

Abstract
Proteins of the Rad51 family play a key role in homologous recombination by carrying out DNA strand exchange. Here, we present the methodology and the protocols for the 4-strand exchange between gapped circular DNA and homologous linear duplex DNA promoted by human Rad51 and Escherichia coli RecA orthologs. This reaction includes formation of joint molecules and their extension by branch migration in a polar manner. The presented methodology may be used for reconstitution of the medial-to-late stages of homologous recombination in vitro as well as for investigation of the mechanisms of branch migration by helicase-like proteins, e.g., Rad54, BLM, or RecQ1.
1. INTRODUCTION
The recombinases of the Rad51 family, which include eukaryotic Rad51 and Dmc1, bacterial RecA, and archaeal RadA orthologs, promote DNA strand exchange between homologous DNA molecules (Kowalczykowski, 2015). In this reaction, Rad51–ssDNA filament invades the homologous duplex DNA to produce a joint molecule, in which the incoming ssDNA forms a heteroduplex with the complementary DNA strand displacing the identical strand of the duplex. Rad51 recombinases can promote extension of joint molecules via a kinetically and mechanistically distinct reaction, known as 3-strand branch migration. Experimental data indicate that branch migration is driven by cycles of recombinase dissociation, reassociation, and polymerization on the displaced ssDNA of joint molecules directed toward the ssDNA–dsDNA junction (Rossi, Mazina, Bugreev, & Mazin, 2011). Consequently, the branch migration activity depends on ATP hydrolysis which requires for dissociation of Rad51 recombinases from DNA (Kowalczykowski, 1991). In contrast, the Rad51 DNA strand exchange activity does not require protein dissociation from DNA and can be carried out in the presence of nonhydrolyzable ATP analogs. The polarity of branch migration is determined by the direction of the recombinase polymerization on the displaced ssDNA strand; e.g., it is 5′–3′ for RecA and 3′–5′ for hRad51 (Konforti & Davis, 1992; Kowalczykowski, 1991; Rossi et al., 2011).
When DNA heteroduplex extension reaches the ssDNA–dsDNA junction on the invading tailed dsDNA strand, the 3-stranded joint molecule is converted into a 4-stranded crossover junction, known as the Holliday junction (Fig. 1). Rad51 recombinases can promote migration of Holliday junctions (Cunningham, DasGupta, Shibata, & Radding, 1980; Murayama, Kurokawa, Mayanagi, & Iwasaki, 2008; Murayama, Tsutsui, & Iwasaki, 2011; Robu, Inman, & Cox, 2001; Rossi, Mazina, Bugreev, & Mazin, 2010; West, Cassuto, Mursalim, & Howard-Flanders, 1980), although with a lower efficiency than specialized helicase-like branch migration proteins, e.g., RuvAB or Rad54 (Mazina, Rossi, Deakyne, Huang, & Mazin, 2012). The 4-strand branch migration activity is mechanistically similar to the 3-strand branch migration and strictly depends on ATP hydrolysis.
Fig. 1.

The scheme of 4-strand exchange reaction promoted by hRad51 and E. coli RecA. (A) Joint molecules with the 3′-ssDNA displaced strand are produced by hRad51 using gapped DNA and XhoI-linearized pBS II SK(+) dsDNA. (B) Joint molecules with the 5′-ssDNA displaced strand are produced by RecA using gapped DNA and AlwNI-linearized pBS II SK(+) dsDNA. Curved arrows show the direction of polymerization of hRad51 (3′ → 5′) and RecA (5′ → 3′) on the ssDNA region of gapped DNA to initiate the reactions.
Here, we describe reconstitution of the 4-strand DNA exchange, between circular dsDNA containing a region of ssDNA (gapped DNA) and homologous linear dsDNA (Fig. 1). The DNA strand exchange (invasion) step is initiated at the single-stranded gap to produce joint molecules (σ-structures) that are then extended by 3-strand branch migration. As the reaction proceeds beyond the gap, joint molecules are converted into α-structures that undergo 4-strand branch migration to produce nicked circular DNA and displaced tailed dsDNA products (Fig. 1). We describe the use of human Rad51 and Escherichia coli RecA which promote the 4-strand DNA exchange with the opposite polarities, the 3′–5′ and 5′–3′, respectively (Fig. 1). The described methodology can be applied for studies of branch migration promoted by Rad51 recombinases (Rossi et al., 2011), and also by helicase-like proteins, e.g., Rad54, BLM, or RecQ1 (Mazina et al., 2012).
2. PREPARATION OF DNA SUBSTRATES FOR 4-STRAND DNA EXCHANGE
2.1. Preparation of Gapped DNA Substrate
The preparation of gapped circular DNA takes several days and includes the following steps (Fig. 2): (i) digestion of plasmid DNA with two restriction enzymes to produce a dsDNA fragment that is shorter than the original plasmid DNA by several hundred bp (the size of the future gap); (ii) isolation of the dsDNA fragment by agarose gel electrophoresis, its excision from the gel and electroelution; (iii) denaturation of the purified dsDNA fragment and its annealing to circular ssDNA to produce gapped DNA; (iv) purification of the gapped DNA by agarose gel electrophoresis, followed by gel excision and electroelution.
Fig. 2.

Construction of gapped DNA. (i) pBS II SK(+) plasmid DNA is cleaved with XhoI and AlwNI restriction endonucleases. (ii) The 2065 bp XhoI–AlwNI dsDNA fragment is purified by electrophoresis in agarose gels, then (iii) denatured and annealed to circular ssDNA to produce gapped DNA. (iv) Finally, the gapped DNA is purified by electrophoresis in agarose gels.
Duplex and circular ssDNA may be derived from ssDNA bacteriophages (e.g., M13, φX174), or from phagemids (e.g., pBS II). The phagemids are smaller in size and preferable for preparation of gapped DNA, because the efficiency of DNA elution from agarose gels deceases significantly with the increase in DNA size.
2.1.1. Equipment
Standard agarose gel running setup (Bio-Rad Wide Mini-Sub Cell GT)
AlphaImager 3400 gel documentation station (Alpha Innotech)
Vortex-2 Genie (Scientific Industries)
Low-speed benchtop centrifuge
Heat blocks or water baths set at 37°C and 65°C
UV lamp
Electroelution apparatus: Schleicher&Schuell Elutrap (Whatman)
Micro Bio-Spin 6 columns (Bio-Rad, cat# 732-6221)
50-μL quartz cuvettes (Agilent Technologies, cat# 5062-2496)
2.1.2. Buffers and Reagents
NanoPure water
XhoI restriction endonucleases (NEB)
AlwNI restriction endonucleases (NEB)
Bovine serum albumin (BSA), 20 mg/mL (NEB, cat# B9000S)
Ethanol 200 proof
TAE buffer: 40 mM Tris, 20 mM acetic acid, 1 mM EDTA, pH 8.0
TE buffer: 10 mM Tris–HCl, 1 mM EDTA, pH 8.0
Agarose, Type I-A, Low-EEO (Sigma cat# A0169) for analytical electrophoresis
Agarose, Certified Molecular Biology (Bio-Rad cat# 161-3101) for purification of dsDNA and gapped DNA molecules by electroelution
1-kb DNA ladder Molecular Weight Marker
10 × DNA loading buffer for agarose gels (0.25% bromophenol blue, 50% glycerol, 1 mM EDTA, pH 8.0)
Ethidium bromide staining solution, 2 μg/mL in water
1-Butanol ≥99.4% ACS reagent (Sigma cat# 360465)
10 × annealing buffer: 250 mM Tris-acetate, pH 7.5, 500 mM NaCl
Formamide ≥99.5% (Sigma cat# F9037)
2.1.3. Procedure for Preparation of Linear dsDNA Fragment
Carry out the cleavage of pBS II SK (+) plasmid dsDNA with XhoI and AlwNI restriction endonucleases. Next, separate the 2065 and 896 bp dsDNA fragments (Fig. 2i) by agarose gel electrophoresis. Excise the 2065 bp dsDNA fragment from the gel and electroelute it. Note: load not more than 60 μg of pBS II SK (+) plasmid DNA per a 10 × 15 × 0.5-cm agarose gel. To scale up dsDNA fragment preparation use larger gels or multiple gels.
Take two 1.5-mL Eppendorf test tubes. In each tube, mix 30 μg pBS II SK (+), 30 μL 10 × NEBuffer 2, 3 μL BSA (10 mg/mL), 6 μL XhoI (20 units/μL), and water to 300 μL. Next steps are described for each 300 μL reaction.
Incubate reaction mixture at 37°C for 1 h.
Test completeness of XhoI digestion. To do that, withdraw 1 μL (100 ng of DNA) out of the reaction mixture and combine it with 5 μL TE buffer (pH 8.0) premixed with 1 μL of 10 × DNA loading buffer. Continue the incubation of the remaining 299 μL at 37°C until the test will confirm full digestion.
Load DNA samples onto a 6-cm long 0.8% agarose gel and run analytical gel electrophoresis. Use intact pBS II SK (+) plasmid DNA (100 ng) and a 1-kb DNA ladder as migration markers.
Stain the gel with ethidium bromide for 30 min at room temperature, destain for 30 min in a large volume of water, and visualize using an AlphaImager gel documentation station.
If XhoI digest is incomplete, check completeness of XhoI digestion again after 2 h from the beginning of the reaction. If XhoI digest is complete, heat the reaction at 65°C for 20 min to inactivate XhoI, and then cool it on ice. Prepare XhoI-linearized DNA sample as described in step 3 and keep at −20°C. This sample will be used later as a marker during test for completeness of AlwNI digestion.
Precipitate XhoI-linearized DNA with ethanol. To do that, add 15 μL of 5 M NaCl to 300 μL of reaction mixture (to 300 mM including 50 mM NaCl in 1 × NEB buffer 2), mix well, and then add 800 μL of ethanol. Mix and incubate at −20°C for at least 1 h (or overnight).
Centrifuge at >16,000 g for 30 min at 4°C, discard the supernatant, and dry the pellet. Resuspend the pellet (30 μg) in reaction mixture, containing 261 μL of water and 30 μL of 10 × NEBuffer 4, and then add 9 μL of AlwNI (10 units/μL).
Perform steps 3–8, except that the pellet has to be resuspended in 150 μL of TE buffer (pH 8.0) and proceed to purification of the 2065 bp DNA fragment.
Add 22 μL of DNA loading buffer to each of the two DNA samples, combine them (total volume 344 μL) and load onto a 1.1% agarose gel (10 × 15 × 0.5 cm). Modify a standard 20-well comb using a tape to create a 5-well comb with 2 preparative wells (~50 mm wide) and 3 analytical wells (5 mm wide) (Fig. 3A). Load 7 μL samples in each of the analytical wells (lanes A, C, and E), split the remaining 323 μL between two preparative wells (lanes B and D). Run the gel in TAE buffer at 1.7V/cm for 13–14 h at room temperature. No ethidium bromide should be present in the gel or the running buffer since any residual ethidium bromide in the DNA sample will inhibit subsequent DNA strand exchange.
Following electrophoresis, excise lanes A, C, and E from the gel and stain them with ethidium bromide. Reassemble the gel and visualize it under a UV lamp, revealing two bands. Using lanes A, C, and E as markers, excise the sections of agarose containing the 2065 bp DNA fragment from lanes B and D (dashed white boxes in Fig. 3A).
Place two agarose gel slices into one Schleicher and Schuell Elutrap following the manufacturer’s instructions. The DNA is extracted by electroelution at 150 mA for 4 h using TAE buffer. To increase the yield of extracted DNA do second elution at 150 V for 4 h or at 75 V overnight.
Concentrate the recovered DNA in each eluate (500–930 μL) to ~200 μL by extraction with 1-butanol. To do that, add equal volume of 1-butanol, mix well, and separate two phases by centrifugation in a benchtop centrifuge. Discard the upper phase. Repeat this step until the volume of DNA solution reaches ~200 μL. Precipitate the DNA by adding 1/10 volume of 3 M sodium acetate, pH 7.0, and 2.5 volumes of ethanol. Incubate for at least 1 h (or overnight) at −20°C. Centrifuge at >16,000 × g for 30 min at 4°C, dry the pellets, and resuspend each in 20 μL of TE buffer. Because 1-butanol extraction increases salt concentration in DNA samples, desalting is needed. Combine the DNA from eluate #1 and #2 (~40 μL) and pass it through a Micro-BioSpin 6 column equilibrated with 10 mM Tris–HCl, pH 8.0. Determine the DNA concentration on a spectrophotometer at 260 nm (usually at 1:40 dilution) using 50-μL cuvettes and the ratio of 1 unit OD260 = 50 μg/mL. Good yields are in the range of 20–27 μg of dsDNA (~40% of the theoretical yield).
Check the quality of the purified 2065 bp ds DNA fragment (Fig. 4, lane 3) as described in steps 3 and 5. Run a sample of XhoI–AlwNI digested pBS II K (+) plasmid DNA (100 ng) and a 1-kb DNA ladder along with the purified 2065 bp dsDNA fragment (2 μL or ~100 ng) to judge its purity and concentration.
Fig. 3.
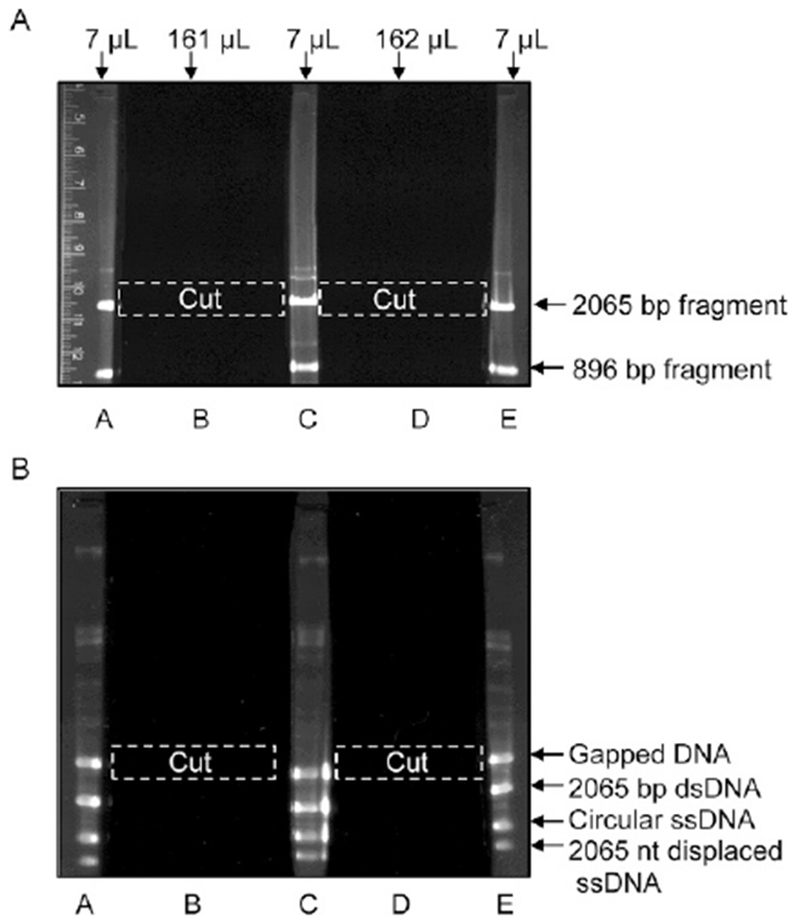
Purification of the 2065 bp XhoI–AlwNI dsDNA fragment of pBS II SK(+) (A) and the gapped DNA (B) by electrophoresis in agarose gels. (A) After electrophoresis, lanes A, C, and E are excised from the gel and stained with ethidium bromide to visualize the DNA bands. Using these bands as markers, the area of the gel with the 2065 bp dsDNA fragment (A) and with the gapped DNA (B) is cut out of lanes B and D (dashed white boxes), and then the DNA is extracted from the gel by electroelution.
Fig. 4.

Analysis of the DNA substrates, intermediates, and gapped DNA product by electrophoresis in a 1.4% agarose gel. Lane 1: 1-kb DNA ladder. Lane 2, 3, and 5: purified circular ssDNA pBS II SK (+) (100 ng), 2065 bp dsDNA (125 ng), and gapped DNA (125 ng), respectively. Lane 4: DNA products of the annealing reaction (415 ng).
2.1.4. Procedure of Preparation of Gapped DNA
Prepared circular ssDNA of pBS II SK (+) as described in Sambrook and Russell (2001). Because pBluescript II can be secreted from E. coli cells in a circular ssDNA form only in the presence of VCSM13 helper phage, a small amount of this phage ssDNA (~6 kb) is always present in the pBluscript II ssDNA preparation.
Perform a trial small-scale annealing reaction (22 μL) to determine the optimum ratio between circular ssDNA and dsDNA fragment for the maximal gapped DNA yield. Typically, 1 μg of linear duplex fragment is mixed with 1, 1.5, and 2 μg of circular pBS II SK (+) ssDNA in 1 × annealing buffer and 50% formamide.
Incubate the mixture for 5 min at 75°C, vortex, spin the tube for 2s in a low-speed benchtop centrifuge just to collect the liquid, and continue the incubation for another 5 min to denature the dsDNA fragment.
Carry out annealing at room temperature for at least 3 h (or overnight).
To analyze DNA after annealing, withdraw 5 μL (450–680 ng) from each reaction mixture, combine with 4 μL TE buffer (pH 8.0) premixed with 1 μL of 10 × DNA sample buffer and load on a 10-cm long 1.4% agarose gel along with circular ssDNA (100 ng) and purified 2065 bp dsDNA fragment (100 ng). DNA samples can be stored at −20°C and used later as a marker (Fig. 4, lane 4) for testing the quality of purified gapped DNA.
Based on the obtained result, prepare a large-scale annealing reaction. Take two 1.5-mL Eppendorf tubes. Typically, in each tube we mix 17 μg of the linear dsDNA fragment, 17 μg of circular ssDNA (~1.4-fold excess in terms of DNA molecules), 37.4 μL 10 × annealing buffer, 188 μL of formamide (99.5%), and water to 374 μL.
Perform annealing reaction as described in steps 2 and 3.
Precipitate DNA in each annealing reaction by adding 34 μL of 3 M sodium acetate, pH 7.0, and 1020 μL of ethanol. Mix and incubate at −20°C for at least 1 h (or overnight).
Centrifuge at >16,000 × g for 30 min at 4°C, dry the pellets, and resuspend each pellet in 150 μL of TE buffer (pH 8.0).
Perform electrophoresis as in Section 2.1.3, step 10, except that percentage of an agarose gel has to be 1.4% to improve the separation of the DNA products.
Following electrophoresis, excise lanes A, C, and E and stain them with ethidium bromide (Fig. 3B). Reassemble the gel and visualize it under a UV lamp, to reveal four major bands. Using DNA in lanes A, C, and E as markers, excise the uppermost band (the gapped DNA) from lanes B and D (dashed white boxes in Fig. 3B). Be precise when cutting out this band, it is easy to pick up the 2065 bp dsDNA that migrates slightly faster than the gapped DNA.
Electroelute the DNA, as in Section 2.1.3, step 12.
Concentrate DNA from the eluates #1 and #2 (500–930 μL) to 100–150 μL with 1-butanol as in Section 2.1.3, step 13.
Determine the final concentration of gapped DNA on a spectrophotometer at 260 nm (usually at dilution 1:10) using 50-μL cuvettes and the ratio 1 OD260 = 45 μg/mL. Good yields are in the range 8–10 μg of gapped DNA.
Check the quality of purified gapped DNA by electrophoresis in a 10-cm long 1.4% agarose gel (Fig. 4, lane 5). Run a sample of the small-scale annealing reaction (5 μL containing 450 ng of DNA) (Fig. 4, lane 4) along with the purified gapped DNA (100 ng) to judge its purity.
2.1.5. Notes
We do not cleave pBS II SK (+) plasmid dsDNA simultaneously with XhoI and AlwNI, although both enzymes are active in NEBuffer 2. First, the mobilities of pBS II SK (+) supercoiled plasmid DNA and 2065 bp dsDNA fragment are similar making it difficult to judge the completeness of digestion. Second, AlwNI is more active in NEBuffer 4. After AlwNI digest, we precipitate DNA with ethanol to remove salt from the sample and to reduce its volume. This allows to have more sharp DNA bands during preparative gel electrophoresis, and therefore to minimize the width of the gel slices. Reduction of reaction volume during AlwNI digest will increase the DNA concentration above 100 μg/mL that results in an incomplete digestion regardless of restriction endonuclease concentration and incubation time. During the gel excision step, be sure to minimize the width of the gel slices, as an excess of agarose will decrease the recovery of DNA. After removing the bands, stain the remainder of the gel with ethidium bromide to determine how successful the band excision was. Minor bands that migrate slower than the gapped DNA likely represent VCSM13 helper phage DNA (Fig. 4, lane 4).
2.2. Labeling of Linear dsDNA
For quantification of the 4-strand reaction, use 32P-labeled DNA joint molecules. Also, these joint molecules need to be suitable for branch migration in either the 5′–3′ or the 3′–5′ direction (Fig. 1). For this purpose, we prepare XhoI- and AlwNI-linearized dsDNA molecules labeled with 32P at the 5′- and 3′-end, respectively.
2.2.1. Equipment
Equipment is as in Section 2.1.1
2.2.2. Buffers and Reagents
Shrimp Alkaline Phosphatase (USB cat# 70092Y)
[γ-32P] ATP 6000 Ci/mmol, 10 mCi/mL (Perkin Elmer cat# BLU002Z250UC)
T4 polynucleotide kinase (NEB cat# M0201S)
Terminal deoxynucleotidyl transferase (NEB cat# M0315S).
[α-32P] dCTP 6000 Ci/mmol, 10 mCi/mL (Perkin Elmer cat# BLU013Z)
Other buffers and reagents are as in Section 2.1.2
2.2.3. Procedure of 32P-Labeling of XhoI-Linearized pBS II SK(+) dsDNA at the 5′-End
The ssDNA region of gapped DNA is defined by the AlwNI and XhoI restriction sites of pBS II SK (+). The ssDNA of the gap runs in a 5′–3′ direction from the AlwNI to the XhoI site (Fig. 1). To produce joint molecules with a 3′-displaced ssDNA, pBS II SK (+) plasmid DNA needs to be digested with XhoI (Fig. 1A). XhoI creates a 5′-protruding DNA end that can readily be 32P-labeled with T4 polynucleotide kinase.
Mix in a tube 10 μg of pBS II SK (+) plasmid DNA, 10 μL of 10 × NEBuffer 2, 10 μL of BSA (1 mg/mL), 2 μL of XhoI (20 units/μL), 2 μL of Shrimp Alkaline Phosphatase (1 unit/μL), and water to 100 μL. The phosphatase removes the 5′ terminal phosphate group left on DNA ends after XhoI digestion, producing the OH-end suitable for incorporation of 32P label.
Prepare XhoI-linearized dsDNA as in Section 2.1.3, steps 2–8, except that the pellet has to be resuspended in 20 μL of Tris–HCl, pH 8.0, to give a DNA concentration ~0.5 μg/μL.
Radiolabel 5 μg of XhoI-linearized pBS II SK (+) DNA in a 40-μL reaction mixture containing 100 μCi of [γ-32P] ATP (10 mCi/mL), 1 × T4 polynucleotide kinase buffer, and 2 units of T4 polynucleotide kinase. Incubate the reaction for 1 h at 37°C, and then inactivate the enzyme for 20 min at 65°C. Remove unincorporated [γ-32P] ATP using a Micro-BioSpin 6 column equilibrated with 10 mM Tris–HCl, pH 8.0, as described by the manufacturer.
Determine the DNA concentration on a spectrophotometer at 260 nm (usually at dilution 1:10) using 50-μL cuvettes and the ratio 1 OD260 = 50 μg/mL.
2.2.4. Procedure of 32P-Labeling of AlwNI-Linearized pBS II SK(+) dsDNA at the 3′-End
Joint molecules can also be constructed with a 5′-end displaced ssDNA strand using pBS II SK (+) linear dsDNA produced by AlwNI (Fig. 1B). AlwNI generates DNA ends with 3′ overhangs, which may be 3′ radio-labeled with terminal deoxynucleotidyl transferase.
Mix in 100 μL reaction 10 μg of pBS II SK (+) plasmid DNA, 10 μL of 10 × NEBuffer 4, and 4 μL of AlwNI (10 units/μL).
Prepare AlwNI-linearized dsDNA as in Section 2.1.3, steps 2–8, except that the pellet has to be resuspended in 20 μL of Tris–HCl, pH 8.0, to give a DNA concentration ~0.5 μg/μL.
Radiolabel 5 μg of AlwNI-linearized pBS II K (+) plasmid DNA in a 40-μL mixture containing 16 μCi of [α-32P] dCTP (10 mCi/mL) and terminal deoxynucleotidyl transferase (10 units) in cacodylate buffer as described by the manufacturer.
Incubate the reaction for 15 min at 37°C, then inactivate the enzyme for 10 min at 75°C. Remove unincorporated [α-32P] dCTP using a Micro-BioSpin 6 column equilibrated with 10 mM Tris–HCl, pH 8.0, as described by the manufacturer.
Determine the DNA concentration on a spectrophotometer at 260 nm (usually at dilution 1:10) using 50-μL cuvettes and the ratio 1 OD260 = 50 μg/mL.
2.2.5. Notes
The number of incorporated nucleotide residues depends on the ratio 3′ DNA ends:dNTP. We use 5.2 pmol of 3′-DNA ends and 2.6 pmol of [α-32P] dCTP. Under these conditions, dCTP and dGTP provide smaller number of incorporated nucleotide residues per a DNA end (1–3 nt) than dATP or dTTP (1–5 nt), and therefore are preferable. [α-32P] ddNTP that allows only a single nucleotide incorporation is even better, though is more expensive.
3. PREPARATION OF JOINT MOLECULES
We use hRad51 and RecA to prepare joint molecules with different polarities, as substrates for the 4-strand branch migration. Optimal conditions for the joint molecule preparation need to be determined for specific stocks of the proteins, gapped DNA, and 32P-labeled linear duplex DNA. For this purpose, conduct small-scale reactions (10 μL), as described in Section 3.1.3, varying the concentrations of DNA and the proteins. Using optimal conditions scale-up the reaction to 90 μL.
3.1. Preparation of 3′-Joint Molecules
3.1.1. Equipment
Typhoon FLA 7000 Phosphor imaging system (GE Healthcare)
Whatman DE81 chromatography paper (GE Healthcare cat# 3658-915) or Amersham Hybond-N+ membrane (GE Healthcare cat# RPN1520B)
Illustra MicroSpin S-400 HR column (GE Healthcare cat# 27-5140-01)
Other equipment is as in Section 2.1.1
3.1.2. Buffers and Reagents
hRad51 was purified as described (Sigurdsson, Trujillo, Song, Stratton, & Sung, 2001)
Human Replication protein A (hRPA) was purified as described (Henricksen, Umbricht, & Wold, 1994)
Proteinase K, 20 mg/mL (Roche cat# 03115828001)
Rad51 strand exchange buffer: 25 mM Tris-acetate, pH 7.5, 275 mM NaCl, 2 mM ATP, 1 mM MgCl2, 1 mM CaCl2, 1 mM DTT, and 100 μg/mL BSA
3 × deproteinization buffer: 4.8 mg/mL proteinase K, 3% SDS, 18% glycerol, and 0.03% bromophenol blue
Other buffers and reagents are as in Section 2.1.2
3.1.3. Procedure of Preparation of 3′-Joint Molecules
hRad51 polymerizes on ssDNA preferentially in the 3′–5′ direction (Fig. 1A) and promotes strand exchange between gapped DNA and linear dsDNA in the same direction, generating the displaced 3′-ssDNA strand.
To prepare joint molecules with a 3′-displaced ssDNA strand (90 μL final volume), mix 20 μM (nt) gapped DNA with 5 μM hRad51 in Rad51 strand exchange buffer. Incubate for 10 min at 37°C, then add 0.4 μM hRPA, incubate for another 10 min, and finally add 20 μM (nt) 32P-labeled XhoI-linearized dsDNA to initiate joint molecule formation. Incubate the mixture for 2 h at 37°C.
Withdraw a 10-μL aliquot to estimate the extent of joint molecule formation. Incubate the remaining 80 μL for another 20 h, and then freeze it in dry ice. Store the 80 μL at −80°C.
Mix the 10 μL aliquot with 5 μL of 3 × deproteinization buffer. Incubate for 15 min at 37°C to deproteinize DNA products.
Separate the reaction products by electrophoresis in a 1.5% agarose gel (10 × 15 × 0.5 cm). Run the gel in TAE buffer at 5V/cm for 1.5 h at room temperature.
Dry the gel on DE81 chromatography paper.
Visualize and quantify 32P-labeled products using Typhoon FLA 7000 Phosphor imaging system. If the yield is in the range of 50%–60%, the joint molecules can be purified (see Section 3.3) and used for testing branch migration activities.
3.2. Preparation of 5′-Joint Molecules
3.2.1. Equipment
Equipment is as in Section 3.1.1.
3.2.2. Buffers and Reagents
RecA protein (USB cat# 70028Z)
ssDNA-binding protein (SSB) (USB cat# 70032Y)
RecA strand exchange buffer: 35 mM Tris–HCl, pH 7.5, 3 mM ATP, 15 mM MgCl2, 2 mM DTT, and 100 μg/mL BSA
Phosphocreatine (Sigma cat# P7936)
Creatine phosphokinase (CalBiochem cat# 2384)
Other buffers and reagents are as in Section 3.1.2
3.2.3. Procedure for Preparation of 5′-Joint Molecules
RecA polymerizes on ssDNA preferentially in the 5′–3′ direction (Fig. 1B) and promotes strand exchange in the 5′–3′ direction, generating the displaced 5′-ssDNA strand.
To prepare joint molecules with a 5′-ssDNA displaced strand (90 μL final volume), mix 20 μM (nt) of gapped DNA with 4 μM RecA in RecA strand exchange buffer supplemented with 10 mM phosphocreatine, and 10 units/mL creatine phosphokinase (an ATP regeneration system). Incubate for 5 min at 37°C, then add 0.33 μM SSB, incubate for another 1 min, and finally add 20 μM (nt) 32P-labeled AlwNI-linearized dsDNA to initiate joint molecule formation. Incubate for 10 min at 37°C.
Withdraw a 10-μL aliquot for the analysis and freeze the remaining 80 μL of the reaction on dry ice. Store at −80°C.
Estimate the extent of joint molecule formation in a 1.5% agarose gel as described in Section 3.1.3, steps 3–6. If the yield is in the range of 50%–60%, the joint molecules can be purified (see Section 3.3) and used for testing branch migration activities.
3.3. Deproteinization and Purification of Joint Molecules
3.3.1. Equipment
Equipment is the same as in Section 3.1.1.
3.3.2. Buffers and Reagents
0.5 M EDTA (pH 8.0)
Other buffers and reagents are as in Section 3.1.2
3.3.3. Procedure of Deproteinization and Purification of Joint Molecules
Following joint molecule formation (by hRad51 or RecA), add 8 μL 10% SDS and 8 μL 20 mg/mL proteinase K to the reaction mixture (80 μL), mix well, and incubate for 15 min at 37°C to deproteinize DNA, and then add 2 μL 98 mM EDTA, pH 8.0, to chelate Ca2+ or Mg2+.
Prepare two S-400 Spin columns by equilibrating them with 30 mM Tris-acetate, pH 7.5, as described by the manufacturer.
Pass deproteinized joint molecules (98 μL) through first S-400 Spin column, and then apply the recovered eluate #1 (98–100 μL) to the second S-400 Spin column.
Add 2–10 mM MgCl2 to the recovered eluate #2 to inhibit spontaneous branch migration and store at −20°C. The concentration of the purified joint molecules (typically 0.5–1 nM molecules) can be calculated by comparing their radioactivity to a known amount of 32P-labeled dsDNA after electrophoresis on the same gel.
3.3.4. Notes
As a control, always determine the extent of protein-independent (spontaneous) joint molecule formation by replacing Rad51/RecA with their storage buffers.
Passage of deproteinized joint molecules through the second S-400 Spin column is essential to remove all SDS traces, which can still be present in the eluate after the first column.
4. BRANCH MIGRATION OF JOINT MOLECULES
4.1. 4-Strand DNA Branch Migration by hRad51
In the presence of Ca2+ and ATP, hRad51 forms a stable filament that promotes formation of joint molecules, but not their branch migration. Thus, the joint molecules will accumulate, reaching the maximum at 22 h. To increase the yield of joint molecules further, 2 mM ATP may be replaced with 2 mM dATP. In the presence of 2 mM dATP, only tiny amounts of branch migration products (nicked circular dsDNA and displaced tailed dsDNA) can be detected after 20 h of incubation (Fig. 5A, lane 3). 4-Strand branch migration is initiated by Ca2+ depletion with EGTA, yielding ~36% of products at 20 h of incubation (Fig. 5A, lane 4).
Fig. 5.

4-strand branch migration of nondeproteinized joint molecules by hRad51 (5 μM) (A) or RecA (4 μM) (B). Reaction scheme is shown in Fig. 1. dsDNA was 32P-labeled. (A) Ca2+ stimulates joint molecule formation but inhibits branch migration by hRad51. The reaction was carried out in the presence of 1 mM Ca2+ and 1 mM Mg2+ for 2 h (lane 2) as described in Section 3.1.3, except that 2 mM ATP was replaced with 2 mM dATP. The reaction mixture was divided between two test tubes. In the first, incubation was continued for another 20 h (lane 3). In the second, EGTA (1.2 mM, pH 8.0) was added to deplete Ca2+ and incubation continued for 20 h (lane 4). (B) RecA promotes strand exchange and 4-strand branch migration under the same reaction conditions as described in Section 3.2.3. Time course of the RecA-promoted 4-strand exchange reaction (lanes 1–5). (C) In the presence of 4 mM Mg2+ and 1 mM ATPγS, RecA (12 μM) promotes joint molecule formation, but not their branch migration (lane 2).
4.1.1. Equipment
Equipment is as in Section 3.1.1
4.1.2. Buffers and Reagents
0.5 M EGTA, pH 8.0
Rad51 branch migration buffer: 30 mM Tris-acetate, pH 7.5, 350 mM NaCl, 2 mM ATP, 10 mM MgCl2, 1 mM DTT, and 100 μg/mL BSA
Other buffers and reagents are as in Section 3.2.2
4.1.3. Procedure for Initiation of hRad51 4-Strand Branch Migration on Nondeproteinized 3′-Joint Molecules
Set up 90 μL of 4-strand exchange reaction as described in Section 3.1.1, step 1. Incubate the mixture for 2 h at 37°C.
Withdraw a 10-μL aliquot to estimate the extent of joint molecule formation.
To initiate branch migration mix the remaining 80 μL with 2 μL of 50 mM EGTA, pH 8.0, and continue incubation at 37°C.
Withdraw 10 μL aliquots at desirable time points and analyze DNA products as in Section 3.1.3, steps 3–6 (Fig. 5A, lane 4).
4.1.4. Procedure for ReInitiation of hRad51 Branch Migration on Purified 3′-Joint Molecules
For hRad51, initiation of 4-strand (forward) branch migration on purified 3′-joint molecules requires 1–10 mM Mg2+, an ATP regeneration system, elevated concentrations of NaCl (350 mM) and hRad51 (10 μM). Ca2+ inhibits the reaction. Purified 5′-joint molecules support only 3-strand (reverse) branch migration that leads to disruption of joint molecules into the original DNA substrates (32P-labeled linear dsDNA and unlabeled gapped DNA).
Mix 0.3–0.5 nM (molecules) 3′-joint molecules in Rad51 branch migration buffer, supplemented with 8 mM phosphocreatine, and 8 units/mL creatine phosphokinase (an ATP regeneration system). Initiate the reaction by the addition of RAD51 (10 μM) and carry it out at 37°C.
Withdraw 10 μL aliquots at desirable time points and analyze DNA products as in Section 3.1.3, steps 3–6.
4.2. 4-Strand DNA Branch Migration by E. coli RecA
RecA forms dynamic filament in the presence of Mg2+, which promotes both strand exchange and branch migration. Branch migration converts joint molecules into 4-strand Holliday junctions and drives them further into nicked circular DNA and displaced tailed DNA products (Fig. 1B). The 10-min reaction time represents a compromise allowing for the largest accumulation of joint molecules (Fig. 5B, lane 2) before the branch migration products become visible (Fig. 5B, lanes 3–5). To produce joint molecules with RecA, but without initiating branch migration, 3 mM ATP can be replaced with 1 mM ATPγS (Fig. 5C, lane 2). In this case, the ATP regeneration system needs to be omitted, RecA concentration increased to 12 μM, MgCl2 concentration decreased to 4 mM, and the reaction time increased from 10 min to 24 h. However, even at 24 h, the yield of RecA-promoted joint molecules in the presence of ATPγS is half as high as in the presence of ATP (30% vs 60%).
4.2.1. Equipment
Equipment is the same as in Section 3.1.1
4.2.2. Buffers and Reagents
RecA branch migration buffer: 30 mM Tris–HCl, pH 7.5, 10 mM MgCl2, 2 mM DTT, and 2 mM ATP
Other buffers and reagents are as in Section 3.2.2
4.2.3. Procedure of Re-Initiation of RecA Branch Migration on Purified 5′-Joint Molecules
For RecA, initiation of 4-strand (forward) branch migration on purified 5′-joint molecules requires the same conditions as the standard 4-strand branch migration. RecA promotes the 4-strand branch migration with the rate approximately 11-fold faster than hRad51.
Purified 3′-joint molecules support only reverse branch migration that leads to disruption of joint molecules into the original DNA substrates (32P-labeled linear dsDNA and unlabeled gapped DNA).
Mix 0.3–0.5 nM (molecules) 5′-joint molecules in RecA branch migration buffer, supplemented with 10 mM phosphocreatine, and 10 units/mL creatine phosphokinase (an ATP regeneration system). Initiate the reaction by the addition of RecA (4 μM) and carry it out at 37°C.
Withdraw 10-μL aliquots at desirable time points and analyze DNA products as in Section 3.1.3, steps 3–6.
4.2.4. Notes
Branch migration of joint molecules can proceed either in the forward (DNA 4-strand branch migration) or the reverse direction (3-strand reaction). This direction is determined by (1) the polarity of the displaced ssDNA strand in the joint molecules and (2) by the intrinsic polarity of branch migration protein. As a control, always determine the extent of protein-independent (spontaneous) branch migration by replacing your branch migration protein with its storage buffer.
5. SUMMARY AND CONCLUSION
Here, we describe the methodology and the protocols for preparation of the joint molecules with the opposite polarities, which can be used to study the mechanisms of branch migration of Holliday junctions promoted by different proteins that play an important role in homologous recombination and DNA repair. The described basic design of these joint molecules can be further modified, e.g., by incorporation the regions of heterology (Mazina et al., 2012). In addition, these joint molecules may serve as substrates for structure-specific endonucleases that are responsible for cleavage of Holliday junctions.
ACKNOWLEDGMENTS
We thank Kritika Hanamshet and Nadish Goyal for the feedback on this protocol. We acknowledge funding from the National Cancer Institute of the National Institutes of Health (NIH) Grant numbers CA188347, P30CA056036 (to A.V.M.) and from Drexel Coulter Program (to A.V.M.) for supporting this work. The authors declare no conflicts of interests.
ABBREVIATIONS
- bp
base pair
- BSA
bovine serum albumin
- dsDNA
double-stranded DNA
- DTT
1,4-Dithiothreitol
- nt
nucleotide
- RPA
replication protein A
- SSB
single-stranded DNA-binding protein
- ssDNA
single-stranded DNA
REFERENCES
- Cunningham RP, DasGupta C, Shibata T, & Radding CM (1980). Homologous pairing in genetic recombination: recA protein makes joint molecules of gapped circular DNA and closed circular DNA. Cell, 20(1), 223–235. [DOI] [PubMed] [Google Scholar]
- Henricksen LA, Umbricht CB, & Wold MS (1994). Recombinant replication protein A: Expression, complex formation, and functional characterization. The Journal of Biological Chemistry, 269(15), 11121–11132. [PubMed] [Google Scholar]
- Konforti BB, & Davis RW (1992). ATP hydrolysis and the displaced strand are two factors that determine the polarity of RecA-promoted DNA strand exchange. Journal of Molecular Biology, 227(1), 38–53. [DOI] [PubMed] [Google Scholar]
- Kowalczykowski SC (1991). Biochemistry of genetic recombination: Energetics and mechanism of DNA strand exchange. Annual Review of Biophysics and Biophysical Chemistry, 20, 539–575. [DOI] [PubMed] [Google Scholar]
- Kowalczykowski SC (2015). An overview of the molecular mechanisms of recombinational DNA repair. Cold Spring Harbor Perspectives in Biology, 7(11), a016410 10.1101/cshperspect.a016410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazina OM, Rossi MJ, Deakyne JS, Huang F, & Mazin AV (2012). Polarity and bypass of DNA heterology during branch migration of Holliday junctions by human RAD54, BLM, and RECQ1 proteins. The Journal of Biological Chemistry, 287(15), 11820–11832. 10.1074/jbc.M112.341347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama Y, Kurokawa Y, Mayanagi K, & Iwasaki H (2008). Formation and branch migration of Holliday junctions mediated by eukaryotic recombinases. Nature, 451(7181), 1018–1021. [DOI] [PubMed] [Google Scholar]
- Murayama Y, Tsutsui Y, & Iwasaki H (2011). The fission yeast meiosis-specific Dmc1 recombinase mediates formation and branch migration of Holliday junctions by preferentially promoting strand exchange in a direction opposite to that of Rad51. Genes and Development, 25(5), 516–527. 10.1101/gad.1997511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robu ME, Inman RB, & Cox MM (2001). RecA protein promotes the regression of stalled replication forks in vitro. Proceedings of the National Academy of Sciences of the United States of America, 98(15), 8211–8218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi MJ, Mazina OM, Bugreev DV, & Mazin AV (2010). Analyzing the branch migration activities of eukaryotic proteins. Methods, 51(3), 336–346. 10.1016/j.ymeth.2010.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi MJ, Mazina OM, Bugreev DV, & Mazin AV (2011). The RecA/RAD51 protein drives migration of Holliday junctions via polymerization on DNA. Proceedings of the National Academy of Sciences of the United States of America, 108(16), 6432–6437. 10.1073/pnas.1016072108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, & Russell DW (Eds.), (2001). In Molecular cloning: A laboratory manual. : Vol. 1 (pp. 1.65–1.68) (3rd ed.). Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Sigurdsson S, Trujillo K, Song B, Stratton S, & Sung P (2001). Basis for avid homologous DNA strand exchange by human Rad51 and RPA. The Journal of Biological Chemistry, 276(12), 8798–8806. [DOI] [PubMed] [Google Scholar]
- West SC, Cassuto E, Mursalim J, & Howard-Flanders P (1980). Recognition of duplex DNA containing single-stranded regions by recA protein. Proceedings of the National Academy of Sciences of the United States of America, 77, 2569–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
FURTHER READING
- Mazina OM, & Mazin AV (2004). Human Rad54 protein stimulates DNA strand exchange activity of hRad51 protein in the presence of Ca2+. The Journal of Biological Chemistry, 279(50), 52042–52051. [DOI] [PubMed] [Google Scholar]