
Fig. 1.
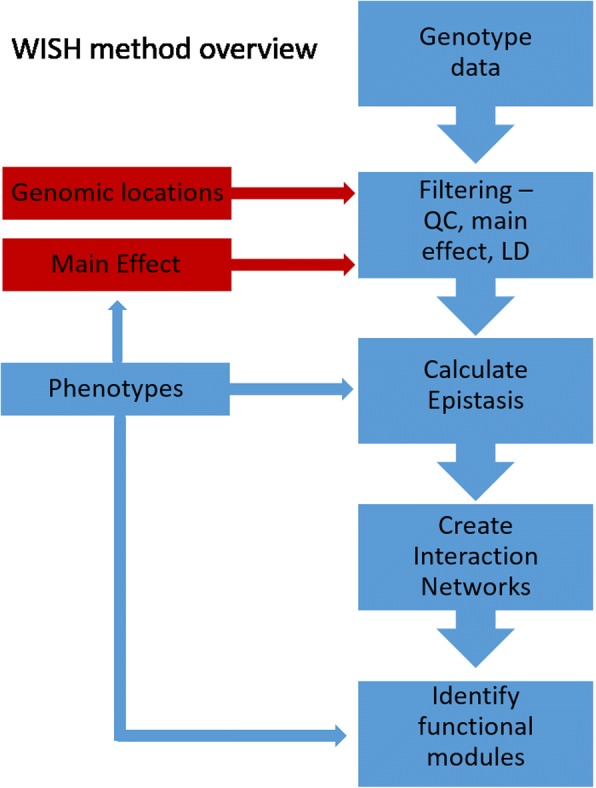
Overview of the package pipeline and workflow. The method only requires phenotypes and genotype data to run. The boxes in red are optional but recommended. The genotype data should be input using the PLINK ped and tped format [14]. Phenotypes can be either continuous or binary. The WISH method can be separated into three overall parts: QC and data filtering, calculation of epistasis and network and module generation. The QC should be similar to a standard GWAS based on call rates and minor allele frequency. An additional step can be done to filter based on LD, which is built into the package. The calculation of epistasis is the most computationally heavy part and is fully parallelized. The network and module construction part is based on converting the epistatic coefficients into correlations and running the WGCNA pipeline, which is integrated into the WISH package