10.1038/ni.2077. [DOI] [PubMed] [Google Scholar]
- 22.Richard DM, Dawes MA, Mathias CW, Acheson A, Hill-Kapturczak N, Dougherty DM. L-tryptophan: basic metabolic functions, behavioral research and therapeutic indications. Int J Tryptophan Res. 2009;2:45–60. doi: 10.4137/ijtr.s2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beadle GW, Mitchell HK, Nyc JF. Kynurenine as an intermediate in the formation of nicotinic acid from tryptophane by neurospora. Proc Natl Acad Sci USA. 1947;33:155–158. doi: 10.1073/pnas.33.6.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pantouris G, Serys M, Yuasa HJ, Ball HJ, Mowat CG. Human indoleamine 2,3-dioxygenase-2 has substrate specificity and inhibition characteristics distinct from those of indoleamine 2,3-dioxygenase-1. Amino Acids. 2014;46:2155–2163. doi: 10.1007/s00726-014-1766-3. [DOI] [PubMed] [Google Scholar]
- 25.Batabyal D, Yeh SR. Human tryptophan dioxygenase: a comparison to indoleamine 2,3-dioxygenase. J Am Chem Soc. 2007;129:15690–15701. doi: 10.1021/ja076186k. [DOI] [PubMed] [Google Scholar]
- 26.Murray MF. The human indoleamine 2,3-dioxygenase gene and related human genes. Curr Drug Metab. 2007;8:197–200. doi: 10.2174/138920007780362509. [DOI] [PubMed] [Google Scholar]
- 27.Yuasa HJ, Ball HJ, Ho YF, Austin CJ, Whittington CM, Belov K, et al. Characterization and evolution of vertebrate indoleamine 2, 3-dioxygenases IDOs from monotremes and marsupials. Comp Biochem Physiol B Biochem Mol Biol. 2009;153:137–144. [PubMed] [Google Scholar]
- 28.Stone TW, Darlington LG. Endogenous kynurenines as targets for drug discovery and development. Nat Rev Drug Discov. 2002;1:609–620. doi: 10.1038/nrd870. [DOI] [PubMed] [Google Scholar]
- 29.Ladomersky E, Zhai L, Gritsina G, Genet M, Lauing KL, Wu M, et al. Advanced age negatively impacts survival in an experimental brain tumor model. Neurosci Lett. 2016;630:203–208. doi: 10.1016/j.neulet.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Baren N, Van den Eynde BJ. Tryptophan-degrading enzymes in tumoral immune resistance. Front Immunol. 2015;6:1–9. doi: 10.3389/fimmu.2015.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhai L, Spranger S, Binder DC, Gritsina G, Lauing KL, Giles FJ, et al. Molecular pathways: targeting IDO1 and other tryptophan dioxygenases for cancer immunotherapy. Clin Cancer Res. 2015;21:5427–5433. doi: 10.1158/1078-0432.CCR-15-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ott M, Litzenburger UM, Rauschenbach KJ, Bunse L, Ochs K, Sahm F, et al. Suppression of TDO-mediated tryptophan catabolism in glioblastoma cells by a steroid-responsive FKBP52-dependent pathway. Glia. 2015;63:78–90. doi: 10.1002/glia.22734. [DOI] [PubMed] [Google Scholar]
- 33.Knox WE. Two mechanisms which increase in vivo the liver tryptophan peroxidase activity: specific enzyme adaptation and stimulation of the pituitary adrenal system. Br J Exp Pathol. 1951;32:462–469. [PMC free article] [PubMed] [Google Scholar]
- 34.Hassanain HH, Chon SY, Gupta SL. Differential regulation of human indoleamine 2,3-dioxygenase gene expression by interferons-gamma and -alpha. Analysis of the regulatory region of the gene and identification of an interferon-gamma-inducible DNA-binding factor. J Biol Chem. 1993;268:5077–5084. [PubMed] [Google Scholar]
- 35.Taylor MW, Feng GS. Relationship between interferon-gamma, indoleamine 2,3-dioxygenase, and tryptophan catabolism. FASEB J. 1991;5:2516–2522. [PubMed] [Google Scholar]
- 36.Yoshida R, Hayaishi O. Induction of pulmonary indoleamine 2,3-dioxygenase by intraperitoneal injection of bacterial lipopolysaccharide. Proc Natl Acad Sci USA. 1978;75:3998–4000. doi: 10.1073/pnas.75.8.3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fujigaki S, Saito K, Sekikawa K, Tone S, Takikawa O, Fujii H, et al. Lipopolysaccharide induction of indoleamine 2,3-dioxygenase is mediated dominantly by an IFN-gamma-independent mechanism. Eur J Immunol. 2001;31:2313–2318. doi: 10.1002/1521-4141(200108)31:8<2313::aid-immu2313>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 38.Babcock TA, Carlin JM. Transcriptional activation of indoleamine dioxygenase by interleukin 1 and tumor necrosis factor alpha in interferon-treated epithelial cells. Cytokine. 2000;12:588–594. doi: 10.1006/cyto.1999.0661. [DOI] [PubMed] [Google Scholar]
- 39.Mellor AL, Baban B, Chandler PR, Manlapat A, Kahler DJ, Munn DH. Cutting edge: CpG oligonucleotides induce splenic CD19+ dendritic cells to acquire potent indoleamine 2,3-dioxygenase-dependent T cell regulatory functions via IFN Type 1 signaling. J Immunol. 2005;175:5601–5605. doi: 10.4049/jimmunol.175.9.5601. [DOI] [PubMed] [Google Scholar]
- 40.Braun D, Longman RS, Albert ML. A two-step induction of indoleamine 2,3 dioxygenase (IDO) activity during dendritic-cell maturation. Blood. 2005;106:2375–2381. doi: 10.1182/blood-2005-03-0979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prendergast GC, Metz R, Muller AJ, Merlo LM, Mandik-Nayak L. IDO2 in immunomodulation and autoimmune disease. Front Immunol. 2014;5:585. doi: 10.3389/fimmu.2014.00585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478:197–203. doi: 10.1038/nature10491. [DOI] [PubMed] [Google Scholar]
- 43.Vogel CFA, Goth SR, Dong B, Pessah IN, Matsumura F. Aryl hydrocarbon receptor signaling mediates expression of indoleamine 2,3-dioxygenase. Biochem Biophys Res Commun. 2008;375:331–335. doi: 10.1016/j.bbrc.2008.07.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bankoti J, Rase B, Simones T, Shepherd DM. Functional and phenotypic effects of AhR activation in inflammatory dendritic cells. Toxicol Appl Pharmacol. 2010;246:18–28. doi: 10.1016/j.taap.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mautino MR, Metz RA, Jaipuri F, Waldo J, Kumar S, Marcinowicz-Flick A, et al. Abstract 1633: novel specific- and dual- tryptophan-2,3-dioxygenase (TDO) and indoleamine-2,3-dioxygenase (IDO) inhibitors for tumor immunotherapy. Cancer Res. 2014;74:1633. [Google Scholar]
- 46.Boyland E, Williams DC. The estimation of tryptophan metabolites in the urine of patients with cancer of the bladder. Biochem J. 1955;60:v. [PubMed] [Google Scholar]
- 47.Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281:1191–1193. doi: 10.1126/science.281.5380.1191. [DOI] [PubMed] [Google Scholar]
- 48.Platten M, Ho PP, Youssef S, Fontoura P, Garren H, Hur EM, et al. Treatment of autoimmune neuroinflammation with a synthetic tryptophan metabolite. Science. 2005;310:850–855. doi: 10.1126/science.1117634. [DOI] [PubMed] [Google Scholar]
- 49.Friberg M, Jennings R, Alsarraj M, Dessureault S, Cantor A, Extermann M, et al. Indoleamine 2,3-dioxygenase contributes to tumor cell evasion of T cell-mediated rejection. Int J Cancer. 2002;101:151–155. doi: 10.1002/ijc.10645. [DOI] [PubMed] [Google Scholar]
- 50.Grohmann U, Fallarino F, Puccetti P. Tolerance, DCs and tryptophan: much ado about IDO. Trends Immunol. 2003;24:242–248. doi: 10.1016/s1471-4906(03)00072-3. [DOI] [PubMed] [Google Scholar]
- 51.Wainwright DA, Dey M, Chang A, Lesniak MS. Targeting Tregs in malignant brain cancer: overcoming IDO. Front Immunol. 2013;4:116. doi: 10.3389/fimmu.2013.00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wainwright DA, Sengupta S, Han Y, Lesniak MS. Thymus-derived rather than tumor-induced regulatory T cells predominate in brain tumors. Neuro Oncol. 2011;13:1308–1323. doi: 10.1093/neuonc/nor134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Malchow S, Leventhal DS, Nishi S, Fischer BI, Shen L, Paner GP, et al. Aire-dependent thymic development of tumor-associated regulatory T cells. Science. 2013;339:1219–1224. doi: 10.1126/science.1233913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ye J, Qiu J, Bostick JW, Ueda A, Schjerven H, Li S, et al. Aryl hydrocarbon receptor preferentially marks and promotes gut regulatory T cells. Cell Rep. 2017;21:2277–2290. doi: 10.1016/j.celrep.2017.10.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med. 1999;189:1363–1372. doi: 10.1084/jem.189.9.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Seymour RL, Ganapathy V, Mellor AL, Munn DH. A high-affinity, tryptophan-selective amino acid transport system in human macrophages. J Leukoc Biol. 2006;80:1320–1327. doi: 10.1189/jlb.1205727. [DOI] [PubMed] [Google Scholar]
- 57.Hayashi T, Mo JH, Gong X, Rossetto C, Jang A, Beck L, et al. 3-Hydroxyanthranilic acid inhibits PDK1 activation and suppresses experimental asthma by inducing T cell apoptosis. Proc Natl Acad Sci USA. 2007;104:18619–18624. doi: 10.1073/pnas.0709261104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Munn DH. Blocking IDO activity to enhance anti-tumor immunity. Front Biosci. 2012;4:734–745. doi: 10.2741/e414. [DOI] [PubMed] [Google Scholar]
- 59.Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest. 2007;117:1147–1154. doi: 10.1172/JCI31178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mellor AL, Munn DH. Ido expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. 2004;4:762–774. doi: 10.1038/nri1457. [DOI] [PubMed] [Google Scholar]
- 61.Pallotta MT, Orabona C, Volpi C, Vacca C, Belladonna ML, Bianchi R, et al. Indoleamine 2,3-dioxygenase is a signaling protein in long-term tolerance by dendritic cells. Nat Immunol. 2011;12:870–878. doi: 10.1038/ni.2077. [DOI] [PubMed] [Google Scholar]
- 62.Wainwright DA, Balyasnikova IV, Chang AL, Ahmed AU, Moon K-S, Auffinger B, et al. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin Cancer Res. 2012;18:6110–6121. doi: 10.1158/1078-0432.CCR-12-2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhai L, Ladomersky E, Dostal CR, Lauing KL, Swoap K, Billingham LK, et al. Non-tumor cell IDO1 predominantly contributes to enzyme activity and response to CTLA-4/PD-L1 inhibition in mouse glioblastoma. Brain Behav Immun. 2017;62:24–29. doi: 10.1016/j.bbi.2017.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu Y, Liang X, Yin X, Lv J, Tang K, Ma J, et al. Blockade of IDO-kynurenine-AhR metabolic circuitry abrogates IFN-gamma-induced immunologic dormancy of tumor-repopulating cells. Nat Commun. 2017;8:15207. doi: 10.1038/ncomms15207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang L, Li L, Lemos H, Chandler PR, Pacholczyk G, Baban B, et al. Cutting edge: DNA sensing via the STING adaptor in myeloid dendritic cells induces potent tolerogenic responses. J Immunol. 2013;191:3509–3513. doi: 10.4049/jimmunol.1301419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lemos H, Mohamed E, Huang L, Ou R, Pacholczyk G, Arbab AS, et al. STING promotes the growth of tumors characterized by low antigenicity via IDO activation. Cancer Res. 2016;76:2076–2081. doi: 10.1158/0008-5472.CAN-15-1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. 2015;11:1018–1030. doi: 10.1016/j.celrep.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12:860–875. doi: 10.1038/nrc3380. [DOI] [PubMed] [Google Scholar]
- 69.Ravishankar B, Liu H, Shinde R, Chaudhary K, Xiao W, Bradley J, et al. The amino acid sensor GCN2 inhibits inflammatory responses to apoptotic cells promoting tolerance and suppressing systemic autoimmunity. Proc Natl Acad Sci USA. 2015;112:10774–10779. doi: 10.1073/pnas.1504276112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ravishankar B, Shinde R, Liu H, Chaudhary K, Bradley J, Lemos HP, et al. Marginal zone CD169+ macrophages coordinate apoptotic cell-driven cellular recruitment and tolerance. Proc Natl Acad Sci USA. 2014;111:4215–4220. doi: 10.1073/pnas.1320924111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ravishankar B, Liu H, Shinde R, Chandler P, Baban B, Tanaka M, et al. Tolerance to apoptotic cells is regulated by indoleamine 2,3-dioxygenase. Proc Natl Acad Sci USA. 2012;109:3909–3914. doi: 10.1073/pnas.1117736109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sharma MD, Shinde R, McGaha TL, Huang L, Holmgaard RB, Wolchok JD, et al. The PTEN pathway in Tregs is a critical driver of the suppressive tumor microenvironment. Sci Adv. 2015;1:e1500845. doi: 10.1126/sciadv.1500845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Solito S, Pinton L, Damuzzo V, Mandruzzato S. Highlights on molecular mechanisms of MDSC-mediated immune suppression: paving the way for new working hypotheses. Immunol Invest. 2012;41:722–737. doi: 10.3109/08820139.2012.678023. [DOI] [PubMed] [Google Scholar]
- 74.Yu J, Du W, Yan F, Wang Y, Li H, Cao S, et al. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol. 2013;190:3783–3797. doi: 10.4049/jimmunol.1201449. [DOI] [PubMed] [Google Scholar]
- 75.Zhang H, Maric I, DiPrima MJ, Khan J, Orentas RJ, Kaplan RN, et al. Fibrocytes represent a novel MDSC subset circulating in patients with metastatic cancer. Blood. 2013;122:1105–1113. doi: 10.1182/blood-2012-08-449413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jitschin R, Braun M, Buttner M, Dettmer-Wilde K, Bricks J, Berger J, et al. CLL-cells induce IDOhi CD14+HLA-DRlo myeloid-derived suppressor cells that inhibit T-cell responses and promote TRegs. Blood. 2014;124:750–760. doi: 10.1182/blood-2013-12-546416. [DOI] [PubMed] [Google Scholar]
- 77.Holmgaard RB, Zamarin D, Li Y, Gasmi B, Munn DH, Allison JP, et al. Tumor-expressed IDO recruits and activates MDSCs in a Treg-dependent manner. Cell Rep. 2015;13:412–424. doi: 10.1016/j.celrep.2015.08.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mellor AL, Sivakumar J, Chandler P, Smith K, Molina H, Mao D, et al. Prevention of T cell-driven complement activation and inflammation by tryptophan catabolism during pregnancy. Nat Immunol. 2001;2:64–68. doi: 10.1038/83183. [DOI] [PubMed] [Google Scholar]
- 79.Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J Immunother Cancer. 2014;2:3. doi: 10.1186/2051-1426-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fallarino F, Grohmann U, Hwang KW, Orabona C, Vacca C, Bianchi R, et al. Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol. 2003;4:1206–1212. doi: 10.1038/ni1003. [DOI] [PubMed] [Google Scholar]
- 81.Zhai L, Ladomersky E, Lauing KL, Wu M, Genet M, Gritsina G, et al. Infiltrating T cells increase IDO1 expression in glioblastoma and contribute to decreased patient survival. Clin Cancer Res. 2017;23:6650–6660. doi: 10.1158/1078-0432.CCR-17-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chamuleau ME, van de Loosdrecht AA, Hess CJ, Janssen JJ, Zevenbergen A, Delwel R, et al. High INDO (indoleamine 2,3-dioxygenase) mRNA level in blasts of acute myeloid leukemic patients predicts poor clinical outcome. Haematologica. 2008;93:1894–1898. doi: 10.3324/haematol.13113. [DOI] [PubMed] [Google Scholar]
- 83.Brandacher G, Perathoner A, Ladurner R, Schneeberger S, Obrist P, Winkler C, et al. Prognostic value of indoleamine 2,3-dioxygenase expression in colorectal cancer: effect on tumor-infiltrating T cells. Clinical Cancer Res. 2006;12:1144–1151. doi: 10.1158/1078-0432.CCR-05-1966. [DOI] [PubMed] [Google Scholar]
- 84.Astigiano S, Morandi B, Costa R, Mastracci L, D'Agostino A, Ratto GB, et al. Eosinophil granulocytes account for indoleamine 2,3-dioxygenase-mediated immune escape in human non-small cell lung cancer. Neoplasia. 2005;7:390–396. doi: 10.1593/neo.04658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Suzuki Y, Suda T, Furuhashi K, Suzuki M, Fujie M, Hahimoto D, et al. Increased serum kynurenine/tryptophan ratio correlates with disease progression in lung cancer. Lung Cancer. 2010;67:361–365. doi: 10.1016/j.lungcan.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 86.Feder-Mengus C, Wyler S, Hudolin T, Ruszat R, Bubendorf L, Chiarugi A, et al. High expression of indoleamine 2,3-dioxygenase gene in prostate cancer. Eur J Cancer. 2008;44:2266–2275. doi: 10.1016/j.ejca.2008.05.023. [DOI] [PubMed] [Google Scholar]
- 87.Okamoto A, Nikaido T, Ochiai K, Takakura S, Saito M, Aoki Y, et al. Indoleamine 2,3-dioxygenase serves as a marker of poor prognosis in gene expression profiles of serous ovarian cancer cells. Clin Cancer Res. 2005;11:6030–6039. doi: 10.1158/1078-0432.CCR-04-2671. [DOI] [PubMed] [Google Scholar]
- 88.Takao M, Okamoto A, Nikaido T, Urashima M, Takakura S, Saito M, et al. Increased synthesis of indoleamine-2,3-dioxygenase protein is positively associated with impaired survival in patients with serous-type, but not with other types of, ovarian cancer. Oncol Rep. 2007;17:1333–1339. [PubMed] [Google Scholar]
- 89.Ino K, Yoshida N, Kajiyama H, Shibata K, Yamamoto E, Kidokoro K, et al. Indoleamine 2,3-dioxygenase is a novel prognostic indicator for endometrial cancer. Br J Cancer. 2006;95:1555–1561. doi: 10.1038/sj.bjc.6603477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sakurai K, Enomoto K, Amano S, Kimura T, Sugito K, Kimizuka K, et al. [Study of indoleamine 2,3-dioxygenase expression in patients of esophageal squamous cell carcinoma] Gan To Kagaku Ryoho. 2004;31:1780–1782. [PubMed] [Google Scholar]
- 91.Riesenberg R, Weiler C, Spring O, Eder M, Buchner A, Popp T, et al. Expression of indoleamine 2,3-dioxygenase in tumor endothelial cells correlates with long-term survival of patients with renal cell carcinoma. Clin Cancer Res. 2007;13:6993–7002. doi: 10.1158/1078-0432.CCR-07-0942. [DOI] [PubMed] [Google Scholar]
- 92.Ishio T, Goto S, Tahara K, Tone S, Kawano K, Kitano S. Immunoactivative role of indoleamine 2,3-dioxygenase in human hepatocellular carcinoma. J Gastroenterol Hepatol. 2004;19:319–326. doi: 10.1111/j.1440-1746.2003.03259.x. [DOI] [PubMed] [Google Scholar]
- 93.Pan K, Wang H, Chen MS, Zhang HK, Weng DS, Zhou J, et al. Expression and prognosis role of indoleamine 2,3-dioxygenase in hepatocellular carcinoma. J Cancer Res Clin Oncol. 2008;134:1247–1253. doi: 10.1007/s00432-008-0395-1. [DOI] [PubMed] [Google Scholar]
- 94.Piras F, Colombari R, Minerba L, Murtas D, Floris C, Maxia C, et al. The predictive value of CD8, CD4, CD68, and human leukocyte antigen-D-related cells in the prognosis of cutaneous malignant melanoma with vertical growth phase. Cancer. 2005;104:1246–1254. doi: 10.1002/cncr.21283. [DOI] [PubMed] [Google Scholar]
- 95.Zhai L, Ladomersky E, Lauing KL, Wu M, Genet M, Gritsina G, et al. Infiltrating T cells increase IDO1 expression in glioblastoma and contribute to decreased patient survival. Clin Cancer Res. 2017;23:6650–6660. doi: 10.1158/1078-0432.CCR-17-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Röhrig UF, Majjigapu SR, Vogel P, Zoete V, Michielin O. Challenges in the Discovery of Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitors. J Med Chem. 2015;58:9421–9437. doi: 10.1021/acs.jmedchem.5b00326. [DOI] [PubMed] [Google Scholar]
- 97.Iversen TZ, Engell-Noerregaard L, Ellebaek E, Andersen R, Larsen SK, Bjoern J, et al. Long-lasting disease stabilization in the absence of toxicity in metastatic lung cancer patients vaccinated with an epitope derived from indoleamine 2,3 dioxygenase. Clin Cancer Res. 2014;20:221–232. doi: 10.1158/1078-0432.CCR-13-1560. [DOI] [PubMed] [Google Scholar]
- 98.Soliman HH, Jackson E, Neuger T, Dees CE, Harvey DR, Han H, et al. A first in man phase I trial of the oral immunomodulator, indoximod, combined with docetaxel in patients with metastatic solid tumors. Oncotarget. 2014;5:8136–8146. doi: 10.18632/oncotarget.2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Perez RP, Riese MJ, Lewis KD, Saleh MN, Daud A, Berlin J, et al. Epacadostat plus nivolumab in patients with advanced solid tumors: preliminary phase I/II results of ECHO-204. J Clin Oncol. 2017;35:3003–3003. [Google Scholar]
- 100.Siu LL, Gelmon K, Chu Q, Pachynski R, Alese O, Basciano P, et al. Abstract CT116: BMS-986205, an optimized indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor, is well tolerated with potent pharmacodynamic (PD) activity, alone and in combination with nivolumab (nivo) in advanced cancers in a phase 1/2a trial. Cancer Res. 2017;77:CT116–CT116. [Google Scholar]
- 101.Soliman HH, Jackson E, Neuger T, Dees EC, Harvey RD, Han H, et al. A first in man phase I trial of the oral immunomodulator, indoximod, combined with docetaxel in patients with metastatic solid tumors. Oncotarget. 2014;5:8136–8146. doi: 10.18632/oncotarget.2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Soliman HH, Minton SE, Han HS, Ismail-Khan R, Neuger A, Khambati F, et al. A phase I study of indoximod in patients with advanced malignancies. Oncotarget. 2016;7:22928–22938. doi: 10.18632/oncotarget.8216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Spira AI, Hamid O, Bauer TM, Borges VF, Wasser JS, Smith DC, et al. Efficacy/safety of epacadostat plus pembrolizumab in triple-negative breast cancer and ovarian cancer: Phase I/II ECHO-202 study. J Clin Oncol. 2017;35:1103–1103. [Google Scholar]
- 104.Gangadhar TC, Hamid O, Smith DC, Bauer TM, Wasser JS, Luke JJ, et al. Preliminary results from a phase I/II study of epacadostat (incb024360) in combination with pembrolizumab in patients with selected advanced cancers. J Immunother Cancer. 2015;3:O7. [Google Scholar]
- 105.Tang T, Gill HS, Ogasawara A, Tinianow JN, Vanderbilt AN, Williams SP, et al. Preparation and evaluation of L- and D-5-[18F]fluorotryptophan as PET imaging probes for indoleamine and tryptophan 2,3-dioxygenases. Nucl Med Biol. 2017;51:10–17. doi: 10.1016/j.nucmedbio.2017.05.001. [DOI] [PubMed] [Google Scholar]
- 106.Xin Y, Cai H. Improved radiosynthesis and biological evaluations of L- and D-1-[18F]fluoroethyl-tryptophan for pet imaging of ido-mediated kynurenine pathway of tryptophan metabolism. Mol Imaging Biol. 2017;19:589–598. doi: 10.1007/s11307-016-1024-z. [DOI] [PubMed] [Google Scholar]
- 107.Lob S, Konigsrainer A, Schafer R, Rammensee HG, Opelz G, Terness P. Levo- but not dextro-1-methyl tryptophan abrogates the IDO activity of human dendritic cells. Blood. 2008;111:2152–2154. doi: 10.1182/blood-2007-10-116111. [DOI] [PubMed] [Google Scholar]
- 108.Löb S, Königsrainer A, Zieker D, Brücher BDM, Rammensee H-G, Opelz G, et al. IDO1 and IDO2 are expressed in human tumors: levo- but not dextro-1-methyl tryptophan inhibits tryptophan catabolism. Cancer Immunol Immunother. 2009;58:153–157. doi: 10.1007/s00262-008-0513-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.