

Long intergenic non-coding RNAs have been shown to play important roles in cancer. However, because lincRNAs are a relatively new class of RNAs compared to protein-coding mRNAs, the mutational landscape of lincRNAs has not been as extensively studied. Here we characterize expressed somatic nucleotide variants within lincRNAs using 12 cancer RNA-Seq datasets in TCGA. We build machine-learning models to discriminate somatic variants from germline variants within lincRNA regions (AUC 0.987). We build another model to differentiate lincRNA somatic mutations from background regions (AUC 0.72) and find several molecular features that are strongly associated with lincRNA mutations, including copy number variation, conservation, substitution type and histone marker features.
1. Introduction
Long intergenic non-coding RNAs (lincRNAs) have been shown to play important roles in many diseases, including cancer. The expression of thousands of lincRNAs are deregulated in cancer, and many lincRNAs have been proposed as biomarkers for tumor tissues and patient prognosis [1]–[3]. There is also strong evidence that lincRNAs may serve as drivers of tumorigenesis, cause drug resistance or cause metastasis [4]–[7]. Mutations in cancer driver genes lead to a series of downstream events, including gene expression changes [8], [9]. However, because lincRNAs are a relatively new class of non-coding RNAs compared to protein coding mRNAs, the mutational landscape of lincRNAs and their impact on gene expression, have not been extensively studied.
While most people use exome-Seq to investigate somatic mutations, the coverage on lincRNA regions is very limited. Furthermore, several previous studies have shown that expressed somatic nucleotide variations (eSNVs) can be robustly called from RNA-Seq data [10]–[12]. Therefore, to interrogate the effects of lincRNA mutations, we used the RNA-Seq data from The Cancer Genome Atlas (TCGA), analyzing 6118 patient samples from 12 cancer datasets.
Due to the fact that most RNA-Seq samples do not contain normal controls, we constructed a Random Forest model on exome-Seq data to differentiate eSNVs and germline variants, and then extrapolated this model to the RNA-Seq eSNVs. Subsequently, we interrogated the features related to eSNVs within lincRNAs. We find several molecular features that are strongly associated with lincRNA mutations, including copy number variation, conservation, substitution type and histone marker features.
2. Methods
2.1. TCGA Datasets
We used 12 cancer datasets from TCGA with a total of 6118 primary tumor samples in this study. These datasets include bladder urothelial carcinoma (BLCA, n=406), breast invasive carcinoma (BRCA, n=1084), head and neck squamous cell carcinoma (HSNC, n=514), kidney renal clear cell carcinoma (KIRC, n=525), liver hepatocellular carcinoma (LIHC, n=364), low grade glioma (LGG, n=513), lung adenocarcinoma (LUAD, n=512), lung squamous cell carcinoma (LUSC, n=498), ovarian serous cystadenocarcinoma (OV, n=300), stomach adenocarcinoma (STAD, n=414), prostate adenocarcinoma (PRAD, n=491) and thyroid carcinoma (THCA, n=497). RNA-Seq fastq files were downloaded using GeneTorrent program from the UCSC Cancer Genomics Hub (https://cghub.ucsc.edu). Additional TCGA samples were downloaded from NCBI Genomic Data Commons Data Portal (https://gdc-portal.nci.nih.gov) using the GDC data transfer tool.
2.2. Predicting germline and somatic mutations
The exome sequencing variant calls, including somatic and germline variants, were downloaded for 7 TCGA datasets (BLCA, HNSC, KIRC, LGG, LIHC, LUAD, PRAD and STAD). A Random Forest model was built to classify somatic vs. germline variants, from the exome sequencing data from TCGA. In this model, the class labels were derived as 1 – somatic mutation and 0 – germline mutation, determined by the paired exome-seq data.
The Xgboost package in R was used (version 0.6–0) with 1000 trees. Five features were used in the building of this model: mutation frequency across the entire cohort (frequency), dbsnp (whether an SNV occurred at a position annotated by the dbSNP database), fa.tumor (the estimate allele ratio of the SNV in the tumor exome sample), conservation (PhyloP conservation score from the UCSC genome browser) and transversion (whether the SNV was a transition or transversion mutation). These features were chosen in order to be independent of the subsequent models. The performance was evaluated using 5-fold cross-validation.
2.3. Expressed somatic nucleotide variations (eSNVs)
Raw read data were downloaded from UCSC Cancer Genomics Hub in the fastq format. Reads were first aligned to the hg19 genome reference using STAR aligner [13] in two-pass mode. Aligned BAM files were sorted using ReorderSam function in Picard-tools (http://broadinstitute.github.io/picard) and reads were split based on splicing junctions using SplitNCigarReads function in Genome Analysis Toolkit (GATK)[14]. Reads were then processed through duplicate removal, INDEL realignment and base recalibration, following standard protocols. Variant calling was performed using GATK’s Haplotype caller. Data processing was performed on the high performance computing cluster of University of Hawaii. To further reduce potential false positive calls, variants were filtered based on SNV clusters and read strand bias following recommendations from the developers. To identify lincRNA specific eSNVs, variants associated with lincRNAs based on the lncipedia 4.0 reference [6] were used for analysis.
2.4. Predictive models to classify eSNVs from background nucleotide sites
We constructed classification models in order to predict eSNVs from germline variants for each cancer type. The class labels for this model were 1 - a eSNVs determined in lincRNA regions from the RNA-Seq data (based on the results of the first model) and 0 - background “negative” eSNVs, i.e., random non-mutated locations on expressed lincRNAs in each RNA-Seq sample. The models were built on balanced datasets. The molecular features in these models include conservation, copy number variation, histone marker features, nucleotide composition features, location on exon junctions and transcription start and end sites. Three algorithms were employed on these datasets: logistic regression with ridge regularization (LR), a fast linear classification algorithm using the glmnet R package (version 2.0-5); a neural network classifier using Tensorflow (version 1.1.0); and Gradient Boosted Trees [15], a fast non-linear tree-based classifier using the xgboost R package (version 0.6-0).
To evaluate each model, the datasets were split into 80% training and 20% testing. AUC was calculated as the performance metric on the testing sets. The Gradient Boosted Trees models were evaluated and the Gain value of each feature was computed, to determine feature importance. In an ensemble forest model (Random Forest or Gradient Boosted Trees), Gain is the average improvement of performance of the model on each tree branch, split by the features in the ensemble forest [15].
3. Results
3.1. Computational pipeline accurately predicts genetic variation in tumor RNA-Seq samples
We selected 6118 primary tumor RNA-Seq samples from 12 TCGA datasets and implemented a pipeline for calling mutations from bulk RNA-Seq data described in the methods section (Figure 1). To verify the quality of the results, we compared the variant calls from exome sequencing in paired exome and RNA-Seq sample datasets. On average, 80% of the expressed somatic nucleotide variants (eSNVs) found in RNA-Seq data were also found in the exome sequencing variant calls, within the exome-seq read regions. This high concordance of eSNVs detected by RNA-Seq relative to exome-seq is better than what others showed for the same samples using different analysis platforms (~50%) [16], suggesting that our eSNV calls from the RNA-Seq are reliable.
Figure 1.
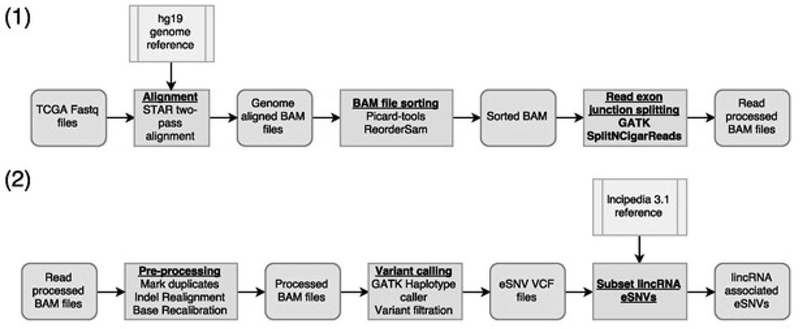
Workflow describing the pipeline for the raw data processing, calling eSNVs and calculating expression data. (1) RNA-Seq alignment and read pre-processing. (2) Re-alignment, re-calibration and lincRNA variant calling.
3.2. A Random Forest model differentiates somatic and germline mutations
Because RNA-Seq samples, from which we called eSNVs, usually do not have matched normal samples, directly determining somatic or germline status of a variant through these RNA-Seq samples is not possible. However, since most eSNVs from RNA-Seq overlap with the SNVs detected through exome sequencing in the protein-coding genes, we then aimed to predict the somatic or germline origin of these variants in RNA-Seq using a Random Forest model trained on the exome-seq data. Exome-sequencing data are preferred “gold-standard” training data, as these data had paired normal and tumor samples (and therefore SNVs could be accurately differentiated from germline variants). We built a random forest model classifying the somatic mutations versus the germline mutations based upon five features: frequency (mutation frequency across the samples in a dataset), dbsnp (whether the mutation is documented in the NCBI dbSNP database), FA.tumor (the fraction of the alternate allele in the tumor sample), conservation (PhyloP conservation score) and transversion (whether the mutation is a transversion or a transition mutation). This model had an AUC of 0.988 on the exome sequencing data and an AUC of 0.987 based on RNA-Seq data respectively (Figure 2A). By comparison, the logistic model had slightly lower AUCs of 0.979 and 0.985. We therefore decided to use the results of random forest model for the following sections. Mutation frequency, dbsnp and FA.tumor features have relatively high importance scores relevant to the outcome, with values of 0.485, 0.291, and 0.184 (Figure 2B). Conservation and transversion do not present as important features in this model (Figure 2B).
Figure 2.
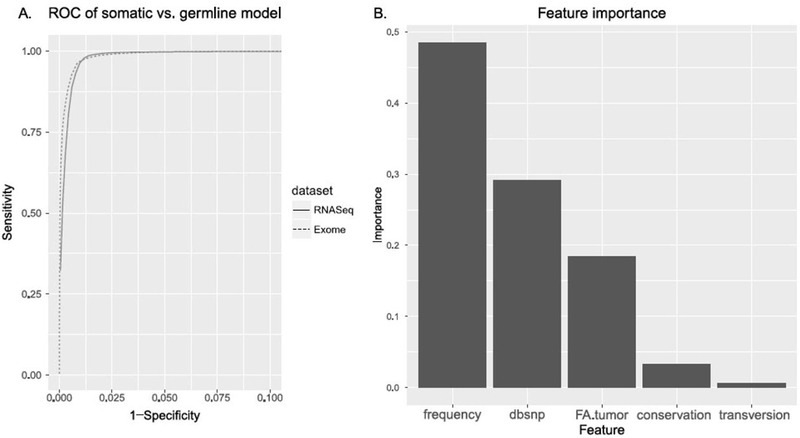
A – Receiver operating characteristic (ROC) curve showing the performance of the random forest models for differentiating somatic and germline mutations in the exome-sequencing data. B – Feature importance based on the random forest model.
Secondly, we then applied this model to the 12 RNA-Seq datasets, and selected eSNVs that are highly confident as either somatic (posterior probability > 0.97) or germline variants (posterior probability < 0.03). Using these thresholds, 1.25 million somatic mutations were detected in protein-coding genes and 94,700 were detected in lincRNAs. For germline variants, 170 million protein coding variants were detected and 15.5 million lincRNA variants were detected. We calculated the density of lincRNAs genome-wide, relative to the lincRNA exon density. There are many regions of enriched lincRNA eSNVs throughout the genome (Figure 3).
Figure 3.

Circos plot of the normalized lincRNA eSNV mutation density. For each cancer type, the number of lincRNA eSNVs was binned using window sizes of 100,000 across the genome. The bin count was then normalized by the lincRNA exon density in the corresponding transcriptomic region. The outer layer shows the human genome cytogenetics. The middle layer shows the eSNV density within lincRNA regions. The inner layer shows the exon density of the lincRNA transcriptome.
There are some regions that have an increased frequency of lincRNA eSNVs. The top four regions included chr2p11.2, chr14q32.33, chr22q11.22 and chr3q29. In particular, chr2p11.2 is known to be heavily associated with breast cancer [17]. Sahin et al. found that copy number imbalances in chr2p11.2 had a significant effect on breast screening and detection. They also found that the imbalance had a significant effect on disease free survival. However, they were not able to determine any association with protein coding genes. These results suggest that the association of this region with cancer phenotypes could be due to lincRNA mutations.
3.3. A machine learning model predicts mutation likelihood on nucleotide positions
Next, we wanted to determine which nucleotide positions were more likely to have somatic mutations. For each of the 12 TCGA cancer types, we constructed a classification model to predict eSNV likelihood within lincRNAs. Similarly, we also built models using the germline variants using the same features. We applied three machine learning algorithms to each dataset: logistic regression (a linear classifier), a neural networks (a flexible non-linear classifier) and gradient boosted trees (a fast tree-based non-linear classifier). In each dataset, the boosted trees model performed considerably better than the neural network and logistic regression models. The neural network models generally performed better than the logistic regression. Across all 12 TCGA datasets, Boosted Trees had an average AUC of approximately 0.72 for eSNVs and 0.89 for germline variants (Figure 4). By comparison, the logistic regression models had AUCs of 0.68 and 0.77 for eSNVs and germline variants respectively.
Figure 4.

Model performance differentiating somatic eSNVs and germline variants from background in lincRNAs for the 12 TCGA datasets, using the Gradient boosted Trees machine learning algorithm.
3.4. Molecular features correlated with somatic eSNVs differ from germline variants
To evaluate the importance of each feature in the two models (somatic vs. germline), we used the Gain measure, which calculates the average increase in performance for each feature in every tree in the Boosted Trees ensemble. For the 12 lincRNA eSNV models, conservation followed by copy number variation (cnv_pos and cnv_promoter) are the most important features (Figure 5A). For the germline variant models, copy number variation does not have a high feature importance score (Figure 5B). For the eSNV models, on average, the third most important feature is tranversion – the type of mutation.
Figure 5.

A. Heatmap of feature importance for lincRNA eSNVs. Feature importance was calculated using the Gain measure, which evaluates the average increase in accuracy at a feature’s node splitting in each tree. B. Comparison of feature importance between lincRNA eSNVs and germline variants.
Several histone features show importance in specific datasets (Figure 5A and 5B). We measured histone methylation levels at two locations: the promoter regions of each lincRNA and the position of the eSNV. Promoter methylation signatures are relatively less important than methylation signatures at the eSNV position. For kidney renal cell carcinoma and prostate cancer, H3k04me3 (histone 3 trimethylation signature) position information is the most important histone modification feature. H3k4me3 and H3k36me3 histone methylation are both important for liver cancer. In addition, nucleotide composition upstream or downstream of the eSNV are not as important as the nucleotide mutation site, with C/G nucleotides being much less likely to be mutated. Exon junction and transcription start and stop sites features (TSS and TES) are among the least important features, suggesting that there was neither enrichment nor depletion of eSNVs on splice junctions and the two ends of lincRNA transcripts.
4. Discussion
In this study, we show that machine learning models can accurately separate a portion of highly confident eSNVs from germline variants, using exome sequencing data for training. Since paired normal samples are often not available (as in the case of older FFPE DNA samples [18], or RNA-Seq expression samples in the present case) this investigation has wide applications. Based on the random forest model (Figure 2), mutation frequency, dbsnp and allele frequency are important features in predicting which variants are somatic. dbSNP variants are those commonly found in population germlines, and therefore are much less likely to be somatic.
Similarly, it has been noted that variants that have low allele frequency are likely to be cancer mutations, and may even play important roles in cancer development [19]. Thus, variants found to have 100% allele frequency in the tumor samples are unlikely to be somatic mutations, as normal sample contamination is usually present [20]. Furthermore, even if normal sample contamination were removed, tumor samples often contain multiple populations that may have different alleles and mutational profiles [21]. Therefore, it is unlikely for a somatic mutation to have an allele frequency of 1.
The models predicting eSNVs from the background nucleotide positions showed strong performance (Figure 4), suggesting that some nucleotide positions within lincRNA are more likely to gain somatic mutations than other positions. Comparing the different classification algorithms, the logistic regression performed worse than the non-linear Boosted Trees algorithm, suggesting that that the prediction of lincRNA may be complex and non-linear.
Interestingly, conservation is the most important feature in the lincRNA somatic model. Although conservation scores are determined through evolutionary homology, it has been shown that conservation correlates with somatic mutation hot spots [22]. The germline models for lincRNAs, in contrast, scored conservation slightly higher. This may be expected, as conservation itself is a direct measure of the likelihood of variation through a species’ germline lineage.
The second most important feature for most lincRNA somatic models was cnv_pos, followed by cnv_promoter (i.e., copy number variation at the mutation position and promoter, determined by a microarray on a corresponding DNA TCGA sample). Previous studies have found that many somatic gene mutations are significantly correlated with copy number alterations in cancer, including EGFR and KRAS [23]. However, although many genes were found to be correlated, on a global scale, many genes did not reach significance [23]. As may be expected, copy number variation was much more important in the somatic eSNV model, compared to the germline model, as copy number variations themselves are somatic alterations, and should not alter the original germline genomic state.
The next most important feature for the lincRNA somatic model was transversion (whether a mutation was a transversion – 1, or transition mutation – 0). Transition somatic mutations, particularly C>T transitions, are more frequent than transversion somatic mutations [24].
However, for particular tumor types and even specific genes (e.g., p53 somatic mutations), the prevalence of transversions may be higher than transitions [24], [25]. This suggests the type of mutation may potentially be important in determining a mutation’s biological importance.
For the datasets with matched tissue cell line histone data, histone features related to the lincRNA sites were determined to have a significant effect on the prediction of eSNVs sites. Previous studies have found that chromatin modifications had a major effect on regional mutation rates in cancer cells [26]. Since histone methylation and acetylation status determines the 3-dimensional conformation and openness of genomic regions, differences in histone modifications between regions may change the exposure of a region to mutagenic forces in a tumor.
While using RNA-Seq to perform mutation calling is an interesting idea to couple SNVs with expression data, false negatives may arise due to the fact that many lincRNAs and transcripts are lowly expressed or not expressed at all in certain tissues or conditions [27]. On the other hand, false positives may also be introduced as RNA splicing of transcripts could cause additional read misalignment to the genome reference [12].
Additionally, since expression data and eSNVs both come from RNA-Seq and require the presence of expressed transcripts to produce reads for measurement, expression and eSNVs are inherently coupled, at a technical level. A gene that is not expressed will also not have any detected mutations. This suggests that there may be bias towards regions of high read coverage and therefore high expression.
However, within the TCGA RNA-Seq datasets, the majority of eSNVs detected that lie within exome probe boundaries, are also detected in exome-sequencing variant calling from the same patients. Previous studies have found that, from the same patient, the concordance between sequencing platforms and variant calling software to be about 50% [16]. This suggests that the false positives from the eSNV RNA-Seq pipeline are much less of an issue than other technical factors, such as the choice of sequencing platform.
The sparsity of SNPs and SNVs in a genome suggests that individual sites may not be able to be definitively predicted with high certainty. Biologically, this is a result of the stochastic nature of somatic point mutations. However, individual genes, lincRNAs, genomic regions, or possibly individual exons or sections of lincRNAs may be predicted as more or less likely to be mutated, relative to other exons or genes.
5. Conclusion
In this study, we generated two types of models: first, a Random Forest model to differentiate germline and somatic mutations, and second, a Gradient Boosted Trees model that finds lincRNAs nucleotide positions that are more likely to contain mutations. Additionally, we have explored the eSNV landscape and found regions across the genome that have an increase in lincRNA mutations, such as chr2p11.2. This is an important step in finding the biological significance of lincRNAs that are susceptible to somatic mutations in cancer.
6. References
- [1].Ching T et al. , “Pan-Cancer Analyses Reveal Long Intergenic Non-Coding RNAs Relevant to Tumor Diagnosis, Subtyping and Prognosis,” EBioMedicine, 2016. [DOI] [PMC free article] [PubMed]
- [2].Ching T, Masaki J, Weirather J, and Garmire LX, “Non-coding yet non-trivial: a review on the computational genomics of lincRNAs,” BioData Min, vol. 8, no. 1, p. 1, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Prensner JR and Chinnaiyan AM, “The emergence of lncRNAs in cancer biology,” Cancer Discov, vol. 1, no. 5, pp. 391–407, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Zarate R, Boni V, Bandres E, and Garcia-Foncillas J, “MiRNAs and LincRNAs: Could They Be Considered as Biomarkers in Colorectal Cancer?,” Int. J. Mol. Sci, vol. 13, no. 1, pp. 840–865, January 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zhou X, Chen J, and Tang W, “The molecular mechanism of HOTAIR in tumorigenesis, metastasis, and drug resistance,” Acta Biochim. Biophys. Sin, vol. 46, no. 12, pp. 1011–1015, December 2014. [DOI] [PubMed] [Google Scholar]
- [6].Yang Y, Li H, Hou S, Hu B, Liu J, and Wang J, “The Noncoding RNA Expression Profile and the Effect of lncRNA AK126698 on Cisplatin Resistance in Non-Small-Cell Lung Cancer Cell,” PLOS ONE, vol. 8, no. 5, May 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lanzós A et al. , “Discovery of Cancer driver long noncoding RNAs across 1112 tumour genomes: new candidates and distinguishing features,” Sci. Rep, vol. 7, p. 41544, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gonzalez-Perez A et al. , “IntOGen-mutations identifies cancer drivers across tumor types,” Nat. Methods, vol. 10, no. 11, pp. 1081–1082, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Tamborero D et al. , “Comprehensive identification of mutational cancer driver genes across 12 tumor types,” Sci. Rep, vol. 3, October 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Peng Z et al. , “Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome,” Nat. Biotechnol, vol. 30, no. 3, pp. 253–260, 2012. [DOI] [PubMed] [Google Scholar]
- [11].Piskol R, Ramaswami G, and Li JB, “Reliable identification of genomic variants from RNA-seq data,” Am. J. Hum. Genet, vol. 93, no. 4, pp. 641–651, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tang X et al. , “The eSNV-detect: a computational system to identify expressed single nucleotide variants from transcriptome sequencing data,” Nucleic Acids Res, p. gku1005, 2014. [DOI] [PMC free article] [PubMed]
- [13].Dobin A et al. , “STAR: ultrafast universal RNA-seq aligner,” Bioinformatics, vol. 29, no. 1, pp. 15–21, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].McKenna A et al. , “The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data,” Genome Res, vol. 20, no. 9, pp. 1297–1303, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chen T and Guestrin C, “Xgboost: A scalable tree boosting system,” in Proceedings of the 22Nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, 2016, pp. 785–794. [Google Scholar]
- [16].O’Rawe J et al. , “Low concordance of multiple variant-calling pipelines: practical implications for exome and genome sequencing,” Genome Med, vol. 5, no. 3, p. 1, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sahin AA, Edgerton ME, Murray JL, and Bondy M, “Copy Number Imbalances between Screen-and Symptom-Detected Breast Cancers and Impact on Disease-Free Survival,” 2011. [DOI] [PMC free article] [PubMed]
- [18].Meric-Bernstam F et al. , “A Decision Support Framework for Genomically Informed Investigational Cancer Therapy,” J. Natl. Cancer Inst, vol. 107, no. 7, p. djv098, July 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Costello M et al. , “Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation,” Nucleic Acids Res, p. gks1443, 2013. [DOI] [PMC free article] [PubMed]
- [20].Cibulskis K et al. , “Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples,” Nat. Biotechnol, vol. 31, no. 3, pp. 213–219, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Heppner GH, Dexter DL, DeNucci T, Miller FR, and Calabresi P, “Heterogeneity in drug sensitivity among tumor cell subpopulations of a single mammary tumor,” Cancer Res, vol. 38, no. 11 Part 1, pp. 3758–3763, 1978. [PubMed] [Google Scholar]
- [22].Walker R et al. , “Evolutionary conservation and somatic mutation hotspot maps of p53: correlation with p53 protein structural and functional features,” Oncogene, vol. 18, no. 1, pp. 211–218, 1999. [DOI] [PubMed] [Google Scholar]
- [23].Ding L et al. , “Somatic mutations affect key pathways in lung adenocarcinoma,” Nature, vol. 455, no. 7216, pp. 1069–1075, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kandoth C et al. , “Mutational landscape and significance across 12 major cancer types,” Nature, vol. 502, no. 7471, pp. 333–339, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hollstein M, Sidransky D, Vogelstein B, and Harris CC, “p53 mutations in human cancers,” Science, vol. 253, no. 5015, pp. 49–54, 1991. [DOI] [PubMed] [Google Scholar]
- [26].Schuster-Böckler B and Lehner B, “Chromatin organization is a major influence on regional mutation rates in human cancer cells,” nature, vol. 488, no. 7412, p. 504, 2012. [DOI] [PubMed] [Google Scholar]
- [27].Cabili MN et al. , “Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses,” Genes Dev, vol. 25, no. 18, pp. 1915–1927, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]