
FIGURE 2.
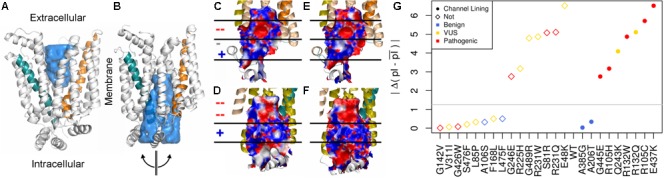
Conservation of inner pore properties. Despite the relatively modest sequence identity across the GLUT family, structural properties, such as the electrostatic distribution around and within the ligand-binding pocket, are conserved. We show our (A) outward-facing and (B) inward-facing GLUT10 models with the binding pocket filled in. In the outward-facing conformation, a large cavity is accessible from the extracellular side, but the inner binding site is fully occluded. A semi-transparent blue surface fills the solvent accessible cavity. Helix 4 (teal) and helix 10 (orange) are colored similar to previous figures. After transitioning to the inward-facing conformation, an opposite relationship is observed. Considering the inward-facing conformation, we split the structure in half according to the two helical bundled described above and color each interior surface by electrostatic potential (red negative, white neutral, and blue positive). Pealing the structure in half reveals that the interior of the pore for (C,D) GLUT10 and (E,F) GLUT1 are highly similar, despite their low sequence conservation (24%) and that GLUT1 was not used in the MSA for constructing our GLUT10 model (Supplementary Table S2). Horizontal lines and positive (+) or negative (–) annotation indicate regions where electrostatics switch consistently between GLUT1 and GLUT10. (G) Pathogenic variants tend to alter charge segregation of the channel lining. We first selected residue positions that make up the channel interior lining in either conformation. Each variant’s change to the electrostatic character of the channel interior was quantified using a simple pI-based score; see the section “Materials and Methods” section. A gray line marks the maximum change possible among uncharged amino acids.