
FIG. 1.

A toy protein design problem in which conformational ensembles (A) and optimal mutations (B) must be computed at three residues. Residues of the fibronectin F1 module (Fn, blue ribbon) and of a fragment of Staphylococcus aureus fibronectin binding protein A (FNBPA-5, green ribbon) are shown (PDB id: 2RL0). Side-chain conformations, labeled with amino acid identity, are also shown per residue. (A) Previous provable methods require a fully defined sequence to compute an SS 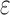 -approximation bound on binding affinity [i.e., a
-approximation bound on binding affinity [i.e., a 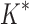 score, Eq. (2)]. (B) A key innovation in this article is the MS bound for binding affinity in protein design. An MS bound is a provable bound on the binding affinity of a partial sequence. Unassigned residues, whose amino acid identities are not defined by the partial sequence, adopt side-chain conformations from multiple amino acids, shown as the blue, purple, pink, and light blue ensembles. Thus, an MS bound is a provable upper bound on the binding affinity of all sequences containing that partial sequence, and is obtained without computing any SS bounds. (The full analysis of the Fn:FNBPA-5 design problem is described in Section 4.3). MS, multi-sequence; SS, single sequence.
score, Eq. (2)]. (B) A key innovation in this article is the MS bound for binding affinity in protein design. An MS bound is a provable bound on the binding affinity of a partial sequence. Unassigned residues, whose amino acid identities are not defined by the partial sequence, adopt side-chain conformations from multiple amino acids, shown as the blue, purple, pink, and light blue ensembles. Thus, an MS bound is a provable upper bound on the binding affinity of all sequences containing that partial sequence, and is obtained without computing any SS bounds. (The full analysis of the Fn:FNBPA-5 design problem is described in Section 4.3). MS, multi-sequence; SS, single sequence.