

Abstract
Metaflammation is responsible for several metabolic syndromes, such as type 2 diabetes. However, the mechanisms by which metabolic disorders trigger metaflammation remain unclear. We identified a cell type-specific downregulation of CD1d expression in M2 macrophages during the progression of obesity prior to the onset of inflammation in visceral adipose tissues. A reduction in CD1d expression influenced the ability of M2 macrophages to present antigens and caused a change in antigen-presenting cells from M2 macrophages to M1 macrophages. With CD1d conditional knockout (KO) mice, we further demonstrated that natural killer T (NKT) cell activation by M2 macrophages inhibited metaflammation and insulin resistance by promoting Th2 responses and M2 polarization in visceral adipose tissues of obese mice, whereas NKT cell activation by M1 macrophages exacerbated metaflammation and insulin resistance by promoting Th1 responses and inhibiting M2 polarization. Our results suggest that an M2-specific reduction of CD1d is an initiating event that switches NKT cell-mediated immune responses and disrupts the immune balance in visceral adipose tissues in obese mice.
Keywords: CD1d, macrophage, metaflammation, NKT cells
INTRODUCTION
Obesity-associated chronic inflammation, metaflammation, is critically linked to insulin resistance and type 2 diabetes. Specifically, inflammation in adipose tissue has a vital role in the development of insulin resistance.1,2 Considerable progress has been made in understanding the mechanisms of metaflammation in adipose tissue. Metabolic disorders cause the activation of inflammatory pathways, such as NOD, TLR, NFκB and NLRP3. Moreover, the accumulation of proinflammatory cells in the adipose tissue of obese mice, such as classically activated macrophages (M1) and CD8+ T cells, further promotes metaflammation. There are also abundant alternatively activated macrophages (M2), ILC2, eosinophils and Tregs in the adipose tissue of lean animals, which inhibit inflammation by secreting the Th2 cytokines IL4, IL10 and IL13.3,4 Thus, the onset of metaflammation is a result of an imbalance between anti- and proinflammatory responses. The underlying mechanisms that disrupt the immune balance remain unclear.
Macrophages are major sources of mediators that cause insulin resistance in adipose tissues.5,6 Alteration in macrophage composition is an early event that indicates metaflammation. M2 macrophages have anti-inflammatory functions and predominantly comprise the adipose tissue macrophages in lean animals.3,4 Eosinophils, which are sustained by ILC2 in adipose tissues, promote the polarization of M2 macrophages by secreting IL4.7,8 Obesity increases the proportion of M1 macrophages, which promote metaflammation and insulin resistance by releasing TNFα, IL1β and IL6. The Th1 cytokines IFNγ and TNFα from CD8+ T cells, NK cells and adipocytes promote the polarization of M1 macrophages.3,9 In addition, saturated free fatty acid and microbial-derived LPS in obese mice also promote M1 polarization via TLR4 activation.10 The cells and molecules that regulate macrophage polarization have been demonstrated; however, the mechanisms by which metabolic disorders change adipose macrophage composition remain unclear.
Natural killer T (NKT) cells recognize CD1d-presented lipid antigens. As innate-like T lymphocytes and tissue-resident cells,11,12 NKT cells participate in the first-line of host defense. Furthermore, NKT cells have been shown to regulate inflammatory diseases, including non-alcoholic fatty liver disease,13 alcoholic steatohepatitis13,14 and asthma.15,16 The roles of NKT cells in the regulation of metaflammation have been reported by several labs. However, the results are controversial.17,18,19,20 Some groups indicate a protective role of iNKT cells via the promotion of Th2 responses and accumulation of M2 macrophages and Tregs.18,19,21,22 However, another group demonstrated a pathogenic role of iNKT cells via the promotion of Th1 responses.20 The reasons for these discrepancies are not clear. NKT cells may secrete both Th1 and Th2 cytokines. However, the type of antigen-presenting cells (APCs), type of lipid antigens and NKT subsets would all contribute to biased immune responses.23,24,25 Here, we determined that M2-specific downregulation of CD1d expression in adipose tissues of obese mice influenced the antigen-presenting capability of M2 macrophages and caused changes in APCs from M2 to M1 macrophages. The activation of NKT cells by M2 macrophages or M1 macrophages resulted in differently polarized cytokine responses. Using CD1d conditional knockout (KO) mice, we showed that NKT cells had opposite roles (protective vs pathogenic) in the regulation of metaflammation and insulin resistance during the progression of obesity as a result of the change in APCs. Thus, the M2-specific reduction of CD1d expression in the adipose tissues of obese mice switched the NKT cell-mediated immune responses from Th2 to Th1 and disrupted the balance between the anti- and proinflammatory responses.
MATERIALS AND METHODS
Mice
WT mice were purchased from the Model Animal Research Center of Nanjing University. Cd1d1 conditional knockout mice and Vα14 Tg.cxcr6gfp/+ mice have previously been described and were provided by Dr Albert Bendelac.23,26 All mice were male and on the C57BL/6J background. In general, the male mice were fed a normal chow diet (NCD) (10 kcal% fat, Research Diets, New Brunswick, NJ, USA) or high fat diet (HFD) (60 kcal% fat, Research Diets, New Brunswick, NJ, USA) from 7 weeks old. The mice were housed under specific pathogen-free conditions. All animal procedures were approved by the USTC Institutional Animal Care and Use Committee. Moreover, all experiments were performed in accordance with the approved guidelines.
Preparation of stromal vascular cells
Visceral adipose tissue (VAT) was minced and centrifuged to remove erythrocytes and subsequently digested with collagenase type II (Sigma-Aldrich, St Louis, MO, USA). Cell suspensions were then passed through a 100-μm stainless steel mesh and centrifuged to remove floating adipocytes. The cell pellets were resuspended in 40% Percoll and centrifuged. Red blood cells were lysed by erythrocyte lysis buffer (Solarbio, Beijing, China).
Cell stimulation assays
Cells were cultured in RPMI-1640 medium supplemented with 10% FBS and 50 μm β-mercaptoethanol. M2 macrophages and M1 macrophages from NCD mice and HFD mice (50 000 cells per well) were sorted and cocultured independently with sorted iNKT cells (90 000 cells per well) in the presence of α-GalCer (1.5 μg/ml, Avanti Polar Lipids, Alabaster, AL, USA) or DMSO (vehicle control) for 24 h. To investigate the capability of adipose iNKT cells to produce cytokines, iNKT cells were sorted from the VATs of Lyz2 Δ/Δ mice, Cd11c Δ/Δ mice and littermate controls on NCD or HFD and stimulated (2000 cells per well) with PMA (50 ng/ml) and ionomycin (1 μm) overnight. Cytokines in the supernatants were detected using CBA kits (BD Biosciences, Franklin Lakes, NJ, USA).
Glucose tolerance tests and insulin tolerance tests
For glucose tolerance tests (GTTs), the mice were injected (i.p.) with 2 g glucose (Sigma-Aldrich, St Louis, MO, USA) per kg body weight after 8-h fasts. Insulin tolerance tests were performed by injecting (i.p.) 0.75 U insulin (Thermo Fisher Scientific, Waltham, MA, USA) per kg body weight after 6-h fasts.
Western blot
The mice were injected with insulin (1 U/kg) 10 min prior to the removal of visceral adipose tissues, livers or muscles. Antibodies against IRS1 (CST, Danvers, MA, USA), pSer307-IRS1 (Santa Cruz Biotech, Dallas, TX, USA), AKT (CST), pThr308-AKT (CST) and GAPDH (Proteintech, Chicago, IL, USA) were used to measure protein levels via western blot.
Flow cytometry analysis
Stromal vascular cells (SVCs) were blocked with anti-CD16/32 (1 μg/ml) and subsequently stained with monoclonal antibodies on ice. For intracellular CD1d measurements, surface CD1d was blocked with purified anti-CD1d (2.5 μg/ml), and intracellular CD1d was stained with anti-CD1d-PE after fixation and permeabilization. To measure the ability of APCs to present antigen, mice were injected with α-GalCer (5 μg per mice) or PBS buffer overnight, and the antibody L363 was used to detect the CD1d-α-GalCer complex on the cell surface. The monoclonal antibodies were as follows: purified anti-CD16/32 (93), anti-CD11c (N418), anti-CD90.2 (30-H12), anti-CD11b (M1/70), anti-CD206 (C068C2), anti-NK1.1 (PK136), anti-CD1d (1B1), anti-B220 (RA3-6B2), anti-Siglec-F (E50-2440), anti-CD4 (GK1.5), anti-TCRβ (H57-597), anti-CD8α (53-6.7), anti-F4/80 (BM8) and anti-NK1.1 (PK136). All antibodies were purchased from BioLegend (San Diego, CA, USA). CD1d-PBS57 tetramer was provided by the NIH Tetramer Core Facility. Cells were washed and acquired using a FACSVerse flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA), and the data were analyzed with FlowJo 7.6 software (TreeStar, Ashland, OR, USA).
Confocal microscopy
To investigate the cell type that presented α-GalCer, Vα14 Tg.cxcr6gfp/+ mice on NCD or HFD were injected with α-GalCer (5 μg/mice) or PBS buffer for 12 h, and VATs were extracted and minced into 1–2 mm pieces for staining. To investigate the cell types that presented endogenous antigen, GFP+ iNKT cells were enriched from the livers and spleens of Vα14 Tg.cxcr6gfp/+ mice with anti-CD4-PE and anti-PE MicroBeads and were transferred (5 million per mouse) to NCD mice and HFD mice i.p. for 8 h. The purity of iNKT cells among GFP+ cells was ~83% (Supplementary Table S3). VATs were extracted from the recipient mice and minced into 1–2 mm pieces. Adipose tissues were blocked with 5% BSA in PBS buffer and subsequently stained with anti-CD11c-PE and anti-F4/80-APC. For tetramer staining, adipose tissues were initially stained with CD1d-PBS57 tetramers-PE for 2 h on a circular shaker at room temperature, followed by staining with anti-CD3-APC on a circular shaker at 4 °C overnight. Unload CD1d tetramer-PE was used as a negative control. Tissue samples were fixed and mounted with ProLong Gold anti-fade reagent (Thermo Fisher Scientific) prior to obtaining images with an inverted confocal microscope Zeiss LSM 710 (Carl Zeiss, Oberkochen, Germany).
Quantitative RT-PCR analysis
The primers used are listed in the Supplemental Experimental Procedures.
Statistical analysis
Error bars represent the s.e.m. The significance of differences between two groups was determined by Student’s t-test.
RESULTS
M2-specific reduction of CD1d expression occurs prior to the onset of metaflammation in visceral adipose tissues of obese mice
In our studies, 16 weeks of HFD feeding significantly increased the body weight and caused insulin resistance as indicated by the GTTs and insulin tolerance tests (ITTs) (Supplementary Table S1). In these mice, metaflammation was identified in the VATs but not in the livers (Supplementary Table S1). The onset of liver metaflammation required prolonged HFD feeding and was detected after 26 weeks (Supplementary Table S1e). Thus, adipose tissue was the primary tissue that contributed to systemic insulin resistance. In VATs, CD1d was expressed in M2 macrophages (CD11b+, F4/80+, CD11c−, CD206+), M1 macrophages (CD11b+, F4/80+, CD11c+, CD206−) and dendritic cells (DCs, CD11c+, CD11b−). Moreover, the surface CD1d on the M2 macrophages was 3- to 5-fold higher than on the M1 macrophages and DCs (Figure 1a). Neutrophils and eosinophils expressed 10-fold less CD1d than the M1 macrophages (Figure 1a). Furthermore, DCs and macrophages are well established to be more effective in presenting antigens than neutrophils and eosinophils. Thus, M2 macrophages, M1 macrophages and DCs, in contrast to neutrophils and eosinophils, were potential APCs that activated NKT cells in VATs. Interestingly, the 16-week HFD induced a reduction of the CD1d expression in the M2 macrophages in VATs, whereas no difference in the CD1d expression was identified in the M1 macrophages or DCs (Figures 1b and c). A similar M2-specific reduction of the CD1d expression was identified in subcutaneous adipose tissues (SATs; Figures 1d and e). In the spleens and livers, HFD did not downregulate the CD1d expression in the main APCs, including splenic DCs, red pulp macrophages and Kupffer cells (Figure 1f). To investigate the relationship between M2-specific CD1d downregulation and metaflammation, we compared the kinetics of CD1d reduction, M1/M2 ratio elevation and inflammatory gene expression in the VATs of HFD mice. Eight-week HFD feeding substantially reduced the CD1d expression in the M2 macrophages, whereas the M1/M2 ratio, TNFα and IL1β expressions were significantly increased after the 12-week HFD feeding (Figure 1g). Eight-week HFD feeding only exhibited a minor effect on the M1/M2 ratio. Altogether, our results indicated that obesity induced an M2-specific CD1d reduction in VATs, which occurred prior to the onset of metaflammation.
Figure 1.
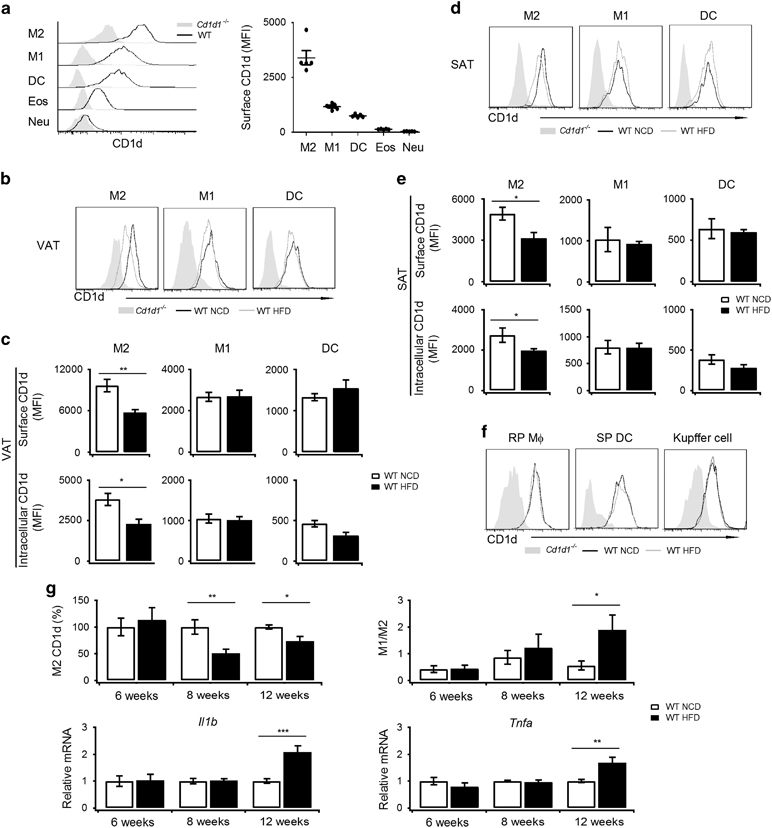
M2-specific reduction of CD1d occurs prior to the onset of metaflammation. (a) CD1d surface expression in the indicated cells in VATs was measured via FACS (black line). Cd1d1 −/− mice were used as negative controls (gray solid). Mean fluorescence intensity is presented on the right (n=5 mice per group); Cd1d1 −/− mice were used to measure the non-specific staining of CD1d, which was subtracted as background. (b–f) CD1d expression in mice on NCD or HFD for 16 weeks. CD1d surface expression on the indicated cells in VATs (b), SATs (d), spleens and livers (f) was measured via FACS (NCD, black line; HFD, gray line). Cd1d1 −/− mice were used as negative controls (gray solid). Mean fluorescence intensity of surface and intracellular CD1d in the indicated cells in VATs (c) or SATs (e) (NCD, white bar; HFD, black bar); Cd1d1 −/− mice were used to measure the non-specific staining of CD1d, which was subtracted as background. The data are pooled from two independent experiments with 5 to 6 mice per group. (g) Surface CD1d on M2 macrophages, M1/M2 ratio, and mRNA level of Il1β and Tnfα in VATs of mice on NCD or HFD for 6 weeks, 8 weeks and 12 weeks (n=5–8 mice per group; NCD, white bar; HFD, black bar). Data are represented as the mean±the s.e.m. *P<0.05, **P<0.01, ***P<0.001. HFD, high fat diet; NCD, normal chow diet; SATs, subcutaneous adipose tissues; VATs, visceral adipose tissues.
M2-specific reduction of CD1d expression changes the type of APCs from M2 to M1
CD1d molecules present lipid antigens to NKT cells. An M2-specific reduction of CD1d expression would influence the ability of M2 macrophages to present antigens. To investigate the antigen-presenting capability of APCs in VATs, mice were injected with the lipid-antigen α-galactosylceramide (α-GalCer), and the antibody L363 was used to detect the CD1d-α-GalCer complex on the cell surface. Surprisingly, adipose DCs presented substantially less α-GalCer than macrophages (Figures 2a and b). Moreover, M2 macrophages were more effective in antigen presentation than M1 macrophages in lean mice (Figures 2a and b). Consistent with the reduction of CD1d expression, decreased antigen presentation was identified in M2 macrophages in the mice on HFD (Figure 2a). Moreover, the ability to present α-GalCer was not influenced by HFD in M1 macrophages or DCs. The cell type-specific reduction of antigen presentation may alter the type of APCs. To further determine the type of APCs that activate NKT cells in vivo, we investigated the colocalizations between iNKT cells and distinct APCs in response to α-GalCer. Vα14 Tg.cxcr6gfp/+ mice have previously been used to investigate the location of iNKT cells in vivo.12,27 Approximately 80–90% of GFP+ cells are iNKT cells in the livers and spleens of these mice, whereas the percentage was substantially lower (<50%) in VATs. Thus, GFP could not be used to indicate iNKT cells in adipose tissues, with the exception of in the presence of the exogenous antigen α-GalCer, which induces interactions between iNKT cells and APCs. VATs were stained with fluorescent antibodies against CD11c and F4/80 to distinguish candidate APCs, M2, M1 and DCs. A few F4/80+ CD11c− M2 macrophages were dispersed in the VATs of the mice fed NCD, whereas F4/80+ CD11c+ M1 macrophages and F4/80− CD11c+ DCs were less observed. GFP+ cells and APCs showed no colocalization in the lean mice without α-GalCer (Figure 2c). α-GalCer recruited GFP+ iNKT cells to M2 macrophages in the mice fed on NCD (Figure 2c). In the mice fed on HFD, GFP+ iNKT cells were recruited to M1 macrophages by α-GalCer (Figure 2c); however, there were abundant M2 macrophages in VATs (Figure 2d). These results indicated that the type of APCs that presented exogenous antigen was changed in the HFD mice as a result of the reduced antigen-presenting capability of M2 macrophages. We subsequently investigated the type of APCs that presented endogenous antigen to NKT cells in obese mice. GFP+ iNKT cells were isolated from the spleen and liver of Vα14 Tg. cxcr6gfp/+ mice and transferred to recipient mice on NCD or HFD (Figure 2e). Most of these GFP+ cells were CD3+ tetramer+ in the VATs of the recipient mice, which indicates iNKT cells (Figure 2f). The majority of the iNKT cells (62%) were not colocalized with the stained APCs in the mice fed on NCD (Figures 2g and i). Interestingly, an 8-week HFD resulted in the accumulation of M2 macrophages in VATs and increased the colocalizations between iNKT cells and M2 macrophages (up to 68%, Figures 2g and i). Increased iNKT-M1 colocalizations were also identified at a lower percentage (15%). At the later stage of obesity (16 weeks on HFD), the colocalizations of iNKT cells with M1 macrophages increased to 52% (Figures 2g and i). However, the colocalizations between iNKT cells and M2 macrophages declined to 30% (Figures 2g and i). Furthermore, the colocalizations between the GFP+ cells and macrophages depended on the CD1d expression. In mice, the cd1d1 gene was deleted in macrophages and DCs (Supplementary Table S4), and iNKT-macrophage colocalizations induced by an HFD were not observed (Figure 2h). These results further proved that the GFP+ cells colocalized with macrophages were iNKT cells. Moreover, the colocalizations between iNKT cells and macrophages were dependent on antigen recognition in the HFD mice in contrast to the NCD mice. Moreover, iNKT-M2 colocalizations decreased substantially faster than the decrease in M2 macrophages (Figure 2j). Thus, a changed macrophage composition could not explain the change in APCs. A reduction of CD1d expression in M2 macrophages contributed to the change in APCs. Furthermore, colocalizations between iNKT cells and DCs were rarely identified in our studies (Figure 2i). DCs were substantially less effective in antigen presentation than macrophages (Figures 2a and b). These findings excluded the role of DCs in NKT cell activation in VATs.
Figure 2.
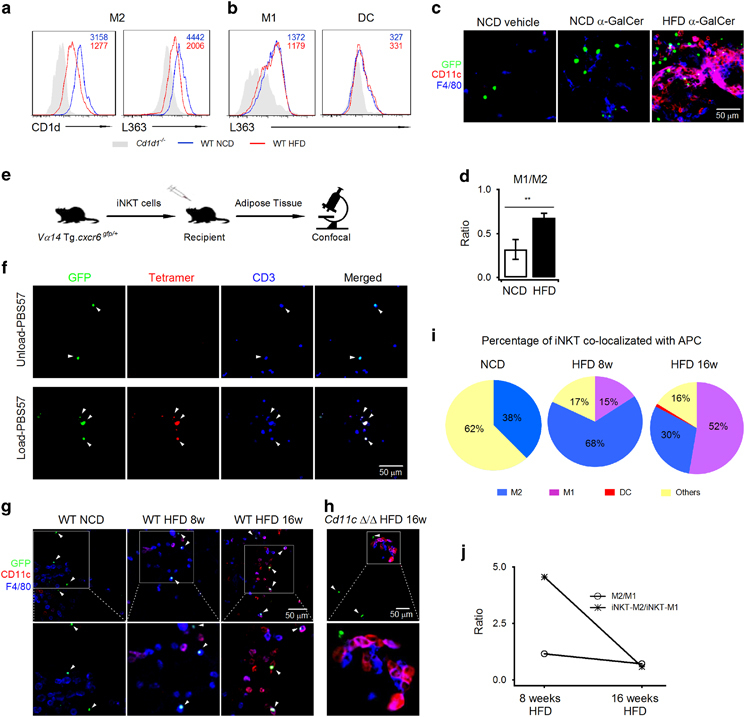
Change in APCs from M2 macrophages to M1 macrophages during progression of obesity. (a, b) Surface CD1d (a) and CD1d-α-GalCer (a, b) on the indicated cells in VATs of mice on NCD (blue line, n=4) or HFD (red line, n=3) for 16 weeks. Cd1d1 −/− mice were used as negative controls (gray solid). The mean fluorescence intensity of CD1d or the CD1d-α-GalCer complex was indicated in each panel. (c) α-GalCer induced colocalizations between iNKT cells (green) and M2 macrophages (blue), M1 macrophages (purple) or DCs (red) in VATs of Vα14 Tg.cxcr6gfp/+ mice on NCD or HFD for 12 weeks (n=3–4 mice per group). Scale bar, 50 μm. (d) M1/M2 ratio in VATs of mice in c. (e, f) Transferred iNKT cells were detected by tetramer staining in VATs of recipient mice. Scale bar, 50 μm. (g) Colocalizations between transferred iNKT cells (green) and M2 macrophages (blue), M1 macrophages (purple) or DCs (red) in VATs of recipient mice on NCD or HFD for the indicated times. Scale bar, 50 μm. The data were representative of three to four mice per group. (h) Colocalizations between transferred iNKT cells and APCs deficient in CD1d expression. Scale bar, 50 μm. (i) Percentages of iNKT cells colocalized with the indicated cells in g. (j) Ratio of iNKT cells that interacted with M2 macrophages/iNKT cells that interacted with M1 macrophages and the ratio of M2/M1 in VATs of recipient mice on HFD for 8 weeks or 16 weeks. Cell numbers of different macrophages were counted in images presented in (g). Data are represented as the mean±the s.e.m. APCs, antigen-presenting cells; HFD, high fat diet; NCD, normal chow diet; SATs, subcutaneous adipose tissues; VATs, visceral adipose tissues.
Opposite roles for NKT cells in the regulation of glucose tolerance are determined by types of APCs
To investigate the influence of APCs on NKT cell-mediated immune responses, M2 macrophages and M1 macrophages sorted from NCD mice, and HFD mice were cocultured independently with iNKT cells in the presence of α-GalCer. Compared with M1 macrophages, M2 macrophages induced the production of more IL4 and IL13 and less IFNγ by iNKT cells. Moreover, the production of cytokines was completely inhibited by the absence of α-GalCer or the presence of anti-CD1d antibody, which indicates their dependence on antigen presentation (Figure 3a). Thus, iNKT cell activation by M2 macrophages polarized iNKT cells toward Th2 responses, whereas M1 macrophages polarized iNKT cells toward Th1 responses. When obesity changed the type of APCs in vivo, it would also change the functions of NKT cells. To further prove this hypothesis, we crossed Cd1d1 fl/fl mice with Lyz2 cre mice or Cd11c cre mice to delete CD1d expression in different APCs. In the Lyz2 Δ/Δ mice, the CD1d expression was deficient in M2 macrophages. In the Cd11c Δ/Δ mice, the expression of CD1d was ablated in both M2 and M1 macrophages, as well as in DCs (Supplementary Table S4). However, DCs were not effective APCs in the activation of NKT cells in VATs (Figures 2b and i). Thus, the differences identified between the Lyz2 Δ/Δ mice and Cd11c Δ/Δ mice were caused by the deficiency of CD1d expression in M1 macrophages. The deletion of CD1d in target APCs was not 100% in the Lyz2 Δ/Δ mice and Cd11c Δ/Δ mice; however, it was sufficient to inhibit the activation of NKT hybridoma cells (Supplementary Table S4). Previous studies have demonstrated that conditional CD1d deletion in APCs would not influence the functions and development of iNKT cells.23 Both Lyz2 Δ/Δ mice and Cd11c Δ/Δ mice fed on NCD exhibited normal macrophage composition and iNKT cell development in adipose tissue (Supplementary Table S4). Moreover, in both NCD mice and HFD mice, conditional CD1d deletion did not change the Th1/Th2 cytokine profiles of adipose iNKT cells, as indicated by the production of cytokines after PMA plus ionomycin stimulation (Figures 3b and c). Interestingly, impaired glucose tolerance tests (GTTs) were observed in Lyz2 Δ/Δ mice on HFD as early as 8 weeks; however, these impairments disappeared after 20 weeks (Figure 3d). By contrast, improved GTTs were observed in Cd11c Δ/Δ mice on HFD after 16 and 20 weeks but not after 8 weeks (Figure 3d). These results indicated opposite roles of NKT cells in the regulation of blood glucose in obese mice through interactions with distinct APCs. NKT cell activation by M2 macrophages protected obese mice against glucose intolerance, whereas NKT cell activation by M1 macrophages exacerbated glucose intolerance in obese mice. Furthermore, the kinetic differences suggested sequential interactions between NKT cells and different macrophage populations in obese mice. M2 macrophages were primary APCs that activated NKT cells at an early stage of obesity, whereas M1 macrophages were dominant APCs that activated NKT cells at a late stage of obesity.
Figure 3.
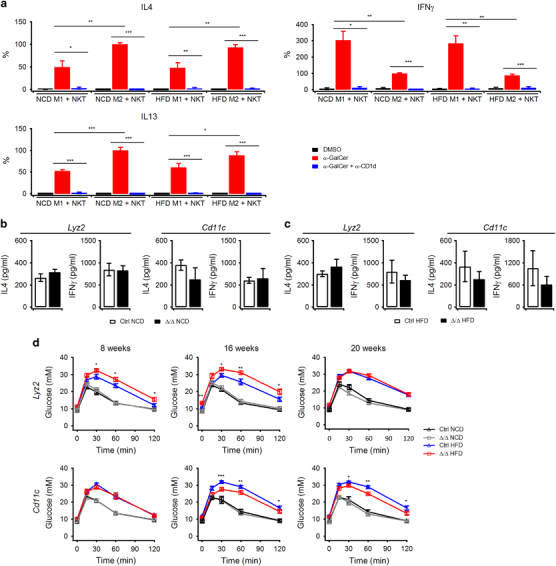
Opposite roles for NKT cells in the regulation of glucose homeostasis are determined by types of APCs. (a) Cytokines produced by iNKT cells cocultured with M2 or M1 macrophages in the presence of α-GalCer (1.5 μg/ml), DMSO (vehicle control) or anti-CD1d antibody (5 μg/ml). The data are representative of three experiments. (b, c) Cytokines produced by adipose iNKT cells from NCD mice (b) or HFD mice (c) after stimulation with PMA and ionomycin (littermate controls, white bar; Lyz2 Δ/Δ and Cd11c Δ/Δ, black bar). The data were representative of three independent experiments. (d) Mice of the indicated genotypes were on NCD or HFD for 8 weeks, 16 weeks or 20 weeks, and underwent a glucose tolerance test (n=8–15 pooled mice per group; littermate controls, black triangle; conditional KO mice, gray square). Data are represented as the mean±the s.e.m. *P<0.05, **P<0.01, ***P<0.001. APCs, antigen-presenting cells; HFD, high fat diet; NCD, normal chow diet.
Inhibiting NKT–M2 interactions reduces M2 polarization and induces metaflammation and insulin resistance in obese mice
Lyz2 Δ/Δ mice on HFD had similar body weights, visceral adipose tissue weights, and SVC numbers per gram of fat as their littermate controls (Figures 4a–c); however, they showed slightly increased serum insulin and exacerbated insulin resistance as indicated by slower glucose clearance in ITTs (Figures 4d and e). Consistently, increased phosphorylation of Ser307-IRS1 and decreased phosphorylation of Thr308-AKT after insulin injection were identified in the VATs of the Lyz2 Δ/Δ mice on HFD (Figure 4f). Moreover, altered pSer307-IRS1 and pThr308-AKT after insulin injection were also detected in the livers and muscles of the Lyz2 Δ/Δ mice on HFD (Supplementary Table S6), which indicates an influence of CD1d deletion on systemic insulin resistance. To further investigate how NKT cells regulate insulin resistance through interactions with M2 macrophages, we isolated SVCs from VATs and compared the proportions of immune cells. Elevated M1 to M2 ratios were identified in the Lyz2 Δ/Δ mice (Figure 4g). Moreover, a cell number analysis also indicated a significant decline of M2 macrophages (Figure 4h). By contrast, the frequencies of DC, NKT, CD8 T, Treg, ILC2, eosinophils and NK cells were unchanged (Figure 4i). Furthermore, we identified substantially decreased IL10 and Arg1 and elevated IFNγ and iNOS at the mRNA levels in the VATs of the Lyz2 Δ/Δ mice (Figure 4j). IL10 has been reported to promote M2 polarization.4 Decreased IL10 and elevated IFNγ may account for the diminished M2 macrophages. Thus, interfering with interactions between NKT cells and M2 macrophages exacerbates insulin resistance by inhibiting M2 polarization and promoting metaflammation in the VATs of HFD-fed mice.
Figure 4.
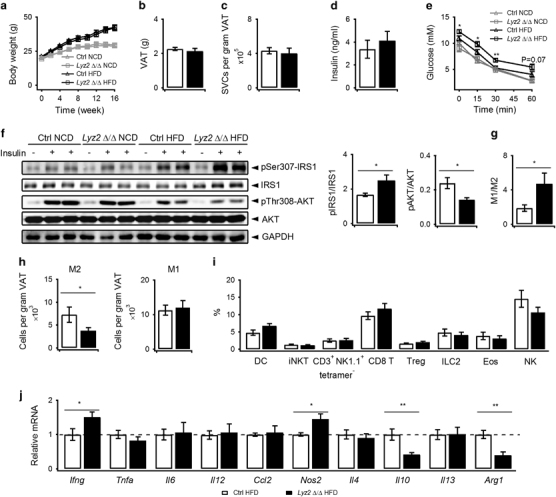
CD1d deletion in M2 macrophages inhibits M2 polarization and promotes metaflammation. (a) Body weights of Lyz2 Δ/Δ mice (square) and littermate controls (triangle) were monitored for 16 weeks on NCD or HFD (n=9–12 pooled mice per group). (b–d) Weights of VAT (n=13–14 pooled mice per group), SVC numbers per gram of VAT (n=6 pooled mice per group), and serum insulin concentrations (n=11 pooled mice per group) in Lyz2 Δ/Δ mice (black bar) and littermate controls (white bar) were measured after 16 weeks on HFD. (e) Lyz2 Δ/Δ mice (square) and littermate controls (triangle) were fed on NCD or HFD for 16 weeks and underwent an insulin tolerance test (ITT, n=7 pooled mice per group). (f) Levels of IRS1, IRS1 phospho-Ser307 (pSer307-IRS1), AKT and AKT phosphor-Thr308 (pThr308-AKT) with or without insulin injection in VATs of mice fed on NCD or HFD for 14–16 weeks were detected via western blot (left). Ratios of pIRS1/IRS1 and pAKT/AKT with insulin in HFD mice are presented on the right (littermate controls, white bar; Lyz2 Δ/Δ mice, black bar; n=4 pooled mice per group). (g–i) Proportions (g n=12–15 pooled mice per group; i n=12–15 or n=5–10 pooled mice per group) and absolute numbers (h n=5–7 pooled mice per group) of the indicated cells in VATs after 16 weeks on HFD (littermate controls, white bar; Lyz2 Δ/Δ mice, black bar). (j) mRNA levels of cytokines, chemokines and macrophage markers in VATs after 16 weeks on HFD (n=12–14 pooled mice per group; littermate controls, white bar; Lyz2 Δ/Δ mice, black bar). Data are represented as the mean±the s.e.m. *P<0.05, **P<0.01. HFD, high fat diet; NCD, normal chow diet; SVC, stromal vascular cell; VATs, visceral adipose tissues.
Inhibiting NKT–M1 interactions promotes M2 polarization and inhibits metaflammation and insulin resistance in obese mice
In Cd11c Δ/Δ mice, CD1d deletion also had no influence on body weights, visceral adipose tissue weights and SVC numbers per gram of fat after feeding on HFD for 16 weeks (Figures 5a–c). However, the Cd11c Δ/Δ mice exhibited lower serum insulin concentrations, faster glucose clearance, decreased phosphorylation of Ser307-IRS1 and increased phosphorylation of Thr308-AKT after insulin injection (Figures 5d–f). These results demonstrated ameliorated insulin resistance in Cd11c Δ/Δ mice on HFD. Moreover, altered pSer307-IRS1 and pThr308-AKT were also detected in the livers and muscles of the Cd11c Δ/Δ mice on HFD after insulin injection (Supplementary Table S6). These results indicate an influence of CD1d deletion on systemic insulin resistance. Furthermore, the M1 to M2 ratios decreased (Figure 5g), and the numbers of M2 macrophages significantly increased (Figure 5h) in the Cd11c Δ/Δ mice on HFD. No change in DC, NKT, CD8 T, Treg, ILC2, eosinophils and NK cells was identified (Figure 5i). Consistent with elevated M2 macrophages, increased IL4, IL13 and Arg1, and decreased TNFα and iNOS mRNA levels were also identified in these mice (Figure 5j). The different outcomes between the Lyz2 Δ/Δ mice and Cd11c Δ/Δ mice were caused by CD1d deletion in M1 macrophages. Altogether, our results suggested that the inhibition of NKT–M1 interactions improved insulin resistance by promoting M2 polarization and inhibiting metaflammation in VATs of HFD mice.
Figure 5.
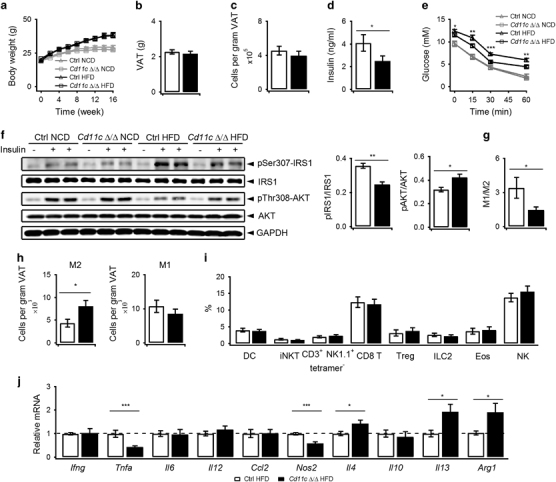
CD1d deletion in M1 macrophages promotes M2 polarization and inhibits metaflammation. Body weights of Cd11c Δ/Δ mice (square) and littermate controls (triangle) were monitored for 16 weeks on NCD or HFD (n=8–9 pooled mice per group). (b–d) Weights of VATs (n=10–13 pooled mice per group), SVC numbers per gram of VAT (n=10–13 pooled mice per group), and serum insulin concentrations (n=12–14 pooled mice per group) in Cd11c Δ/Δ mice (black bar) and littermate controls (white bar) were measured after 16 weeks on HFD. (e) Cd11c Δ/Δ mice (square) and littermate controls (triangle) were fed on NCD or HFD for 16 weeks and underwent an ITT (n=17–20 pooled mice per group). (f) Levels of IRS1, IRS1 phospho-Ser307 (pSer307-IRS1), AKT and AKT phosphor-Thr308 (pThr308-AKT) with or without insulin injection in VATs of mice fed on NCD or HFD for 16 weeks were detected via western blot (left). Ratios of pIRS1/IRS1 and pAKT/AKT with insulin in HFD mice are presented on the right (littermate controls, white bar; Cd11c Δ/Δ mice, black bar; n=4 pooled mice per group). (g, h) Proportions (g, n=13–19 pooled mice per group; i, n=13–19 or n=5–8 pooled mice per group) and absolute numbers (h, n=12–17 pooled mice per group) of the indicated cells in VATs after 16 weeks on HFD (littermate controls, white bar; Cd11c Δ/Δ mice, black bar). (j) mRNA levels of cytokines, chemokines, and macrophage markers in VATs after 16 weeks on HFD (n=10–18 pooled mice per group; littermate controls, white bar; Cd11c Δ/Δ mice, black bar). Data are represented as the mean±the s.e.m. *P<0.05, **P<0.01, ***P<0.001. HFD, high fat diet; ITT, insulin tolerance test; NCD, normal chow diet; SVC, stromal vascular cell; VATs, visceral adipose tissues.
DISCUSSION
Together, our studies demonstrated the mechanisms by which metabolic disorders caused the imbalance of anti- and proinflammatory responses. NKT cell activation by M2 macrophages was induced by metabolic disorders, which promoted M2 polarization and inhibited metaflammation. However, metabolic disorders induced an M2-specific reduction of CD1d expression at a later stage of obesity, which influenced the antigen-presenting capability of M2 macrophages and caused a change in APCs from M2 to M1 macrophages. As a consequence, the activation of NKT cells by M1 macrophages promoted Th1 responses and inhibited M2 polarization. This impaired M2 polarization further favored M1–NKT interactions. Thus, the reduction of CD1d expression in M2 macrophages is a critical event that switches NKT cell-mediated immune responses and disrupts the immune balance.
CD1d is widely expressed in vivo on both hematopoietic and nonhematopoietic cells. Controlling CD1d expression is one approach to modulate NKT cell-mediated immune responses. In addition to our studies, the downregulation of CD1d in epithelial cells or APCs has been reported in inflammatory bowel disease,28,29 tumor30 and infection disease,31,32 and was responsible for the pathogenesis of diseases. The mechanisms that regulate CD1d expression in a cell type-specific manner during the progression of obesity are not fully understood, and further studies are required. In addition to CD1d expression, antigen presentation and the type of antigens also influence the type of APCs and NKT cell-mediated immune responses.33 Inflammatory signals are reported to promote the synthesis of endogenous antigens for NKT cells.34,35 Moreover, lipidomic studies have shown different lipid profiles in obese objects, particularly for phospholipids and sphingolipids.36,37,38 Furthermore, interactions between NKT cells and macrophages in VATs were induced by obesity and depended on CD1d expression (Figures 2g and h), which suggests a potential accumulation of endogenous antigens in obese VATs. The endogenous antigens for NKT cells in VATs have not been identified. Whether M1 macrophages and M2 macrophages present different antigens and whether antigens are synthesized by macrophages or are released by apoptotic adipocytes are not known. Our studies did not exclude that antigen uptake, antigen processing and antigen type would also contribute to the change in APCs during the progression of obesity. The influence of distinct APCs on NKT cell functions has been thoroughly investigated. Moreover, the work indicated that cytokines from activated APCs as feedback influenced NKT cell-mediated immune responses.23 Interactions between NKT cells and macrophages are reciprocal. M1 macrophages have different cytokine profiles than M2 macrophages. It has been shown that the inflammatory cytokines IL12 and IL18 promote IFNγ production from iNKT cells.34,35,39 Thus, M1 macrophages may polarize the NKT cell-mediated responses toward Th1 by releasing IL12 and IL18. Moreover, IL10 from M2 macrophages may inhibit IFNγ production from NKT cells and polarize NKT cell-mediated immune responses toward Th2. In addition to obesity, other stress signals would also disturb the immune balance by modulating CD1d expression and lipid-antigen synthesis. Thus, NKT cells would represent a critical link between stress and the onset of diseases.
Our results demonstrated a protective role of NKT cells in controlling blood glucose at an early stage of obesity and a pathogenic role at a late stage of obesity. These findings explain previous controversial results.17,18,19,20 In obese NKT-deficient mice, exacerbated glucose intolerance and insulin resistance have been reported by several groups. In these studies, NKT cells have been shown to promote Th2 responses, and the accumulation of M2 macrophages and Tregs.18,19,21,22 Moreover, another group indicates that NKT cells promote Th1 responses and insulin resistance in obese mice.20 In their studies, obese NKT-deficient mice exhibited ameliorated glucose intolerance. Our results demonstrated a dynamic switch in NKT cell-mediated immune responses during the progression of obesity. The animal models used in our studies were different from previous studies. CD1d KO and Jα18 KO mice were previously used, in which NKT cells were deficient from birth. These animal models were unable to exhibit alterations in NKT cell functions during the progression of diseases. In addition to previous work that focused on the nature of NKT cells, our studies shed light on the entire cellular networks directly or indirectly regulated by NKT cells. In a comparison of the Lyz2 Δ/Δ mice with the Cd11c Δ/Δ mice on HFD, the latter showed elevated Th2 cytokines (Figures 4j and 5j), which were not induced by direct M2-NKT interactions as a result of the CD1d deficiency in M2 macrophages in the Cd11c Δ/Δ mice (Supplementary Table S4). Thus, NKT–M1 interactions have a broad effect on Th2 response inhibition. Alterations in the type of APCs would influence a number of downstream immune cells. Furthermore, it is well established that NKT cells could regulate the functions of conventional T cells and NK cells,40,41,42 which have been shown to regulate metaflammation in adipose tissue.43,44,45 We did not identify differences in the cell numbers of CD8+ T, Treg, CD3+ tetramer− NK1.1+ cells, NK cells, ILC2 or eosinophils (Figures 4i and 5i); however, their functions may be different. Previous studies have indicated a role of NKT cells in blood glucose regulation in lean mice.18 In our studies, CD1d conditional KO mice on NCD exhibited similar body weights and glucose homeostasis as the littermate controls (Supplementary Table S4). These results were consistent with the rare colocalization between macrophages and iNKT cells in the mice fed on NCD (Figures 2g and i) and suggested that interactions between macrophages and NKT cells were not involved in controlling blood glucose in lean mice. Thus, other cells but not macrophages would be responsible for the activation of NKT cells in the VATs of lean mice. For example, adipocytes have been shown to express CD1d and activate NKT cells.18,46 To understand the reciprocal activation of adipocytes and NKT cells, mice with CD1d deletion in adipocytes would be helpful. We also noted that previous studies have suggested a role of NKT cells in the regulation of liver metaflammation, as well as adipose tissue metaflammation.18,19,20,47 In our studies, the HFD mice only exhibited inflammation in the VATs and not in the livers when we performed the experiments (Supplementary Table S1). Liver inflammation was detected after 26 weeks of HFD feeding. Even in VATs, 12 weeks were required to induce the elevation of the M1/M2 ratio and the expression of inflammatory genes, which was substantially longer than other labs.21 Thus, the rate of disease development in our studies may be slower than other studies. The discrepancy in disease development may be caused by different microbiota between different labs.48,49 Moreover, consistently, no difference in inflammatory gene expression was identified in the livers and muscles of the CD1d conditional KO mice on HFD for 16 weeks (Supplementary Table S5). Thus, adipose tissue inflammation was responsible for the insulin resistance in our studies.
Here, we demonstrated an effect of NKT cells on M2 macrophage polarization but not M1 macrophage polarization. The recruitments of M1 and M2 macrophages are regulated by different mechanisms. Previous studies have suggested that M1 macrophages originate from CCR2+ monocytes in adipose tissues in contrast to M2 macrophages.50 It has been reported that macrophages in tissues could derive from embryonic precursors (tissue-resident macrophages) or bone marrow monocytes.51,52 Different origins of M1 and M2 macrophages may also explain the different CD1d deletion efficiencies in Lyz2 Δ/Δ mice. Whether M2 macrophages in adipose tissue are derived from embryonic precursors remains unknown. Illuminating the origins of adipose macrophages would help to understand the mechanisms that regulate the polarization of M2 macrophages.
Overall, our findings suggest that the M2-specific downregulation of CD1d expression is an initiation signal that switches NKT cell-mediated immune responses and triggers metaflammation. Our results also suggest tissue-resident NKT cells and the CD1d molecule as important players in the regulation of immune homeostasis and early responses to stress signals.
Electronic supplementary material
Acknowledgements
We thank the NIH Tetramer Core Facility for providing us with the CD1d-PBS57 tetramer. This work was supported by the Major State Basic Research Development Program of China (973 Program) 2013CB944902, the National Natural Science Foundation of China 91542203 and 31470859, the Strategic Priority Research Program of the Chinese Academy of Sciences XDA12030201, the Fundamental Research Funds for the Central Universities and Users with Potential 2015HSC-UP018.
Author contributions
HZ and RX contributed equally to the paper. HZ, RX, SZ, SF and ZC performed the experiments. RZ and ZT discussed the experiments. HZ, RX and LB designed the experiments and wrote the manuscript.
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Supplementary Information for this article can be found on the Cellular & Molecular Immunology website 10.1038/cmi.2017.11
References
- 1.Kammoun HL, Kraakman MJ, Febbraio MA. Adipose tissue inflammation in glucose metabolism. Rev Endocr Metab Disord. 2014;15:31–44. doi: 10.1007/s11154-013-9274-4. [DOI] [PubMed] [Google Scholar]
- 2.Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. 2011;121:2111–2117. doi: 10.1172/JCI57132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72:219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- 4.Fujisaka S, Usui I, Bukhari A, Ikutani M, Oya T, Kanatani Y, et al. Regulatory mechanisms for adipose tissue M1 and M2 macrophages in diet-induced obese mice. Diabetes. 2009;58:2574–2582. doi: 10.2337/db08-1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI200319246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu HY, Barnes GT, Yang Q, Tan Q, Yang DS, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–1830. doi: 10.1172/JCI200319451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Molofsky AB, Nussbaum JC, Liang HE, Van Dyken SJ, Cheng LE, Mohapatra A, et al. Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J Exp Med. 2013;210:535–549. doi: 10.1084/jem.20121964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK, et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science. 2011;332:243–247. doi: 10.1126/science.1201475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. 2009;15:914–920. doi: 10.1038/nm.1964. [DOI] [PubMed] [Google Scholar]
- 10.McNelis JC, Olefsky JM. Macrophages, immunity, and metabolic disease. Immunity. 2014;41:36–48. doi: 10.1016/j.immuni.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 11.Thomas SY, Scanlon ST, Griewank KG, Constantinides MG, Savage AK, Barr KA, et al. PLZF induces an intravascular surveillance program mediated by long-lived LFA-1-ICAM-1 interactions. J Exp Med. 2011;208:1179–1188. doi: 10.1084/jem.20102630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lynch L, Michelet X, Zhang S, Brennan PJ, Moseman A, Lester C, et al. Regulatory iNKT cells lack expression of the transcription factor PLZF and control the homeostasis of T(reg) cells and macrophages in adipose tissue. Nat Immunol. 2015;16:85–95. doi: 10.1038/ni.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mathews S, Feng D, Maricic I, Ju C, Kumar V, Gao B. Invariant natural killer T cells contribute to chronic-plus-binge ethanol-mediated liver injury by promoting hepatic neutrophil infiltration. Cell Mol Immunol. 2016;13:206–216. doi: 10.1038/cmi.2015.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bandyopadhyay K, Marrero I, Kumar V. NKT cell subsets as key participants in liver physiology and pathology. Cell Mol Immunol. 2016;13:337–346. doi: 10.1038/cmi.2015.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lisbonne M, Diem S, de Castro Keller A, Lefort J, Araujo LM, Hachem P, et al. Cutting edge: invariant V alpha 14 NKT cells are required for allergen-induced airway inflammation and hyperreactivity in an experimental asthma model. J Immunol. 2003;171:1637–1641. doi: 10.4049/jimmunol.171.4.1637. [DOI] [PubMed] [Google Scholar]
- 16.Wingender G, Rogers P, Batzer G, Lee MS, Bai D, Pei B, et al. Invariant NKT cells are required for airway inflammation induced by environmental antigens. J Exp Med. 2011;208:1151–1162. doi: 10.1084/jem.20102229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu L, Van Kaer L. Contribution of lipid-reactive natural killer T cells to obesity-associated inflammation and insulin resistance. Adipocyte. 2013;2:12–16. doi: 10.4161/adip.22296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schipper HS, Rakhshandehroo M, van de Graaf SF, Venken K, Koppen A, Stienstra R, et al. Natural killer T cells in adipose tissue prevent insulin resistance. J Clin Invest. 2012;122:3343–3354. doi: 10.1172/JCI62739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lynch L, Nowak M, Varghese B, Clark J, Hogan AE, Toxavidis V, et al. Adipose tissue invariant NKT cells protect against diet-induced obesity and metabolic disorder through regulatory cytokine production. Immunity. 2012;37:574–587. doi: 10.1016/j.immuni.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu L, Parekh VV, Gabriel CL, Bracy DP, Marks-Shulman PA, Tamboli RA, et al. Activation of invariant natural killer T cells by lipid excess promotes tissue inflammation, insulin resistance, and hepatic steatosis in obese mice. Proc Natl Acad Sci USA. 2012;109:E1143–E1152. doi: 10.1073/pnas.1200498109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ji Y, Sun S, Xia S, Yang L, Li X, Qi L. Short term high fat diet challenge promotes alternative macrophage polarization in adipose tissue via natural killer T cells and interleukin-4. J Biol Chem. 2012;287:24378–24386. doi: 10.1074/jbc.M112.371807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Satoh M, Andoh Y, Clingan CS, Ogura H, Fujii S, Eshima K, et al. Type II NKT cells stimulate diet-induced obesity by mediating adipose tissue inflammation, steatohepatitis and insulin resistance. PLoS One. 2012;7:e30568. doi: 10.1371/journal.pone.0030568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bai L, Constantinides MG, Thomas SY, Reboulet R, Meng F, Koentgen F, et al. Distinct APCs explain the cytokine bias of alpha-galactosylceramide variants in vivo. J Immunol. 2012;188:3053–3061. doi: 10.4049/jimmunol.1102414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Constantinides MG, Bendelac A. Transcriptional regulation of the NKT cell lineage. Curr Opin Immunol. 2013;25:161–167. doi: 10.1016/j.coi.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sullivan BA, Nagarajan NA, Wingender G, Wang J, Scott I, Tsuji M, et al. Mechanisms for glycolipid antigen-driven cytokine polarization by Valpha14i NKT cells. J Immunol. 2010;184:141–153. doi: 10.4049/jimmunol.0902880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scanlon ST, Thomas SY, Ferreira CM, Bai L, Krausz T, Savage PB, et al. Airborne lipid antigens mobilize resident intravascular NKT cells to induce allergic airway inflammation. J Exp Med. 2011;208:2113–2124. doi: 10.1084/jem.20110522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Geissmann F, Cameron TO, Sidobre S, Manlongat N, Kronenberg M, Briskin MJ, et al. Intravascular immune surveillance by CXCR6+ NKT cells patrolling liver sinusoids. PLoS Biol. 2005;3:e113. doi: 10.1371/journal.pbio.0030113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Colgan SP, Pitman RS, Nagaishi T, Mizoguchi A, Mizoguchi E, Mayer LF, et al. Intestinal heat shock protein 110 regulates expression of CD1d on intestinal epithelial cells. J Clin Invest. 2003;112:745–754. doi: 10.1172/JCI200317241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olszak T, Neves JF, Dowds CM, Baker K, Glickman J, Davidson NO, et al. Protective mucosal immunity mediated by epithelial CD1d and IL-10. Nature. 2014;509:497–502. doi: 10.1038/nature13150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tatsumi T, Takehara T, Yamaguchi S, Sasakawa A, Yamamoto M, Fujita Y, et al. Decreased expressions of CD1d molecule on liver dendritic cells in subcutaneous tumor bearing mice. J Hepatol. 2008;49:779–786. doi: 10.1016/j.jhep.2008.06.011. [DOI] [PubMed] [Google Scholar]
- 31.Yuan W, Dasgupta A, Cresswell P. Herpes simplex virus evades natural killer T cell recognition by suppressing CD1d recycling. Nat Immunol. 2006;7:835–842. doi: 10.1038/ni1364. [DOI] [PubMed] [Google Scholar]
- 32.Sanchez DJ, Gumperz JE, Ganem D. Regulation of CD1d expression and function by a herpesvirus infection. J Clin Invest. 2005;115:1369–1378. doi: 10.1172/JCI200524041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bai L, Sagiv Y, Liu Y, Freigang S, Yu KO, Teyton L, et al. Lysosomal recycling terminates CD1d-mediated presentation of short and polyunsaturated variants of the NKT cell lipid antigen alphaGalCer. Proc Natl Acad Sci USA. 2009;106:10254–10259. doi: 10.1073/pnas.0901228106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brigl M, Tatituri RV, Watts GF, Bhowruth V, Leadbetter EA, Barton N, et al. Innate and cytokine-driven signals, rather than microbial antigens, dominate in natural killer T cell activation during microbial infection. J Exp Med. 2011;208:1163–1177. doi: 10.1084/jem.20102555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brigl M, Bry L, Kent SC, Gumperz JE, Brenner MB. Mechanism of CD1d-restricted natural killer T cell activation during microbial infection. Nat Immunol. 2003;4:1230–1237. doi: 10.1038/ni1002. [DOI] [PubMed] [Google Scholar]
- 36.Goto-Inoue N, Yamada K, Inagaki A, Furuichi Y, Ogino S, Manabe Y, et al. Lipidomics analysis revealed the phospholipid compositional changes in muscle by chronic exercise and high-fat diet. Sci Rep. 2013;3:3267. doi: 10.1038/srep03267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pietilainen KH, Rog T, Seppanen-Laakso T, Virtue S, Gopalacharyulu P, Tang J, et al. Association of lipidome remodeling in the adipocyte membrane with acquired obesity in humans. PLoS Biol. 2011;9:e1000623. doi: 10.1371/journal.pbio.1000623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jove M, Moreno-Navarrete JM, Pamplona R, Ricart W, Portero-Otin M, Fernandez-Real JM. Human omental and subcutaneous adipose tissue exhibit specific lipidomic signatures. FASEB J. 2014;28:1071–1081. doi: 10.1096/fj.13-234419. [DOI] [PubMed] [Google Scholar]
- 39.Nagarajan NA, Kronenberg M. Invariant NKT cells amplify the innate immune response to lipopolysaccharide. J Immunol. 2007;178:2706–2713. doi: 10.4049/jimmunol.178.5.2706. [DOI] [PubMed] [Google Scholar]
- 40.Nakamura T, Sonoda KH, Faunce DE, Gumperz J, Yamamura T, Miyake S, et al. CD4+ NKT cells, but not conventional CD4+ T cells, are required to generate efferent CD8+ T regulatory cells following antigen inoculation in an immune-privileged site. J Immunol. 2003;171:1266–1271. doi: 10.4049/jimmunol.171.3.1266. [DOI] [PubMed] [Google Scholar]
- 41.Carnaud C, Lee D, Donnars O, Park SH, Beavis A, Koezuka Y, et al. Cutting edge: Cross-talk between cells of the innate immune system: NKT cells rapidly activate NK cells. J Immunol. 1999;163:4647–4650. [PubMed] [Google Scholar]
- 42.Fujii S, Shimizu K, Smith C, Bonifaz L, Steinman RM. Activation of natural killer T cells by alpha-galactosylceramide rapidly induces the full maturation of dendritic cells in vivo and thereby acts as an adjuvant for combined CD4 and CD8 T cell immunity to a coadministered protein. J Exp Med. 2003;198:267–279. doi: 10.1084/jem.20030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee BC, Kim MS, Pae M, Yamamoto Y, Eberle D, Shimada T, et al. Adipose Natural Killer Cells Regulate Adipose Tissue Macrophages to Promote Insulin Resistance in Obesity. Cell Metab. 2016;23:685–698. doi: 10.1016/j.cmet.2016.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wensveen FM, Jelencic V, Valentic S, Sestan M, Wensveen TT, Theurich S, et al. NK cells link obesity-induced adipose stress to inflammation and insulin resistance. Nat Immunol. 2015;16:376–385. doi: 10.1038/ni.3120. [DOI] [PubMed] [Google Scholar]
- 45.O’Rourke RW, Meyer KA, Neeley CK, Gaston GD, Sekhri P, Szumowski M, et al. Systemic NK cell ablation attenuates intra-abdominal adipose tissue macrophage infiltration in murine obesity. Obesity. 2014;22:2109–2114. doi: 10.1002/oby.20823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huh JY, Kim JI, Park YJ, Hwang IJ, Lee YS, Sohn JH, et al. A novel function of adipocytes in lipid antigen presentation to iNKT cells. Mol Cell Biol. 2013;33:328–339. doi: 10.1128/MCB.00552-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martin-Murphy BV, You Q, Wang H, De La Houssaye BA, Reilly TP, Friedman JE, et al. Mice lacking natural killer T cells are more susceptible to metabolic alterations following high fat diet feeding. PLoS One. 2014;9:e80949. doi: 10.1371/journal.pone.0080949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang C, Zhang M, Wang S, Han R, Cao Y, Hua W, et al. Interactions between gut microbiota, host genetics and diet relevant to development of metabolic syndromes in mice. ISME J. 2010;4:232–241. doi: 10.1038/ismej.2009.112. [DOI] [PubMed] [Google Scholar]
- 49.Escobedo G, Lopez-Ortiz E, Torres-Castro I. Gut microbiota as a key player in triggering obesity, systemic inflammation and insulin resistance. Rev Invest Clin. 2014;66:450–459. [PubMed] [Google Scholar]
- 50.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117:175–184. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity. 2014;41:21–35. doi: 10.1016/j.immuni.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol. 2014;14:392–404. doi: 10.1038/nri3671. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.