
Figure 1.
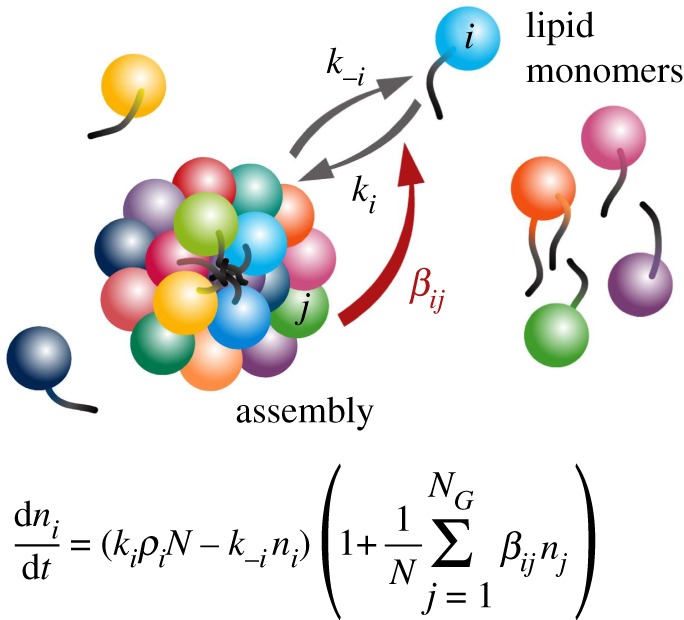
The graded autocatalysis replication domain (GARD) model is based on computer simulations of rigorous chemical behaviour. The model involves a stochastic chemistry simulation based on a set of differential equations as shown. The main reaction step is the entry and exit of an amphiphilic molecule Ai, belonging to a repertoire of NG amphiphile types (represented by different colours), between the environment and an assembly (in this figure exemplified by a small micelle). The variable ni is the count of Ai molecules within the assembly, N = Σni, the total count of all NG species in the assembly, ki and k−i are, respectively, the basal (spontaneous) forward and backward rate constants for Ai, (black arrows), and ρi is the external concentration of Ai. A key aspect, crucial for reaching a kinetically controlled homeostatic growth of the assembly, is the dependence of the reaction rates on the current composition of the assembly. This dependence is controlled by a matrix β, whose elements βij are the rate-enhancement values for internal compounds on the rate of the exchange reaction. The matrix element βij signifies the rate-enhancement parameters for the catalysis exerted by the in-assembly species Aj on the joining and leaving reactions of Ai (red arrow). The matrix elements thus control the dynamics of the mutually catalytic network embodied in the GARD assembly, and its elements are drawn from a probability distribution generated through the RAD model (§4) [65].