

Abstract
Facile intramolecular amination of unactivated alkenes has been achieved by using electricity as a catalyst that helps to generate an intermediate and accelerates formation of cyclic ureas in high yields. Using this method, no metal catalysts were used. During electrolysis, a nitrogen radical was formed at the urea substrate that cyclised with the alkene and generated a terminal carbon radical which further formed a bond with the 2,2,6,6‐tetramethylpiperidine‐N‐oxyl radical (TEMPO). This method of electrolysis not only gives cyclic ureas but also functionalises terminal unactivated alkenes. This method can be considered to be environmentally friendly given that it avoids the issues of toxicity or complicated metal ligands and could therefore be potentially employed in green chemistry.
Keywords: amination, cyclic ureas, electrochemical oxidation, TEMPO, terminal alkenes
Go green: Several reagents and reagent combinations have been developed for the amination and functionalisation of unactivated alkenes. However, most of these are dependent on oxidants and catalysts. Herein, we report a green approach for the intramolecular amination of urea‐tethered alkenes by generating radicals without using expensive catalysts and employing only electricity.
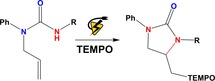
1. Introduction
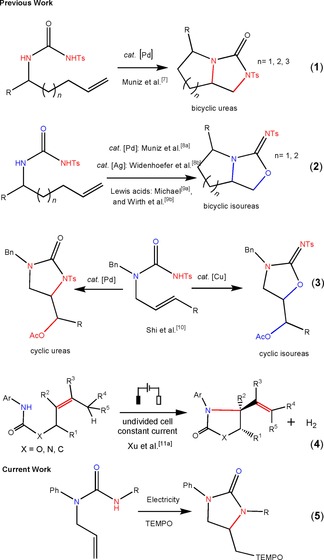
The 1,2‐difunctionalisation of alkenes with heteroatoms is a powerful organic transformation to prepare molecules with diverse new functionalities.1 These functionalised hetero molecules can be used further to investigate prominent biological activities as well as in the synthesis of chiral reagents and natural products. Vicinal difunctionalisation of alkenes and amination reactions mainly depend on transition metal catalysis, hypervalent iodine reagents and photocatalysis.2, 3, 4, 5 The well‐known Sharpless asymmetric aminohydroxylation and dihydroxylation6 catalysed by osmium metal is a famous route for difunctionalisation of alkenes and this method has been followed several decades. Palladium catalysts have been used for intramolecular aminations of unfunctionalised alkenes to the synthesis of bicyclic ureas [Eq. 1].7 Progress has been made in the application of expensive Pt8a and Ag8b catalysts or by using excess amounts of Lewis acids9 [Eq. (2)] to synthesise fused bicyclic isoureas through intramolecular amino‐oxylation of alkenes. In 2015, Shi et al. reported the efficient chemoselective synthesis of ureas and isoureas in high yields by controlling O/N nucleophilic addition in urea‐tethered alkenes by using PhI(OAc)2 as the oxidant in the presence of Pd or Cu catalyst [Eq. (3)].10 Amination and functionalisation of unactivated alkenes in synthesis is a continuing challenge. Several reagents and reagent combinations have been developed for such reactions, but most of them are dependent on oxidants and catalysts. Moreover, electrochemical intramolecular amination of urea‐tethered terminal alkenes has not been reported.11 However, a few cyclisation reactions have been reported employing allylcarbamates and amides as the substrates.11b–11f Xu et al. have been involved in developing sustainable C−N bond‐forming reactions by employing electrochemically generated nitrogen‐centred radical (NCR) intermediates. Difunctionalisation of a variety of alkenes with carbamates/amides and TEMPO affords aminooxygenation products in good yields.11b Furthermore, they envisioned this approach for electrochemical intramolecular oxidative amination reaction of hindered tri‐ and tetrasubstituted alkenes to synthesis alkene‐bearing cyclic carbamates, ureas and lactams [Eq. (4)].11a Wirth et al. developed an electrochemical flow method for the intramolecular oxidative amination of alkenes with TEMPO. They have demonstrated the facile synthesis of amidyl radicals used in intramolecular hydroaminations to produce isoindolinones.11c The electrically catalysed organic reactions occur at the interface of electrodes and an electrolyte. These reactions serve as powerful methods for the synthesis of complex organic molecules as they provide for an easy generation of highly reactive species, even under very mild conditions, and can be consequently applied in green chemistry.12
Cyclic ureas are heterocyclic motifs frequently observed in biologically active molecules.13 They are also involved in important organic transformations and are important precursors for the synthesis of vicinal diamines and amino alcohols.14 Despite their extensive utility, the development of a new method allowing efficient preparation of these heterocycles continues to be a stimulating challenge in organic synthesis. Herein, we have developed an electrochemical method for difunctionalisation of urea‐tethered alkenes and report an economic, environmentally friendly controlled intramolecular amination. This method allows the construction of cyclic ureas in high yields from urea nucleophiles [Eq. (5)]. In this electrochemical reaction, the addition of the anodically generated nitrogen radical15 to a tethered alkene leads to a new terminal carbon radical, which is trapped with 2,2,6,6‐tetramethylpiperidine‐N‐oxyl radical (TEMPO) to afford the final cyclic urea. This electrochemical method precludes the use of stoichiometric quantities of chemical oxidants. The interception of this carbon radical with a trapping reagent (TEMPO)1f would also lead to difunctionalisation of the alkene. Since no second oxidation would be required in such a situation, the reaction might work with an unactivated olefin.
2. Result and Discussion
N‐allylic urea 1 a was used as the first example and submitted to electrochemical difunctionalisation. The reaction was run at 60 °C and a constant current of 10 mA in a three‐necked round‐bottom flask equipped with a reticulated vitreous carbon (RVC) anode and a platinum wire cathode. A solution of 0.1 m Bu4NBF4 in CH3CN/H2O (19:1 v/v) was used as the supporting electrolyte and 1.5 equivalents of TEMPO were included. After 1.8 F mol−1 of electricity was passed through the electrolysis process, the precursor was consumed and the desired product 2 a was isolated in 83 % yield (Scheme 1). 1.1 equivalents of Na2CO3 was used as a base in this reaction.
Scheme 1.

Difunctionalisation of alkenes. Reaction conditions: 1 a (0.35 mmol), TEMPO (0.53 mmol), H2O (0.5 mL), CH3CN (9.5 mL), 60 °C. 2 a was isolated in 83 % yield.
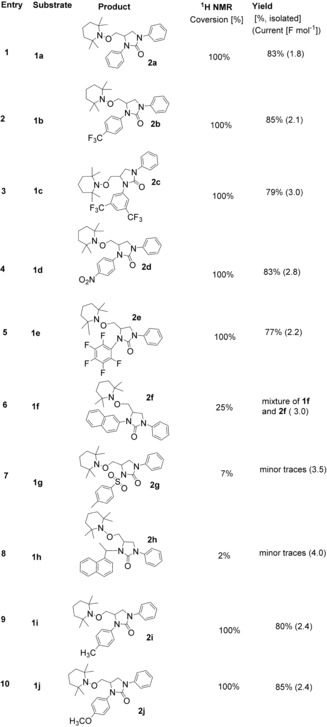
After setting optimised conditions in electrolysis, the substrate scope was examined on the anodically catalysed difunctionalisation of alkenes. The NH‐phenyl ring of 1 a was modified with various functional groups to investigate their effects on alkene difunctionalisation. All substrates were synthesised in high yields (Scheme 2, Figure 1). For their synthesis, allylamine (3, 1.1 equiv) was added to a stirred solution of corresponding aryl isocyanate (4, 1.0 equiv) in dry CH2Cl2 (DCM) at 0 °C and then reaction was allowed to warm to room temperature and stirred for 4 h to obtain the desired urea substrate 1. The substrate bearing functional groups, such as mono‐ and di‐substituted Me, MeO, CF3, NO2, fluoro and benzyl, could proceed smoothly in the electrochemical system (see Table 1, 2 a–e, 2 i,j). The introduction of a tosyl and naphthyl groups, however, led to dissatisfactory results, which was probably caused by the decomposition of the product 2 f–h during the electrolysis (Table 1) or due to the steric hindrance of bulky tosyl and naphthyl groups which block the addition of TEMPO radical to form a bond at the terminal carbon radical of alkenes. The other possible reason could be the failure of 1 f–h to cyclise due to the difficulty in forming the requisite nitrogen radical.
Scheme 2.
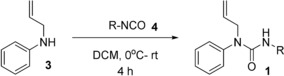
Substrates synthesis (1 a–j). Reaction conditions: aryl isocyanate (1.0 equiv), dry CH2Cl2 (25 mL), allylamine (1.1 equiv), 0 °C to rt, 4 h; yields of isolated products are given in Figure 1.
Figure 1.
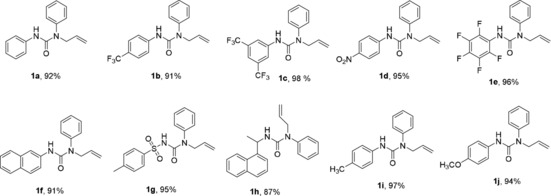
Substrates scope for the synthesis of urea‐tethered alkenes (1 a–j).
Table 1.
Difunctionalisation of alkenes.
|
|
The possible mechanism for the electrosynthesis was proposed using urea substrate R (Scheme 3). The process originates from anodic oxidation of TEMPO to oxoammonium ion, and cathodic reduction of water (H2O) to hydroxide (OH−) and H2. Then deprotonation of substrate R by electrogenerated hydroxide leads to a nitrogen‐containing anion, which is a much better electron donor than its neutral precursor. Single‐electron transfer (SET) between the nitrogen‐containing anion substrate R− and the oxoammonium ion affords the electron‐deficient nitrogen‐centred radical and regenerates the TEMPO radical molecule. Subsequently, the nitrogen radical intermediate can cyclise onto the cyclic urea group to give another radical at the terminal carbon that reacts with the TEMPO radical molecule to form the difunctionalised product P.11f
Scheme 3.
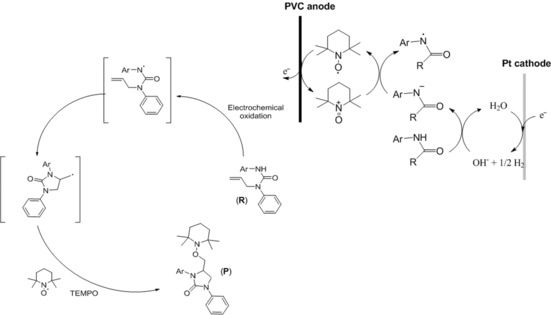
Proposed mechanism for alkene difunctionalisation.
3. Conclusions
In conclusion, this is a facile and green approach to generate a radical at the nitrogen centre without the use of expensive metal catalysts and by using only electricity, which is economically excellent. Then this nitrogen radical on addition to unactivated/unsubstituted terminal alkenes in the presence of TEMPO constitutes a general and mild approach to alkene difunctionalisation and construction of cyclic ureas in high yields. In this procedure, TEMPO plays the dual role of redox facilitator and in radical trapping. This intramolecular amination of urea‐tethered alkenes represents an environmentally friendly method that avoid the issues of toxicity or the complicated ligands of many transition‐metal‐based systems. In this procedure, catalyst‐ and oxidant‐free conditions are used, which deserves significant attention from the viewpoint of green chemistry.
Experimental Section
All the solvent and chemicals were purchased from Sigma Aldrich, Alfa Aesar, Acros Organic and FluoroChem and were used without further purification. All air sensitive reactions were carried out under an argon or nitrogen atmosphere. Thin‐layer chromatography (TLC) was performed on pre‐coated aluminium sheets of Merck silica gel 60 F254 (0.20 mm) and visualised by UV radiation (254 nm). Automated column chromatography was performed on a Biotage® Isolera Four. 1H NMR and 13C NMR spectra were measured on Bruker DPX 300, 400 or 500 apparatus. Mass spectrometric measurements were performed by the EPSRC Mass Spectrometry Facility in Swansea University on a Waters Xevo G2‐S and on a Thermo Scientific LTQ Orbitrap XL machine for high‐resolution mass spectroscopy (HRMS). The electrochemical reactions were carried out in a galvanostatic mode using a GWINSTEK GPR‐30H10D.
General Procedure for the Preparation of Allyl Ureas
To a stirred solution of an aryl isocyanate (1.0 equiv) in dry CH2Cl2 at 0 °C under N2, the corresponding allylamine (1.1 equiv) was added. The reaction was allowed to warm to room temperature and stirred for 4 hours. The reaction mixture was then diluted with CH2Cl2, washed with water, HCl (1 N), NaHCO3 (sat.) and brine, dried with anhydrous MgSO4. The mixture was filtered, and the filtrate was concentrated under vacuum. The residue was purified through silica gel flash chromatography (eluents: hexanes and ethyl acetate).
General Procedure for the Electrolysis
The substrate (0.35 mmol, 1 equiv), TEMPO (0.53 mmol, 1.5 equiv), Bu4NBF4 (1.0 mmol), Na2CO3 (0.39 mmol, 1.1 equiv) were placed in a 25 mL three‐necked round‐bottom flask equipped with a RVC anode and a platinum wire cathode. The flask was flushed with argon. Acetonitrile (9.5 mL) and water (0.5 mL) were added. The resulting mixture was sonicated for 0.5 min. The electrolysis was carried out at 60 °C and a constant current of 10 mA until complete consumption of the substrate (monitored by TLC or 1H NMR). The reaction mixture was cooled to RT. Water (25 mL) and ethyl acetate (25 mL) were added. The phases were separated and the aqueous phase was extracted with ethyl acetate (2×25 mL). The combined organic solution was dried over anhydrous MgSO4 and concentrated under reduced pressure. The residue was chromatographed through silica gel eluting with ethyl acetate/hexanes to give the product. Automated column chromatography was also performed on a Biotage® Isolera Four for purification of few products.
1. ‐Allyl‐1,3‐diphenylurea (1 a)
White crystalline solid, mp=(110–112) °C; 1H NMR (300 MHz, CDCl3) δ=7.43–7.00 (m, 10 H), 6.84 (t, J=6.8 Hz, 1 H), 6.16 (s, 1 H), 5.82 (ddt, J=16.4, 10.5, 6.1 Hz, 1 H), 5.14–4.90 (m, 2 H), 4.21 ppm (d, J=6.1 Hz, 2 H); 13C NMR (101 MHz, Acetone) δ=153.93, 142.06, 140.16, 134.93, 129.80, 128.68, 128.36, 128.15, 127.21, 122.11, 119.27, 118.50, 116.22, 52.09 ppm; ESI HRMS m/z: calcd 252.1263 g mol−1. Observed 253.1352 g mol−1.
2. ‐Allyl‐1‐phenyl‐3‐(4‐(trifluoromethyl)phenyl)urea (1 b)
White crystalline solid, mp=(116–117) °C; 1H NMR (300 MHz, DMSO) δ=8.56 (s, 1 H), 7.70 (d, J=8.7 Hz, 2 H), 7.60 (d, J=8.8 Hz, 2 H), 7.50–7.39 (m, 2 H), 7.31 (ddd, J=8.4, 4.9, 1.6 Hz, 3 H), 5.93 (dtd, J=15.8, 10.6, 5.5 Hz, 1 H), 5.29–5.02 (m, 2 H), 4.37 ppm (d, J=5.5 Hz, 2 H); 13C NMR (75 MHz, DMSO) δ=192.51, 154.51, 144.39, 142.56, 134.95, 129.81, 127.50, 126.82, 125.99, 125.94, 122.55, 122.12, 119.68, 117.10, 52.53 ppm; 19F NMR (376 MHz, Acetone) δ=−62.08 ppm; ESI HRMS m/z [M+H]+calcd 320.1136 g mol−1 Observed 321.1210 g mol−1.
3. ‐Allyl‐3‐(3,5‐bis(trifluoromethyl)phenyl)‐1‐phenylurea (1 c)
White crystalline solid, mp=(128–130) °C; 1H NMR (400 MHz, DMSO) δ=8.83 (s, 1 H), 8.28 (s, 2 H), 7.64 (s, 1 H), 7.54–7.44 (m, 2 H), 7.44–7.33 (m, 3 H), 6.09–5.73 (m, 1 H), 5.24–4.97 (m, 2 H), 4.37 ppm (d, J=5.6 Hz, 2 H); 13C NMR (101 MHz, DMSO) δ=154.40, 142.76, 141.87, 134.70, 131.16, 130.83, 130.51, 130.19, 129.95, 128.01, 127.36, 125.23, 122.52, 119.70, 117.33, 114.81, 114.77, 52.71 ppm; 19F NMR (376 MHz, Acetone) δ=−63.59 ppm; ESI HRMS m/z: calcd 388.1010 g mol−1. Observed 389.1108 g mol−1.
4. ‐Allyl‐3‐(4‐nitrophenyl)‐1‐phenylurea (1 d)
Yellow crystalline solid, mp=(123–125) °C; 1H NMR (400 MHz, Acetone) δ=8.00–7.95 (m, 1 H), 7.89 (s, 1 H), 7.61–7.54 (m, 1 H), 7.38–7.16 (m, 2 H), 5.82 (ddt, J=17.2, 10.2, 6.0 Hz, 1 H), 4.98 (ddq, J=13.2, 10.2, 1.5 Hz, 1 H), 4.23 ppm (dt, J=6.0, 1.4 Hz, 1 H); 13C NMR (75 MHz, Acetone) δ=153.39, 146.67, 141.86, 141.31, 134.29, 129.88, 128.17, 127.61, 124.43, 118.39, 116.62, 52.46 ppm; ESI HRMS m/z: calcd 297.1113 g mol−1. Observed 298.1210 g mol−1
5. ‐Allyl‐3‐(perfluorophenyl)‐1‐phenylurea (1 e)
White crystalline solid, mp=(72–73) °C; 1H NMR (300 MHz, CDCl3) δ=7.43–7.00 (m, 10 H), 6.84 (t, J=6.8 Hz, 1 H), 6.16 (s, 1 H), 5.82 (ddt, J=16.4, 10.5, 6.1 Hz, 1 H), 5.14–4.90 ppm (m, 2 H), 4.21 (d, J=6.1 Hz, 2 H); 13C NMR (75 MHz, CDCl3) δ=153.34, 140.80, 133.24, 130.37, 128.63, 128.38, 118.12, 77.47, 77.04, 76.62, 52.89 ppm; 19F NMR (376 MHz, DMSO) δ=−146.47, −146.52, −146.53, −160.07, −160.13, −160.19, −164.14, −164.20, −164.25, −164.26 ppm; ESI HRMS m/z [M+H]+calcd 342.0792 g mol−1 Observed 343.0866 g mol−1.
6. ‐Allyl‐3‐(naphthalen‐2‐yl)‐1‐phenylurea (1 f)
White crystalline solid, mp=(117–118) °C; 1H NMR (400 MHz, Acetone) δ=7.93 (d, J=2.0 Hz, 1 H), 7.62 (d, J=8.5 Hz, 1 H), 7.60–7.56 (m, 2 H), 7.40–7.14 (m, 9 H), 5.84 (ddt, J=17.2, 10.2, 6.0 Hz, 1 H), 5.07–4.86 (m, 2 H), 4.24 ppm (dt, J=6.0, 1.4 Hz, 2 H); 13C NMR (101 MHz, Acetone) δ=154.02, 142.01, 137.82, 134.89, 134.11, 129.84, 129.82, 128.21, 127.91, 127.40, 127.29, 127.11, 126.08, 123.99, 120.46, 116.28, 114.85, 52.17 ppm; ESI HRMS m/z: calcd 302.1419 g mol−1. Observed 303.1493 g mol−1
N‐(Allyl(phenyl)carbamoyl)‐4‐methylbenzenesulfonamide (1 g)
White crystalline solid, mp=(114–115) °C; 1H NMR (400 MHz, Acetone) δ=8.78 (s, 1 H), 7.78–7.70 (m, 2 H), 7.35–7.08 (m, 7 H), 5.76–5.54 (m, 1 H), 5.02–4.78 (m, 2 H), 4.04 (dt, J=6.1, 1.4 Hz, 2 H), 2.30 ppm (s, 3 H); 13C NMR (126 MHz, Acetone) δ=150.73, 143.89, 140.53, 137.78, 133.43, 129.72, 129.10, 128.28, 128.26, 127.85, 117.12, 52.32, 20.65 ppm.; ESI HRMS m/z: calcd 330.1038 g mol−1. Observed 331.1118 g mol−1
7. ‐(2‐Allylphenyl)‐3‐(1‐(naphthalen‐1‐yl)ethyl)urea (1 h)
Off white liquid; 1H NMR (300 MHz, CDCl3) δ=8.09 (d, J=8.4 Hz, 1 H), 7.75 (d, J=7.8 Hz, 1 H), 7.65 (d, J=8.1 Hz, 1 H), 7.52–7.35 (m, 2 H), 7.34–7.04 (m, 7 H), 5.92–5.66 (m, 2 H), 5.02 (dd, J=2.9, 1.5 Hz, 1 H), 4.97 (dt, J=4.6, 1.4 Hz, 1 H), 4.52 (d, J=7.9 Hz, 1 H), 4.22 (dt, J=6.0, 1.3 Hz, 2 H), 1.46 ppm (d, J=6.8 Hz, 3 H); 13C NMR (75 MHz, CDCl3) δ=155.92, 141.79, 139.63, 134.62, 133.92, 130.99, 129.89, 128.71, 128.23, 127.91, 127.50, 126.28, 125.70, 125.17, 123.65, 122.09, 116.89, 52.32, 46.28, 21.94 ppm.; ESI HRMS m/z: calcd 330.1732 g mol−1. Observed 331.1807 g mol−1
8. ‐Allyl‐1‐phenyl‐3‐(p‐tolyl)urea (1 i)
White crystalline solid, mp=(113–114) °C; 1H NMR (500 MHz, [D6]DMSO) δ=7.89 (s, 1 H), 7.42–7.01 (m, 9 H), 5.88 (m, 1 H), 5.11 (ddq, J=17.1, 10.3, 1.5 Hz, 2 H), 4.31 (dt, J=5.5, 1.4 Hz, 2 H), 2.22 ppm (s, 3 H); 13C NMR (126 MHz, [D6]DMSO) δ=154.80, 143.04, 137.84, 135.34, 131.35, 129.76, 129.12, 127.46, 126.53, 120.44, 116.87, 52.29, 20.79 ppm; ESI HRMS m/z [M+H]+ calcd 266.1419 g mol−1. Observed 367.1509 g mol−1.
9. ‐Allyl‐3‐(4‐methoxyphenyl)‐1‐phenylurea (1 j)
White crystalline solid, mp=(111–112) °C; 1H NMR (500 MHz, [D6]DMSO) δ=7.85 (s, 1 H), 7.42–7.23 (m, 7 H), 6.82–6.79 (m, 2 H), 5.88 (ddd, J=22.6, 10.6, 5.5 Hz, 1 H), 5.11 (ddd, J=13.7, 11.4, 1.3 Hz, 2 H), 4.31 (d, J=5.5 Hz, 2 H) 3.68 ppm (s, 3 H); 13C NMR (126 MHz, [D6]DMSO) δ=154.98, 143.07, 135.41, 133.41, 129.74, 127.49, 126.47, 122.31, 116.83, 113.91, 55.59, 52.28 ppm; ESI HRMS m/z [M+H]+ calcd 282.1368 g mol−1. Observed 283.1452 g mol−1.
10. ,3‐Diphenyl‐4‐(((2,2,6,6‐tetramethylpiperidin‐1‐yl)oxy)methyl)imidazolidin‐2‐one (2 a)
White crystalline solid, mp=(133.4–134.7) °C; 1H NMR (300 MHz, DMSO) δ=7.66–7.46 (m, 4 H), 7.35–7.17 (m, 4 H), 7.05–6.83 (m, 2 H), 4.84–4.50 (m, 1 H), 4.05 (t, J=9.4 Hz, 1 H), 3.81 (dd, J=9.7, 3.4 Hz, 1 H), 3.73–3.62 (m, 2 H), 1.56–0.99 (m, 6 H), 0.94 (s, 1 H), 0.82 (s, 1 H), 0.66 (s, 1 H), 0.59 ppm (s, 1 H). 13C NMR (75 MHz, DMSO) δ=154.88, 140.63, 139.09, 129.18, 128.97, 123.80, 122.69, 121.41, 117.98, 75.55, 59.93, 59.72, 51.60, 45.47, 33.22, 32.54, 19.91, 16.87 ppm. ESI HRMS m/z [M+H]+ calcd 407.2573 g mol−1 Observed 408.2643 g mol−1.
11. ‐Phenyl‐4‐(((2,2,6,6‐tetramethylpiperidin‐1‐yl)oxy)methyl)‐3‐(4 (trifluoromethyl)phenyl)imidazolidin‐2‐one (2 b)
Off white crystalline solid, mp=(135–138) °C; 1H NMR (300 MHz, CDCl3) δ=7.64 (d, J=8.7 Hz, 2 H), 7.56–7.48 (m, 4 H), 7.35–7.27 (m, 2 H), 7.08–6.99 (m, 1 H), 4.61–4.46 (m, 1 H), 4.04 (t, J=9.2 Hz, 1 H), 3.90 (d, J=5.0 Hz, 2 H), 3.79 (dd, J=9.1, 4.3 Hz, 1 H), 1.50–1.11 (m, 6 H), 1.03 (s, 3 H), 0.95 (s, 3 H), 0.82 ppm (s, 6 H), 13C NMR (75 MHz, CDCl3) δ=154.65, 141.84, 139.61, 130.40, 128.98, 128.49, 125.97, 125.92, 125.47, 125.04, 123.41, 122.44, 120.13, 118.48, 118.34, 75.80, 60.03, 51.37, 45.80, 39.57, 33.02, 32.82, 20.03, 19.86, 16.90 ppm, 19F NMR (376 MHz, Acetone) δ=−67.21 ppm. ESI HRMS m/z [M+H]+ calcd 475.2447 g mol−1 Observed 476.2525 g mol−1.
12. ‐(3,5‐Bis(trifluoromethyl)phenyl)‐1‐phenyl‐4‐(((2,2,6,6‐tetramethylpiperidin‐1‐yl)oxy)methyl)imidazolidin‐2‐one (2 c)
White crystalline solid, mp=(151–153) °C; 1H NMR (400 MHz, CDCl3) δ=8.05 (s, 2 H), 7.60–7.45 (m, 3 H), 7.39–7.23 (m, 2 H), 7.13–6.97 (m, 1 H), 4.69–4.49 (m, 1 H), 4.09 (t, J=9.2 Hz, 1 H), 4.01–3.84 (m, 2 H), 3.73 (dt, J=14.9, 7.5 Hz, 1 H), 1.52–1.20 (m, 6 H), 1.01 (s, 3 H), 0.94 (s, 3 H), 0.86 (s, 3 H), 0.72 ppm (s, 3 H), 13C NMR (101 MHz, Acetone) δ=154.27, 141.82, 139.99, 131.57, 131.24, 128.71, 125.01, 122.99, 122.30, 120.21, 120.18, 118.07, 115.49, 115.45, 115.41, 76.90, 59.71, 59.59, 51.19, 45.37, 39.46, 39.45, 32.51, 32.16, 19.21, 19.07, 16.65 ppm, 19F NMR (376 MHz, Acetone) δ=−68.15 ppm. ESI HRMS m/z [M+H]+ calcd 543.2320 g mol−1 Observed 544.2419 g mol−1.
13. ‐(4‐Nitrophenyl)‐1‐phenyl‐4‐(((2,2,6,6‐tetramethylpiperidin‐1‐yl)oxy)methyl)imidazolidin‐2‐one (2 d)
Yellow crystalline solid, mp=(163.5–167) °C; 1H NMR (300 MHz, Acetone) δ=8.13 (d, J=9.3 Hz, 2 H), 7.95 (d, J=9.3 Hz, 2 H), 7.61 (d, J=7.9 Hz, 2 H), 7.26 (t, J=8.0 Hz, 2 H), 6.97 (t, J=7.3 Hz, 1 H), 4.88 (d, J=6.3 Hz, 1 H), 4.20 (t, J=9.3 Hz, 1 H), 4.05 (dd, J=9.9, 3.3 Hz, 1 H), 3.96–3.82 (m, 2 H), 1.47–1.09 (m, 6 H), 0.99 (s, 3 H), 0.88 (s, 3 H), 0.69 ppm (s, 6 H), 13C NMR (101 MHz, Acetone) δ=154.08, 145.84, 142.01, 140.03, 128.72, 124.35, 123.02, 118.76, 118.18, 75.61, 59.80, 59.61, 51.41, 45.44, 39.49, 32.74, 32.13, 29.74, 19.30, 19.23, 16.66, 12.99 ppm. ESI HRMS m/z [M+H]+ calcd 452.2424 g mol−1 Observed 453.2493 g mol−1.
14. ‐(Perfluorophenyl)‐1‐phenyl‐4‐(((2,2,6,6‐tetramethylpiperidin‐1‐yl)oxy)methyl)imidazolidin‐2‐one (2 e)
White crystalline solid, mp=(118–120) °C; 1H NMR (300 MHz, Acetone) δ=7.57–7.50 (m, 2 H), 7.28–7.19 (m, 2 H), 7.00–6.91 (m, 1 H), 4.68–4.51 (m, 1 H), 4.20 (t, J=9.5 Hz, 1 H), 4.05–3.95 (m, 1 H), 3.88 (dd, J=10.3, 3.3 Hz, 1 H), 3.66 (dd, J=9.4, 7.1 Hz, 1 H), 1.34–1.07 (m, 6 H), 0.96 (s, 3 H), 0.92 (s, 3 H), 0.77 (s, 3 H), 0.57 ppm (s, 3 H), 13C NMR (75 MHz, Acetone) δ=153.96, 139.94, 128.74, 122.89, 117.52, 79.52, 59.57, 59.54, 53.12, 45.43, 39.41, 39.32, 32.59, 32.24, 26.63, 18.87, 18.70, 16.66 ppm, 19F NMR (376 MHz, Acetone) δ=−143.50 (d, J=17.8 Hz), −147.06 (d, J=13.8 Hz), −159.14 (t, J=21.0 Hz), −165.54 (s), −165.98 (s). ESI HRMS m/z [M+H]+ calcd 497.2102 g mol−1 Observed 498.2195 g mol−1.
15. ‐Phenyl‐4‐(((2,2,6,6‐tetramethylpiperidin‐1‐yl)oxy)methyl)‐3‐(p‐tolyl)imidazolidin‐2‐one (2 i)
White crystalline solid, mp=(137–138) °C; 1H NMR (500 MHz, DMSO) δ=7.42–7.26 (m, 9 H), 4.60–4.58 (m, 1 H), 4.09 (t, J=7.5 Hz, 1 H), 3.93 (m, 2 H) 3.75 (dt, J=13.3, 6.5 Hz, 1 H), 2.22 (s, 3 H, CH3), 1.30 (m, 6 H), 1.03 (s, 3 H), 0.95 (s, 3 H), 0.82 ppm (s, 6 H). 13C NMR (126 MHz, DMSO) δ=155.11, 140.73, 138.89, 129.44, 123.90, 122.78, 121.27, 117.60, 75.80, 59.72, 51.60, 45.32, 33.59, 32.19, 21.49, 20.34,17.08 ppm. ESI HRMS m/z [M+H]+ calcd 421.2729 g mol−1 Observed 422.2813 g mol−1.
16. ‐(4‐Methoxyphenyl)‐1‐phenyl‐4‐(((2,2,6,6‐tetramethylpiperidin‐1‐yl)oxy)methyl)imidazolidin‐2‐one (2 j)
White crystalline solid, mp=(128–129) °C; 1H NMR (500 MHz, DMSO) δ=7.41–7.28 (m, 7 H), 6.82–6.80 (m, 2 H), 4.54 (m, 1 H), 4.04 (t, J=9.1 Hz, 1 H), 3.91 (d, J=5.6 Hz, 2 H), 3.80 (dd, J=9.2, 3.6 Hz, 1 H), 3.70 (s, 3 H, OCH3) 1.31 (m, 6 H), 1.03 (s, 3 H), 0.95 (s, 3 H), 0.82 ppm (s, 6 H). 13C NMR (126 MHz, DMSO) δ=154.29, 140.43, 138.89, 129.72, 129.44, 123.51, 122.78, 120.54, 118.41, 76.50, 59.99, 55.61, 51.24, 45.32, 33.22, 32.19, 19.63, 16.38 ppm. ESI HRMS m/z [M+H]+ calcd 437.2678 g mol−1 Observed 438.2757 g mol−1.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Marie Skłodowska‐Curie Actions COFUND (Grant No 663830) to N.A. gratefully acknowledged. We thank the Cardiff Chemistry and Welsh Govt for their generous funding to COFUND Fellow (N.A). We thank the Prof. Thomas Wirth from Cardiff School of Chemistry for his continuous technical support. We thank the Higher Education Commission (HEC) Pakistan for IRSIP Fellowship to S.K. We also thank to Aggeliki Vgenopoulou (Erasmus student from Greece) for technical support.
N. Ahmed, S. Khatoon, ChemistryOpen 2018, 7, 576.
References
- 1.
- 1a. Minatti A., MuÇiz K., Chem. Soc. Rev. 2007, 36, 1142–1152; [DOI] [PubMed] [Google Scholar]
- 1b. Chemler S. R., Fuller P. H., Chem. Soc. Rev. 2007, 36, 1153–1160; [DOI] [PubMed] [Google Scholar]
- 1c. Kotov V., Scarborough C. C., Stahl S. S., Inorg. Chem. 2007, 46, 1910–1923; [DOI] [PubMed] [Google Scholar]
- 1d. Jensen K. H., Sigman M. S., Org. Biomol. Chem. 2008, 6, 4083–4088; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1e. McDonald R. I., Liu G., Stahl S. S., Chem. Rev. 2011, 111, 2981–3019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1f. Schultz D. M., Wolfe J. P., Synthesis 2012, 44, 351–361; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1g. Shimizu Y., Kanai M., Tetrahedron Lett. 2014, 55, 3727–3737; [Google Scholar]
- 1h. Courant T., Masson G., J. Org. Chem. 2016, 81, 6945–6952; [DOI] [PubMed] [Google Scholar]
- 1i. Romero R. M., Wçste T. H., MuÇiz K., Chem. Asian J. 2014, 9, 972–983; [DOI] [PubMed] [Google Scholar]
- 1j. Ye K.-Y., Pombar G., Fu N., Sauer G. S., Keresztes I., Lin S., J. Am. Chem. Soc. 2018, 140, 2438–2441; [DOI] [PubMed] [Google Scholar]
- 1k. Ahmed N., Khatoon S., Shirinfar B., ChemElectroChem 2018, 5, 1245–1248; [Google Scholar]
- 1l. Fuller P. H., Kim J.-W., Chemler S. R., J. Am. Chem. Soc. 2008, 130, 17638–17639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Singh A. K., Chawla R., Keshari T., Yadav V. K., Yadav L. D. S., Org. Biomol. Chem. 2014, 12, 8550–8554; [DOI] [PubMed] [Google Scholar]
- 2b. Tang Y., Zhang Y., Wang K., Li X., Xu X., Du X., Org. Biomol. Chem. 2015, 13, 7084–7090; [DOI] [PubMed] [Google Scholar]
- 2c. Wei W., Liu C., Yang D., Wen J., You J., Suo Y., Wang H., Chem. Commun. 2013, 49, 10239–10241; [DOI] [PubMed] [Google Scholar]
- 2d. Taniguchi N., J. Org. Chem. 2015, 80, 7797–7802; [DOI] [PubMed] [Google Scholar]
- 2e. Lu Q., Zhang J., Wei F., Qi Y., Wang H., Liu Z., Lei A., Angew. Chem. Int. Ed. 2013, 52, 7156–7159; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 7297–7300; [Google Scholar]
- 2f. Wang L., Chen M., Qi L., Xu Z., Li W., Chem. Commun. 2017, 53, 2056–2059. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Wang Y., Zhang L., Yang Y., Zhang P., Du Z., Wang C., J. Am. Chem. Soc. 2013, 135, 18048–18051; [DOI] [PubMed] [Google Scholar]
- 3b. Quinn R. K., Schmidt V. A., Alexanian E. J., Chem. Sci. 2013, 4, 4030–4034; [Google Scholar]
- 3c. Bunescu A., Wang Q., Zhu J., Chem. Eur. J. 2014, 20, 14633–14636; [DOI] [PubMed] [Google Scholar]
- 3d. Yi H., Zhang X., Qin C., Liao Z., Liu J., Lei A., Adv. Synth. Catal. 2014, 356, 2873–2877; [Google Scholar]
- 3e. Chatalova-Sazepin C., Wang Q., Sammis G., Zhu J., Angew. Chem. Int. Ed. 2015, 54, 5443–5446; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 5533–5536; [Google Scholar]
- 3f. Liao Z., Yi H., Li Z., Fan C., Zhang X., Liu J., Deng Z., Lei A., Chem. Asian J. 2015, 10, 96–99; [DOI] [PubMed] [Google Scholar]
- 3g. Li L., Huang M., Liu C., Xiao J.-C., Chen Q.-Y., Guo Y., Zhao Z.-G., Org. Lett. 2015, 17, 4714–4717; [DOI] [PubMed] [Google Scholar]
- 3h. Jian W., Ge L., Jiao Y., Qian B., Bao H., Angew. Chem. Int. Ed. 2017, 56, 3650–3654; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 3704–3708; [Google Scholar]
- 3i. Tlahuext-Aca A., Garza-Sanchez A. G., Glorius F., Angew. Chem. Int. Ed. 2017, 56, 3708–3711; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 3762–3765; [Google Scholar]
- 3j. Zhao J., Jiang M., Liu J., Adv. Synth. Catal. 2017, 359, 1626–1630; [Google Scholar]
- 3k. Kou X., Li Y., Wu L., Zhang X., Yang G., Zhang W., Org. Lett. 2015, 17, 5566–5569; [DOI] [PubMed] [Google Scholar]
- 3l. Martínez C., Pérez E. G., Iglesias A., Escudero-Adán E. C., Muñiz K., Org. Lett. 2016, 18, 2998–3001. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Yamamoto D., Soga M., Ansai H., Makino K., Org. Chem. Front. 2016, 3, 1420–1424; [Google Scholar]
- 4b. Zhang T.-S., Xiong Y.-J., Hao W.-J., Zhu X.-T., Wang S.-L., Li G., Tu S.-J., Jiang B., J. Org. Chem. 2016, 81, 9350–9355; [DOI] [PubMed] [Google Scholar]
- 4c. Bag R., Sar D., Punniyamurthy T., Org. Biomol. Chem. 2016, 14, 3246–3255. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Hartmann M., Li Y., Studer A., J. Am. Chem. Soc. 2012, 134, 16516–16519; [DOI] [PubMed] [Google Scholar]
- 5b. Sun X., Li X., Song S., Zhu Y., Liang Y.-F., Jiao N., J. Am. Chem. Soc. 2015, 137, 6059–6066; [DOI] [PubMed] [Google Scholar]
- 5c. J. A. Gurak, Jr. , Yang K. S., Liu Z., Engle K. M., J. Am. Chem. Soc. 2016, 138, 5805–5808; [DOI] [PubMed] [Google Scholar]
- 5d. Jiang H., He Y., Cheng Y., Yu S., Org. Lett. 2017, 19, 1240–1243; [DOI] [PubMed] [Google Scholar]
- 5e. Ye L., Lo K.-Y., Gu Q., Yang D., Org. Lett. 2017, 19, 308–311; [DOI] [PubMed] [Google Scholar]
- 5f. Wei W., Liu X., Yang D., Dong R., Cui Y., Yuan F., Wang H., Tetrahedron Lett. 2015, 56, 1808–1811; [Google Scholar]
- 5g. Yuan Z., Wang H.-Y., Mu X., Chen P., Guo Y.-L., Liu G., J. Am. Chem. Soc. 2015, 137, 2468–2471; [DOI] [PubMed] [Google Scholar]
- 5h. Zeng K., Chen L., Chen Y., Liu Y., Zhou Y., Au C.-T., Yin S.-F., Adv. Synth. Catal. 2017, 359, 841–847; [Google Scholar]
- 5i. Li H., Shan C., Tung C.-H., Xu Z., Chem. Sci. 2017, 8, 2610–2615; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5j. Zaranek M., Witomska S., Patroniak V., Pawluc P., Chem. Commun. 2017, 53, 5404–5407; [DOI] [PubMed] [Google Scholar]
- 5k. Yang Y., Song R., Quyang X., Wang C., Li J., Luo S., Angew. Chem. Int. Ed. 2017, 56, 7916–7919; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 8024–8027. [Google Scholar]
- 6.
- 6a. Sharpless K. B., Patrick D. W., Truesdale L. K., Biller S. A., J. Am. Chem. Soc. 1975, 97, 2305–2307; [Google Scholar]
- 6b. Muñiz K., Chem. Soc. Rev. 2004, 33, 166–174; [DOI] [PubMed] [Google Scholar]
- 6c. Li G. G., Chang H. T., Sharpless K. B., Angew. Chem. Int. Ed. Engl. 1996, 35, 451–454; [Google Scholar]; Angew. Chem. 1996, 108, 449–452; [Google Scholar]
- 6d. Sharpless K. B., Chong A. O., Oshima K., J. Org. Chem. 1976, 41, 177–179; [Google Scholar]
- 6e. Jacobsen E. N., Marko I., Mungall W. S., Schroeder G., Sharpless K. B., J. Am. Chem. Soc. 1988, 110, 1968–1970. [Google Scholar]
- 7.
- 7a. Muñiz K., Hövelmann C. H., Streuff J., J. Am. Chem. Soc. 2008, 130, 763–773; [DOI] [PubMed] [Google Scholar]
- 7b. Streuff J., Hövelmann C. H., Nieger M., Muñiz K. J., J. Am. Chem. Soc. 2005, 127, 14586–14587. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Muñiz K., Iglesias A., Fang Y., Chem. Commun. 2009, 0, 5591–5593; [DOI] [PubMed] [Google Scholar]
- 8b. Li H., Widenhoefer R. A., Tetrahedron 2010, 66, 4827–4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Cochran B. M., Michael F. E., Org. Lett. 2008, 10, 5039–5042; [DOI] [PubMed] [Google Scholar]
- 9b. Farid U., Wirth T., Angew. Chem. Int. Ed. 2012, 51, 3462–3465; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 3518–3522. [Google Scholar]
- 10. Rao W.-H., Yin X.-S., Shi B.-F., Org. Lett. 2015, 17, 3758–3761. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Xiong P., Xu H.-H., Xu H.-C., J. Am. Chem. Soc. 2017, 139, 2956–2959; [DOI] [PubMed] [Google Scholar]
- 11b. Xu F., Zhu L., Zhu S., Yan X., Xu H.-C., Chem. Eur. J. 2014, 20, 12740–12744; [DOI] [PubMed] [Google Scholar]
- 11c. Folgueiras-Amador A. A., Philipps K., Guilbaud S., Poelakker J., Wirth T., Angew. Chem. Int. Ed. 2017, 56, 15446–15450; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 15648–15653; [Google Scholar]
- 11d. Xu H.-C., Campbell J. M., Moeller K. D., J. Org. Chem. 2014, 79, 379–391; [DOI] [PubMed] [Google Scholar]
- 11e. Zhu L., Xiong P., Mao Z.-Y., Wang Y.-H., Yan X., Lu X., Xu H.-C., Angew. Chem. Int. Ed. 2016, 55, 2226–2229; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2266–2269; [Google Scholar]
- 11f. Hou Z.-W., Mao Z.-Y., Zhao H.-B., Melcamu Y. Y., Lu X., Song J., Xu H.-C., Angew. Chem. Int. Ed. 2016, 55, 9168–9172; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 9314–9318. [Google Scholar]
- 12.
- 12a. Mçhle S., Zirbes M., Rodrigo E., Gieshoff T., Wiebe A., Waldvogel S. R., Angew. Chem. Int. Ed. 2018, 57, 6018–6041; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 6124–6149; [Google Scholar]
- 12b. Wiebe A., Gieshoff T., Mçhle S., Rodrigo E., Zirbes M., Waldvogel S. R., Angew. Chem. Int. Ed. 2018, 57, 5594–5619; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 5694–5721; [Google Scholar]
- 12c. Yan M., Kawamata Y., Baran P. S., Chem. Rev. 2017, 117, 13230–13319; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12d. Jiang Y., Xu K., Zeng C., Chem. Rev. 2018, 118, 4485–4540; [DOI] [PubMed] [Google Scholar]
- 12e. Schille B., Giltzau N. O., Francke R., Angew. Chem. Int. Ed. 2018, 57, 422–426; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 429–433; [Google Scholar]
- 12f. Tian C., Massignan L., Meyer T. H., Ackermann L., Angew. Chem. Int. Ed. 2018, 57, 2383–2387; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 2407–2411; [Google Scholar]
- 12g. Wang H., Zhang J., Tan J., Xin L., Li Y., Zhang S., Xu K., Org. Lett. 2018, 20, 2505–2508; [DOI] [PubMed] [Google Scholar]
- 12h. Zhang S., Li L., Wang H., Li Q., Liu W., Xu K., Zeng C., Org. Lett. 2018, 20, 252–255. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Barbachyn M. R., Ford C. W., Angew. Chem. Int. Ed. 2003, 42, 2010–2023; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 2056–2070; [Google Scholar]
- 13b. Mukhtar T. A., Wright G. D., Chem. Rev. 2005, 105, 529–542; [DOI] [PubMed] [Google Scholar]
- 13c. López O., Maya I., Ulgar V., Robina I., Fuentes J., Fernández-Bolaños J. G., Tetrahedron Lett. 2002, 43, 4313–4316. [Google Scholar]
- 14.
- 14a. Bergmeier S. C., Tetrahedron 2000, 56, 2561–2576; [Google Scholar]
- 14b. Anastasi C., Crowe M. A., Powner M. W., Sutherland J. D., Angew. Chem. Int. Ed. 2006, 45, 6176–6179; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 6322–6325. [Google Scholar]
- 15.
- 15a. Esker J. L., Newcomb M. in Advances in Heterocyclic Chemistry, Vol. 58 (Ed.: A. R. Katritzky), Academic Press, New York, 1993, pp. 1–45; [Google Scholar]
- 15b. Fallis A. G., Brinza I. M., Tetrahedron 1997, 53, 17543–17594; [Google Scholar]
- 15c. Stella L. in Radicals in Organic Synthesis, Vol. 2 (Eds.: P. Renaud, M. P. Sibi), Wiley-VCH, Weinheim, 2001, pp. 407–426; [Google Scholar]
- 15d. Zard S. Z., Chem. Soc. Rev. 2008, 37, 1603–1618; [DOI] [PubMed] [Google Scholar]
- 15e. Baralle A., Baroudi A., Daniel M., Fensterbank L., Goddard J.-P., Lacote E., Larraufie M.-H., Maestri G., Malacria M., Ollivier C., in Encyclopedia of Radicals in Chemistry, Biology and Materials (Eds.: C. Chatgilialoglu, A. Studer), Wiley, Chichester, 2012, pp. 767–816. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary