

Abstract
Immunomodulatory signaling imposes tight regulations on metabolic programs within immune cells and consequentially determines immune response outcomes. Although the glucocorticoid receptor (GR) has been recently implicated in regulating the function of myeloid-derived suppressor cells (MDSCs), whether the dysregulation of GR in MDSCs is involved in immune-mediated hepatic diseases and how GR regulates the function of MDSCs in such a context remains unknown. Here, we revealed the dysregulation of GR expression in MDSCs during innate immunological hepatic injury (IMH) and found that GR regulates the function of MDSCs through modulating HIF1α-dependent glycolysis. Pharmacological modulation of GR by its agonist (dexamethasone, Dex) protects IMH mice against inflammatory injury. Mechanistically, GR signaling suppresses HIF1α and HIF1α-dependent glycolysis in MDSCs and thus promotes the immune suppressive activity of MDSCs. Our studies reveal a role of GR-HIF1α in regulating the metabolism and function of MDSCs and further implicate MDSC GR signaling as a potential therapeutic target in hepatic diseases that are driven by innate immune cell-mediated systemic inflammation.
Introduction
Hepatic inflammation is one of the most prevalent pathologic responses in a variety of liver diseases.1 Immune-mediated hepatic injury (IMH) is central to the pathogenesis of inflammatory liver diseases, including autoimmune hepatitis and viral hepatitis.2 The acute inflammatory phenotype can be largely attributed to the front-line immune defense, generated by the innate immune system involving Kupffer cells, monocytes, neutrophils and eosinophils.1 Following an initial defensive response through recognizing pathogens and producing pro-inflammatory cytokines, the innate immune system also instructs long-lasting adaptive immunity and amplifies effector responses through a diverse range of mechanisms.3 As such, innate immune cell-mediated liver injury is driven by acute innate inflammation and is further evidenced by a sustained inflammatory damage imposed from the adaptive immune response within the inflamed liver. Mechanistically, the dynamic and complex interactions involving a diverse range of innate immune cells play an instrumental role in driving the pathological progression and therapeutic outcome in hepatic diseases that are driven by innate immune cell-mediated systemic inflammation. Understanding the molecular and cellular interactions behind these processes will not only elucidate the pathogenesis but also implicate new therapeutic targets of liver inflammatory disease.
Myeloid-derived suppressor cells (MDSCs) are morphologically and functionally heterogeneous population of the myeloid-cell progenitors; they constitute a unique component of the immune system and function as negative regulators of the immune response.4 MDSCs are composed of monocytes, macrophages, granulocytes, dendritic cells (DCs) and immature myeloid cells at different stages of differentiation, and they often present as CD11b+Gr1+ in mice and Lin-HLA-DR-CD33+ or CD11b+CD14-CD33+ in humans.4,5,6 Importantly, MDSCs are able to expand and frequently stay in an activated state with increased production of nitrogen and reactive oxygen species in a diverse range of pathological inflammation, including cancer and some infectious or autoimmune disorders.7 Emerging evidence has shown that the development and accumulation of MDSCs in the tumor microenvironment play a critical role in fostering pro-tumoral immune modulation.4 While MDSCs have been most extensively studied in the context of tumors, recent studies also implicate their involvement in several other pathological contexts.8,9 However, the regulation and function of MDSCs in systemic inflammation-driven hepatic injury remains to be defined.
Synthetic glucocorticoid (GC) immunosuppressants, including dexamethasone (Dex), have been widely used in treating inflammatory disorders and are well known for their immunomodulatory effects.10 GCs exert their biological functions largely through regulating the glucocorticoid receptor (GR), which is a member of the nuclear receptor family and possesses transcription-regulatory function.11 Upon ligand binding, the GR dimerizes and translocates into the nucleus, where it can both directly and indirectly regulate the expression of a diverse range of inflammatory and anti-inflammatory genes.12 It is known that the tissue sensitivity to hormone signals is directly related to the levels of circulating cortisol and to the number of GRs found in cells.13 Previous studies have shown that the level of GR protein displays a dynamic change following the challenge of acute stressors and chronic stressors in various liver diseases.14
Our recent studies indicated that the GR signaling in MDSCs might play a critical role in the modulation of allograft immunity through reprogramming T-cell differentiation.15 In light of this finding, we asked whether the dysregulation of GR in MDSCs is involved in innate immune cell-mediated liver diseases and how GR regulates the function of MDSCs. Here, we have revealed the dysregulation of GR expression in MDSCs during immunological hepatic injury (IMH) and found that GR regulates the function of MDSCs through modulating HIF1α-dependent glycolysis. Moreover, pharmacologically targeting GR signaling in MDSCs represents an effective therapeutic approach for systemic inflammation-driven hepatic injury.
Materials and methods
Mice
All animal experiments were approved by the Animal Ethics Committee of Fudan, Shanghai, China, and Beijing Normal University, Beijing, China. CD45.1+ C57BL/6 (B6) mice were obtained from the Center of Model Animal Research at Nanjing University (Nanjing, China). C57BL/6 (CD45.2+) mice were received from Fudan University (Shanghai, China) or Beijing Normal University (Beijing, China) Experimental Animal Center. LysM Cre and HIF1αflox/flox mice on the C57BL/6 background were from The Jackson Laboratory and were further crossed to generate HIF1αflox/flox, LysM Cre mice. All mice were bred and maintained in specific pathogen-free conditions. Sex-matched littermates at 6-8 wks of age were used in the experiments described in this study.
LPS-induced hepatic injury and tolerance model
As reported,16,17 for lipopolysaccharide (LPS) challenge, mice were injected (intraperitoneally (i.p.)) with LPS (5–10 mg/kg body weight). For endotoxin tolerance, mice were injected (i.p.) with LPS (0.1 mg/kg body weight) daily for 4 consecutive days, and then they were re-challenged with LPS (10 mg/kg body weight). Sera were collected 2 and 6 h after LPS challenge to test the levels of tumor necrosis factor-α (TNFα) and/or interleukin-1β (IL-1β) production by flow cytometry (FCM) or ELISA. The levels of AST and ALT were examined by ELISA. The liver, kidney and lung were fixed with formaldehyde for histopathological examinations. Sections (5 μm) were stained with hematoxylin and eosin for the assessment of cellular infiltration and inflammatory injuries.
Flow cytometry
For cell surface marker analysis, cells were stained with antibodies in PBS containing 0.1% (wt/vol) BSA and 0.1% NaN3. The following mAbs were used in the present study: anti-CD11b (M1/70; eBiosciences, Ben Lomond, CA, USA), anti-Gr1 (RB6-8C5; eBiosciences), anti-CD45 (TU116; BD Biosciences, San Diego, CA, USA), anti-GR (EPR4595; Abcam, Cambridge, MA, USA), anti-HIF1α (IC1935P; R&D system, Minneapolis, MN, USA), anti-human HLADR (L243; Biolegend, San Diego, CA, USA), anti-human CD33 (P67.0; Biolegend) and anti-human CD11b (ICRF44; Biolegend).
Intracellular staining was analysed by FCM as described.18 Cells isolated from different organs were re-stimulated with LPS (L2630; Sigma, St Louis, MO, USA) for CD11b+Gr1+ cell analysis for 5 h, and GolgiStop (554724; BD Bioscience) was added for the last 2 h. After surface staining and washing, the cells were immediately fixed with cytofix/cytoperm solution (554714; BD Biosciences) and were stained with anti-TNFα (MP6-XT22; eBiosciences) and anti-IL-10 (JES5-16E3; eBiosciences). For transcriptional factor analysis, after surface staining, the cells were fixed with fixation/permeabilization buffer (00-5523; eBioscience) and stained with anti-GR and anti-HIF1α. FCM data were acquired on a FACSCalibur (Becton Dickinson, San Diego, CA, USA), and data were analysed with FlowJo (TreeStar, San Carlos, CA, USA).
Cell purification and culture
Following cardiac perfusion with PBS, the liver or spleen was aseptically removed and mechanically disrupted between sterile frosted microscope slides as described before.19 Liver or splenic CD11b+Gr1+ cells and lymph node naïve T cells (CD4+TCR+CD25-CD62LhighCD44- cells) were sorted using a FACSAria II (Becton Dickinsion).
Quantitative reverse transcriptase–PCR and immunoblot analysis
Total RNA of MDSCs was extracted with the RNeasy kit (Qiagen, Valenica, CA, USA), and cDNA was synthesized using SuperScript III reverse transcriptase (Invitrogen, Carlsbad, CA, USA). An ABI 7900 real-time PCR system was used for quantitative PCR (qPCR), with primer and probe sets obtained from Applied Biosystems (Carlsbad, CA, USA). The results were analysed using SDS 2.1 software. The expression of each target gene is presented as the ‘fold change’ relative to that of control samples, as described previously.20 Immunoblot analysis was performed as described previously.21 Western blots were performed using specific antibodies: anti-GR (EPR4595; Abcam), anti-p-ErK (Thr202/Tyr204; Cell Signaling Technology, Danfoss, MA, USA), anti-p-JNK (Thr183/Tyr185; Cell Signaling Technology), anti-p-p38MAPK (Thr180/Tyr182; Cell Signaling Technology), anti-β-actin (AC-15; Sigma-Aldrich, St Louis, MO, USA) and anti-HIF1α (IC1935P; R&D system).
Functional assay of CD11b+Gr1+cells
The depleting anti-Gr-1 mAb (RB6-8C5; 0.5 mg) was used to deplete the CD11b+Gr1+ cells in vivo through i.p. injection into recipients at days -1 prior to the induction of hepatic injury. For the adoptive transfer experiments for CD11b+Gr1+ cells, donor B6 (CD45.2+) mice were challenged with LPS. Two days later, CD11b+Gr1+ cells were sorted from the liver of hepatic injury mice, and ~1 × 106 cells were i.v. injected into syngeneic (CD45.1+) B6 mice. One day after adoptive transfer, the recipient mice were challenged with LPS injection.
Glycolytic flux analysis
Glycolysis of MDSCs was determined by measuring the detritiation of [3-3H]-glucose as reported previously.19 In brief, 1 uCi [3-3H]-glucose (PerkinElmer) was added to the cell culture medium for 2 h. The medium was collected and moved to microcentrifuge tubes containing 50 μl 5N HCL. The microcentrifuge tubes were then placed in 20-ml scintillation vials containing 0.5 ml water, and the vials were capped and sealed. 3H-H2O was separated from unmetabolized [3-3H]-glucose by evaporation for 24 h at room temperature.
NO production assay
After incubating equal volumes of culture supernatant or serum (100 μl) with Greiss reagent, the absorbance at 550 nm was measured using a microplate reader (Bio-Rad, Hercules, CA, USA), as described previously.22
GR receptor knockdown with shRNA
Gene-specific short hairpin RNA (shRNA) sequences were cloned into the lentiviral shRNA expression plasmids (pMagic4.1; Sbo-bio, Shanghai, China) to construct the GR targeting shRNA and scramble control. Lentiviruses were produced and harvested from culture supernatant of 293 T cells (CL1032; Abgent, San Diego, CA, USA) transfected with shRNA vector. Sorted CD11b+Gr1+ cells were infected with recombinant lentivirus, GFP+ cells were sorted using a FACSAria II (Becton Dickinson), and the GR expression was confirmed using qPCR.
Statistical analyses
All data are presented as the mean±s.d. Student’s unpaired t-test was applied for comparison of means to compare differences between groups. Comparison of the survival curves was performed using the Log-Rank (Mantel-Cox) test. A P-value (alpha-value) of <0.05 was considered to be statistically significant.
Results
MDSCs display reciprocal changes in GR expression in hepatic injury or the tolerance IMH mouse model
Our recent studies implicated that GR signaling in MDSCs plays an important role in modulating allograft immunity.15 Here, we sought to extend these findings to evaluate the role of GR signaling and MDSCs in the context of systemic inflammation-driven hepatic injury. In this regard, we established a hepatic injury IMH model through inducing an acute and strong LPS response (LPS shock)23,24 and a tolerance IMH model through a consecutive induction of a moderate LPS response (LPS tolerance)21,22 (Figures 1a and b). The alteration of serum ALT and AST levels in these two models (Figure 1c) confirms the status of hepatic injury and tolerance. Next, we sought to determine whether there are any alterations in MDSCs and to determine the expression of GR in MDSCs in IMH models. As shown in Figure 1d, the CD11b+Gr1+ MDSCs were significantly increased in the LPS-induced tolerance model, but not in the LPS-induced hepatic injury model, when compared with the control group. Consistent with the above findings, the protein and mRNA levels of GR in MDSCs that were isolated from the LPS-induced tolerant mice were higher than in the control, while they were lower in MDSCs that were isolated from the LPS-induced hepatic injury mice (Figure 1e and Supplementary Figure S1). Altogether, these data indicate that the reciprocal expression alteration of GR in MDSCs is regulated in the context of innate immune cell-mediated hepatic injury or tolerance.
Figure 1.
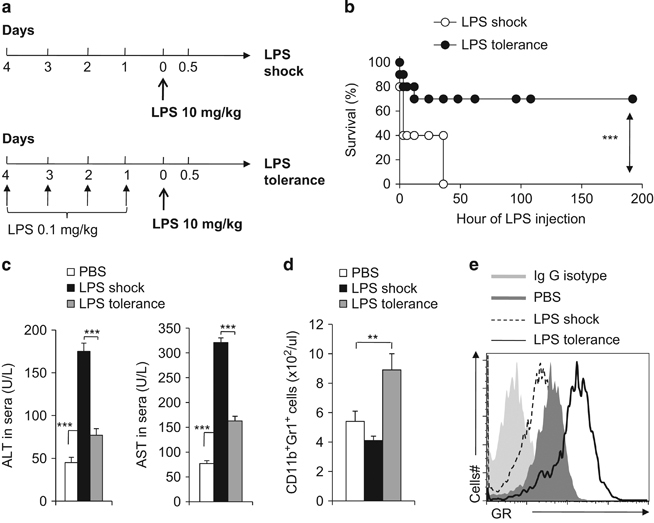
The alternation of myeloid-derived suppressor cell (MDSC) glucocorticoid-receptor in immune-mediated hepatic injuries. (a) diagram of the experimental model for LPS tolerance and for LPS shock induction; the mouse survival curve is plotted in b. In the indicated murine models, the serum ALT and AST levels were determined with ELISA (c); the blood CD11b+Gr1+ cells were analysed by FACS (d); FACS analysis of GR expression in CD11b+Gr1+ cells (e). Data are representative of three independent experiments (n=4–10). **P<0.01 and ***P<0.001 compared with the indicated groups. GR, glucocorticoid receptor; LPS, lipopolysaccharide.
Dex enhances MDSC GR expression and protects mice against systemic inflammation-driven hepatic injury
The significant downregulation of GR in MDSCs upon LPS-shock led us to test the idea of enhancing GR expression as a potential approach for modulating MDSC function and pathological outcomes in IMH. To this end, we applied Dex, a potent agonist of GR, in the IMH model. In agreement with earlier findings,15 Dex treatment increased GR expression at both the mRNA and protein level in the CD11b+Gr1+ cells that were isolated from mice following LPS shock (Figure 2a and Supplementary Figure S2). Strikingly, Dex treatment offered a remarkable protection against LPS-induced lethality in the IMH mouse model (Figure 2b). The histological analysis of liver, lung and kidney that were isolated from IMH mice and the analysis of serum levels of cytokines in IMH mice further confirmed that Dex treatment could ameliorate the LPS-induced immunological injuries and diminish the proinflammatory TNFα and IL-1β production in serum (Figures 2c and d and Supplementary Figure S3A). To further determine the impact of Dex treatment on MDSCs, we isolated the CD11b+Gr1+ cells from experimental animals and examined their suppressive activities on T-cell proliferation and their intracellular cytokine production ex vivo. In addition to significantly potentiating the suppressive activities of MDSCs, Dex treatment reciprocally diminished and increased the proinflammatory TNFα expression and anti-inflammatory IL-10 expression in MDSCs (Figures 2e and f and Supplementary Figure S3B and C). Next, we asked whether MDSCs are responsible for mediating the protection against hepatic injuries in Dex-treated animals. To this end, we depleted MDSCs by injecting anti-Gr1 mAb25 and then followed the survival of mice upon LPS challenge. Efficient depletion of CD11b+Gr1+ cells was confirmed by FACS (data not shown). Importantly, the depletion of Gr1+ cells abolished the protection offered by Dex treatment in IMH mice (Supplementary Figure S4). Conversely, the adoptive transfer of CD11b+Gr1+ MDSCs that were isolated from Dex but not from PBS-treated IMH mice provided a significant protection against LPS-induced lethality in recipient mice (Figure 2g). Collectively, these data suggest that Dex treatment elevates the expression of GR in MDSCs and protects mice against immune-mediated hepatic injuries.
Figure 2.
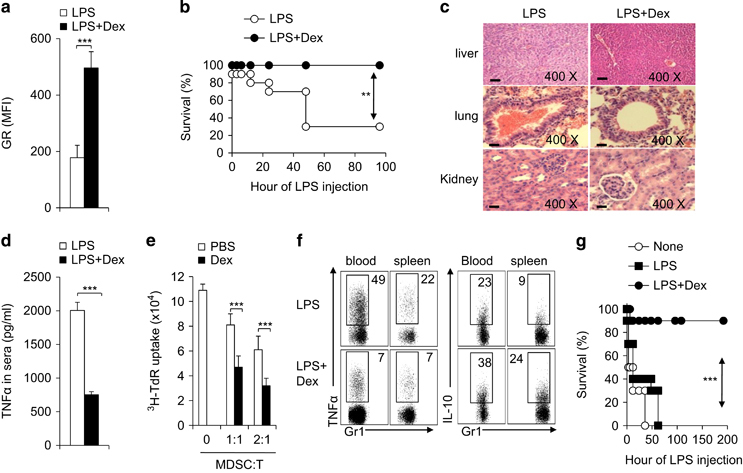
Dex treatment upregulates GR expression, potentiates MDSC activities and protects mice against immune-hepatic injuries. Age-matched C57BL/6 mice were injected i.p. with PBS (solvent) or Dex (5 mg/kg body weight) daily starting at 6 h before LPS (5 mg/kg) injection. The GR expression on the CD11b+Gr1+ cells from liver 72 h following LPS injection was determined by FACS (a) and the survival curve of experimental mice is plotted (b, n=10). In a separate set of animals, the mice were sacrificed at 72 h following LPS-injection. The pathological changes in mouse liver, lung and kidney were examined by histochemistry (c). TNFα level in serum was determined with ELISA (d). The immunosuppressive activity of myeloid-derived CD11b+Gr1+ cells was determined by mixed lymphocyte reaction (e). The TNFα and IL-10 production in CD11b+Gr1+ cells in blood and spleen was analysed by FACS (f). Three groups of donor C57BL/6 mice were challenged with PBS (None), LPS or LPS plus Dex. After 3 days, a total of 1x106 CD11b+Gr1+ cells were sorted from the liver of indicated mice and adoptively transferred into C57BL/6 recipient mice (10 mice per group) via i.v. Injection. After 10-12 h, all groups of recipient mice were challenged with LPS (10 mg/kg) and mouse survival was followed (g). Data are representative of three (c–e) or four (f) independent experiments (n=3–10). **P<0.01 and ***P<0.001 compared with the indicated groups. GR, glucocorticoid receptor; IL-10, interleukin-10; i.v., intravenous; LPS, lipopolysaccharide; MDSC, myeloid-derived suppressor cell; PBS, phosphate-buffered saline; TNFα, tumor necrosis factor-α.
The elevation of GR expression in MDSCs is required for Dex-mediated protection against IMH
Having shown that Dex induces GR expression in MDSCs and protects mice against immune-mediated hepatic injuries, we next sought to determine the impacts of genetic modulation of GR on MDSC function. For this, we transuded either control shRNA or shRNA that targets GR into CD11b+Gr1+ cells that were isolated from the liver and then examined their cytokine production. The efficient knockdown of GR in MDSCs was confirmed by qPCR (Supplementary Figure S5A). Importantly, knockdown of GR enhanced the production of TNFα, which is indicative of liver injury pathology and inflammation (Supplementary Figures S5B and C). Next, we sought to determine whether the inhibition of GR in MDSCs would counteract the effect of Dex treatment in protecting mice against immune-mediated hepatic injuries in the IMH model. For this, we chose a well-established antagonist of GR, RU-486,26 and applied it to the IMH model, either alone or in combination with Dex. As expected, the RU-486 treatment alone efficiently reduced the GR expression in MDSCs that were isolated from LPS-challenged hepatic injury mice (Figure 3a). Similarly, RU-486 treatment alone significantly exacerbated the pathology of hepatic injury in IMH mice, as indicated by the levels of ALT, TNFα and IL-1β in sera (Figures 3b and c). This increased hepatic injury is likely due to its inhibition of MDSC-suppressive activity (Figure 3d). Importantly, the combination of RU-486 and Dex reversed the effect of Dex on GR expression in MDSCs, serum ALT and TNFα and IL-1β levels, and the suppressive activity of MDSCs (Figures 3a–d). To verify MDSCs as the cellular target of RU486 and Dex in the IMH model, we performed an adoptive-transfer experiment using CD11b+Gr1+ MDSCs that were isolated from mice following LPS-challenge in combination with various treatments, as indicated in Figure 3e. While the adoptive transfer of MDSCs isolated from Dex-treated mice protected recipient mice against pathological lethality, the RU-486 treatment of donor mice abolished such protection (Figure 3e). Together, these finding suggest that Dex or RU-486 treatment reciprocally modulates GR signaling and thus impacts the function of MDSCs during immune-mediated hepatic injury.
Figure 3.
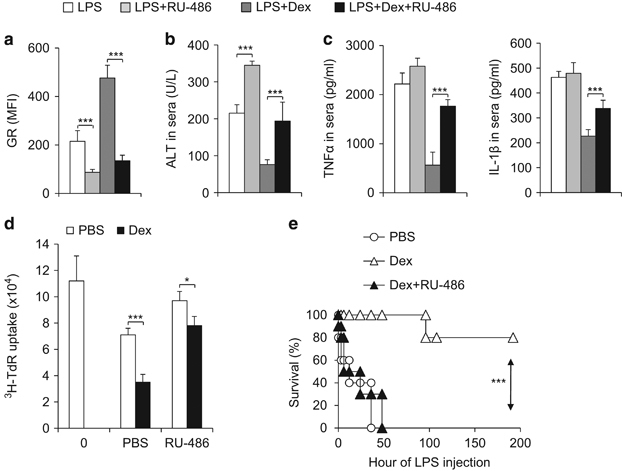
The downregulation of GR expression by RU-486 counteracts the effect of Dex in protecting mice against immune-hepatic injuries. As described in Figure 2, IMH was induced in four groups of age-matched C57BL/6 mice with the indicated treatments. Then, 72 h after LPS challenge, GR expression of CD11b+Gr1+MDSC in liver was analysed by FACS (a) and the level of ALT (b) TNFα and IL-1β (c) in serum were determined by ELISA. The immunosuppressive activity of myeloid-derived CD11b+Gr1+ cells was determined by mixed lymphocyte reaction (d) Three groups of donor C57BL/6 mice were challenged with LPS, LPS plus Dex or LPS plus Dex and RU-486 (10 mg/kg, i.p.). After 3 days, a total of 1 × 106 CD11b+Gr1+ cells were sorted from the liver of indicated mice and adoptively transferred into C57BL/6 recipient mice (10 mice per group) via i.v. injection. After 10–12 h, all groups of recipient mice were challenged with LPS (10 mg/kg), and mouse survival was followed (e) Data are representative of three (a–c) or two (d) independent experiments (a–d; n=3–5). ***P<0.001 compared with the indicated groups. GR, glucocorticoid receptor; IMH, immunological hepatic injury; i.p., intraperitoneal; MDSC, myeloid-derived suppressor cell; LPS, lipopolysaccharide.
The modulation of GR expression in MDSCs impacts LPS-induced immune tolerance
Given the observation of increased GR expression in MDSCs in LPS-induced tolerance mode, we next determined the impact of modulating GR signaling in tolerance. While most of the experimental animals survived through LPS-challenge in the tolerant model, treatment with the GR antagonist RU-486 abolished the tolerance and restored the LPS-induced pathological lethality (Figure 4a). As expected, the RU-486 treatment significantly diminished GR expression in MDSCs isolated from experimental mice (Figure 4b and Supplementary Figure S6) and diminished the suppressive activity of MDSCs (Figure 4c). Consistent with the results of the survival curve (Figure 4a), RU-486 treatment exacerbated the liver injury pathology and inflammation, as indicated by heightened serum ALT, TNFα and IL-1β levels (Figures 4d and e). Taken together, these data suggest that the RU-486 treatment may break immune tolerance through targeting GR in MDSCs.
Figure 4.
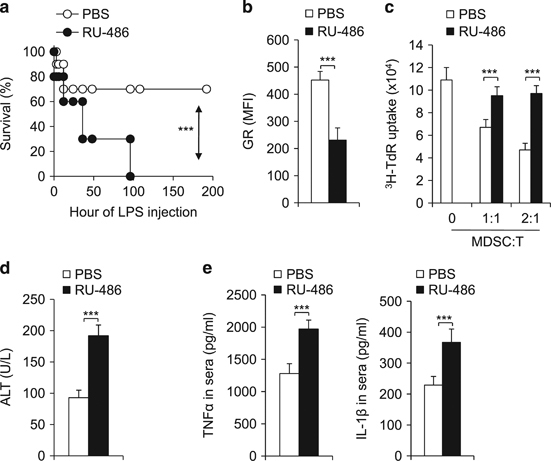
The downregulation of GR expression by RU-486 abolishes immune tolerance in an LPS-mediated tolerance model. Two groups of aged-matched C57BL/6 mice (10 per group) were pretreated with low dose LPS (0.1 mg/kg) daily from day 4 prior to challenge with LPS (10 mg/kg) for LPS tolerance induction. Either PBS or RU-486 was administrated at 6 h before the last LPS challenge. Mouse survival curve is plotted (a). Then, 72 h after the LPS challenge, GR expression of CD11b+Gr1+MDSC in liver was analysed by FACS (b) the immunosuppressive activity of myeloid-derived CD11b+Gr1+ cells was determined by mixed lymphocyte reaction (c) and the levels of ALT (d) TNFα and IL-1β (e) in serum were determined by ELISA. Data are representative of three (b–e) independent experiments (n=3–5). ***P<0.001 compared with the indicated groups. GR, glucocorticoid receptor; IL-1β, interleukin-1β; LPS, lipopolysaccharide; PBS, phosphate-buffered saline; TNFα, tumor necrosis factor-α.
Blocking glycolysis protects mice against IMH
The metabolic shift toward aerobic glycolysis has been recently revealed as a general metabolic feature in myeloid cells upon inflammatory stimulation.27,28 Given that GR signaling has been implicated in the regulation of metabolism in several cellular contexts,29 we next asked how GR signaling impacts MDSC metabolism. For this, we examined the glycolytic activity of MDSCs following LPS-stimulation in the presence of Dex or RU-486. While the Dex treatment increased GR expression and suppressed glycolytic activity in MDSCs, the RU-486 treatment reduced GR expression and enhanced glycolytic activity (Figure 5a). Glucose utilization depends on a chain of reactions catalysed by multiple enzymes, eventually leading to the generation of lactate and to the net production of two ATP molecules as the energy source. The mRNA expression of a set of key glycolytic genes, such as glucose transport-1 (Glut1), gluocose-6-phosphate isomerase (GPI), enolase 1 (Eno1), lactate dehydrogenase-α (LDHα), and monocarboxylic acid transporter member 4 (MCT4), in the MDSCs was therefore determined by qPCR. As shown in Figure 5b, Dex or RU-486 treatment significantly reduced or enhanced the expression of Glut1, Eno1 and MCT4 in MDSCs, respectively. Consistent with the result of pharmacological modulation of GR, the genetic modulation of GR via shRNA knockdown significantly increased the glycolytic activity of MDSCs (Supplementary Figure S7).
Figure 5.
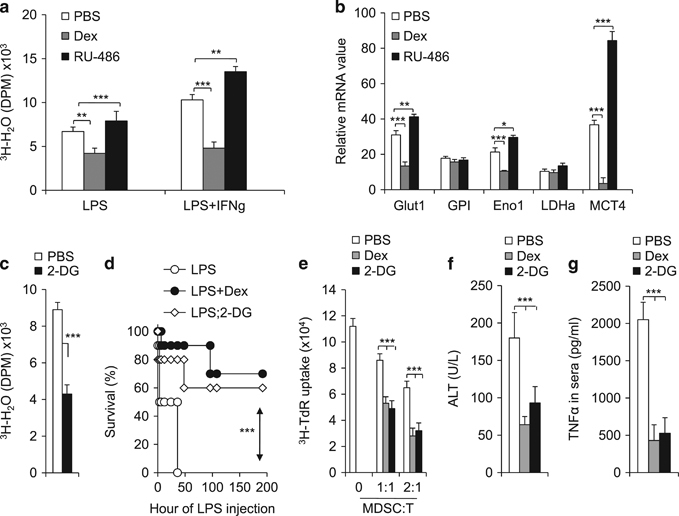
Blocking glycolysis protects mice against immune-hepatic injuries in IMH. As described in Figure 2, IMH was induced in the indicated three groups of age-matched C57BL/6 mice. Then, 12 h after LPS challenge, CD11b+Gr1+ cells were sorted from livers of indicated mice and were stimulated with LPS or LPS+IFN-γ for 10 h. The glycolytic activity (a) or the mRNA expression of indicated glycolytic genes (b) in the indicated groups was measured by the generation of 3H-labeled H2O from [3-3H]-glucose or by qPCR. The liver MDSCs were isolated from IMH mice and were stimulated by LPS for 10 h in the presence of vehicle (PBS) or 1 mmol/l 2-DG. The glycolytic activity was measured as described above (c). As described in Figure 2, IMH was induced in three groups of age-matched C57BL/6 mice with the indicated treatments (LPS 10 mg/kg). The survival curve is plotted (d) Then, 72 h after the LPS challenge, the immunosuppressive activity of myeloid-derived CD11b+Gr1+ cells was determined by mixed lymphocytes reaction (e) and the levels of ALT (f) and TNFα (g) in serum were determined by ELISA. Data are representative of two (a–c) or three (e–g) independent experiments (n=3–10). *P<0.05, **P<0.01 and ***P<0.001 compared with the indicated groups. IMH, immunological hepatic injury; IFN-γ, interferon-γ; LPS, lipopolysaccharide; MDSC, myeloid-derived suppressor cell; qPCR, quantitative PCR; TNFα, tumor necrosis factor-α.
Next, we sought to determine the impact of the glycolysis inhibitor, 2-deoxy-glucose (2-DG), on MDSCs in the IMH model. As expected, 2-DG treatment significantly blocked the glycolytic activity in MDSCs (Figure 5c). Importantly, 2-DG or Dex treatment displayed a comparable protection of mice against LPS-induced pathological lethality (Figure 5d). In addition, both 2-DG and Dex treatment enhanced the suppressive activity of MDSCs (Figure 5e) and ameliorated the liver injury pathology and inflammation, as indicated by reduced ALT and TNFα levels in the serum (Figures 5f and g). Taken together, these data suggest that the glycolysis mechanisms might be related to the GR-mediated regulation of MDSCs in IMH.
HIF1α is the downstream target of GR in MDSCs
The transcription factor hypoxia-inducible factor 1 α (HIF1α) has been implicated as a key regulator of metabolism in cancer cells, inflammatory TH17 cells and myeloid cells.21 We therefore examined the level of HIF1α in MDSCs isolated from IMH mice treated with either Dex or RU-486. While the Dex treatment suppressed the expression of HIF1α (Figure 6a), it did not impact several other key inflammatory signaling pathways in MDSCs, as indicated by the comparable phosphorylation of JNK, ErK and p38MAPK between the two groups (Supplementary Figure S8). Conversely, both the pharmacological inhibition of GR by RU-486 and the genetic knockdown of GR by shRNA enhanced the expression of HIF1α (Figure 6b and Supplementary Figure S9). Importantly, the deficiency of HIF1α significantly ameliorated the liver injury pathology and the pro-inflammatory function of MDSCs and also enhanced MDSC function in the IMH model, as indicated by the reduced ALT serum level, the reduced intracellular TNFα in MDSCs and the enhanced suppression of T cell proliferation (Figures 6c–e and Supplementary Figure S10A and B). In addition, the deficiency of HIF1α led to a significantly prolonged survival in IMH mice (Figure 6f). While the RU-486 treatment exuberated the pathological phenotypes of IMH, HIF1α deficiency abolished the impact of RU-486 (Figures 6c–f).
Figure 6.
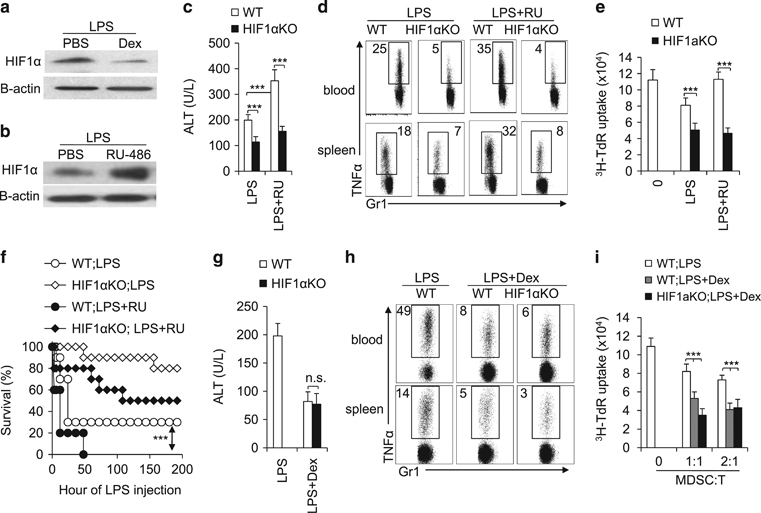
HIF1α is a downstream effector of GR signaling in MDSCs in IMH. The protein level of HIF1α in liver MDSCs that were isolated from IMH mice with the indicated treatments was determined by western blot (a, b). As described in Figure 2, IMH was induced in WT or HIF1αfl/fl, LysM Cre+ mice with the indicated treatments. Then, 72 h after LPS-injection, the ALT level in serum was determined with ELISA (c, g) the TNFα production in CD11b+Gr1+ cells in blood and spleen was analysed by FACS (d, h) and the immunosuppressive activity of myeloid-derived CD11b+Gr1+ cells was determined by mixed lymphocyte reaction (e, i) As described in Figure 2, IMH was induced in age-matched WT or HIF1αfl/fl, LysM Cre+ mice (n=10) with the indicated treatments (LPS 5 mg/kg). The survival curve is plotted (f). Data are representative of two (a–e) or three (g–i) independent experiments (n=3–5). ***P<0.001 compared with the indicated groups. GR, glucocorticoid receptor; IMH, immunological hepatic injury; MDSC, myeloid-derived suppressor cell; LPS, lipopolysaccharide; NS, not significant; TNFα, tumor necrosis factor-α; WT, wild type.
Given that the Dex treatment suppresses the expression of HIF1α (Figure 6a), we reasoned that HIF1α deficiency would not result in any additive effect on the Dex treatment if HIF1α functions as a key target effector of GR signaling. Supporting this idea, the Dex treatment of HIF1α-deficient mice displayed a similar degree of phenotype in liver injury pathology, in inflammatory cytokine production and in the suppressive activity of MDSCs compared to Dex treatment in WT mice (Figures 6g–i; Supplementary Figure S10C and D). Taken together, our results suggest that HIF1α is required for GR-dependent regulation of MDSCs in the IMH model.
GR signaling controls MDSC nitric oxide production
Nitric oxide (NO) production plays a key role in mediating the immunosuppressive activity of MDSCs. We sought to determine whether the modulation of GR signaling impacts NO production in the IMH model. Consistent with the result showing that Dex treatment enhanced MDSC function (Figure 2e), Dex treatment increased the serum level of NO (Figure 7a). While Dex treatment increased the expression of inducible nitric oxide synthase (iNOS), an enzyme that produces NO, it moderately suppressed the expression Arginase I, a substrate-competing enzyme of iNOS, in MDSCs (Figures 7b and c). Conversely, genetic knockdown of GR in MDSCs through shRNA resulted in a reduction of iNOS expression, indicating that the expression of iNOS is dependent on GR signaling (Supplementary Figure S11). Next, we applied L-NMMA, an inhibitor of iNOS, to MDSCs to determine whether blocking NO production would diminish the impact of Dex on the immune suppressive activity of MDSCs. As expected, the L-NMMA treatment abolished NO production (Supplementary Figure S12A). While the treatment of Dex significantly enhanced the immunosuppressive activity of MDSCs, the addition of L-NMMA abolished the effect of Dex on suppressing T-cell proliferation (Figure 7d). Finally, the L-NMMA treatment did not impact the glycolytic activity of MDSCs, excluding the possibility that NO indirectly altered HIF1α and glycolysis in MDSCs (Supplementary Figure S12B). These data suggest that the GR-dependent NO production plays a role in regulating the immune suppressive activity of MDSCs in the IMH model.
Figure 7.
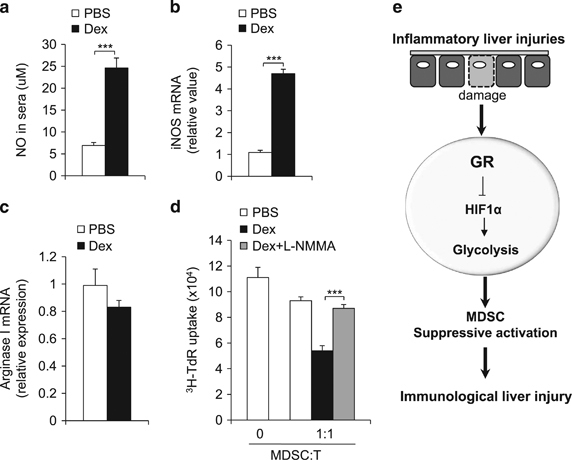
GR signaling modulates NO production in CD11b+Gr1+ MDSCs in IMH. As described in Figure 2, IMH was induced in mice with the indicated treatments. Then, 72 h after LPS-injection, the NO level in serum was determined by Greiss assay (a), the mRNA level of indicated genes in CD11b+Gr1+ cells was determined by qPCR (b,c) and the immunosuppressive activity of myeloid-derived CD11b+Gr1+ cells was determined by mixed lymphocyte reaction (d). Data are representative of three independent experiments (n=4–5). ***P<0.001 compared with the indicated groups. (e) A model summarizes the role of GR signaling in establishing MDSC functions through HIF1α dependent glycolytic pathway. GR, glucocorticoid receptor; IMH, immunological hepatic injury; MDSC, myeloid-derived suppressor cell; qPCR, quantitative PCR.
GR expression is dysregulated in MDSCs isolated from human liver injury patients
By far, we have shown that the dysregulation of GR signaling in MDSCs may play a role in the pathogenesis of the murine IMH model and that the modulation of GR expression in MDSCs may represent an effective therapeutic strategy in treating murine IMH. Next, we sought to extend our finding in the murine IMH model to human patients with immune-mediated liver injuries. In this regard, we collected the peripheral blood from clinically confirmed hepatitis patients and determined the percentage of MDSCs and the expression of GR in MDSCs. As shown in Supplementary Figure S13A, the HLADR-CD33+CD11b+ MDSC percentage and cell number are comparable in hepatitis patients and healthy controls. However, the intracellular staining of GR revealed that the protein level of GR is significantly lower in MDSCs isolated from hepatitis patients compared with MDSCs isolated from healthy controls (Supplementary Figure S13B). Consistent with the expression pattern of HIF1α in MDSCs in the IMH mouse model, MDSCs that were isolated from hepatitis patients displayed an enhanced level of HIF1α compared to MDSCs that were isolated from healthy controls (Supplementary Figure S13C). In summary, our results implicate a GR-HIF1α axis in regulating MDSC metabolism and function. Our studies further suggest that the modulation of GR signaling in MDSCs may represent a novel and valid therapeutic strategy in treating immune-mediated liver injuries (Figure 7e). In addition, the dysregulation of GR signaling in MDSCs may also be present in human immune-mediated liver injury patients.
Discussion
Synthetic glucocorticoids, such as prednisone, dexamethasone, and hydrocortisone, exert their immune modulatory effects in a broad range of immune diseases.10 Glucocorticoids elicit their biological action through the glucocorticoid receptor (GR), which belongs to a nuclear hormone receptor superfamily. In the absence of ligand binding, GR is in complex with chaperons and is retained in the cytosol. Upon glucocorticoid binding, GR is released from chaperon complexes and translocates to the nucleus, where it either directly transactivates targeted gene expression or indirectly modulates gene expression through interacting with other transcription factors.30
GC is also integrated into the complex immune feedback-signaling network that plays a crucial role in fine-tuning immune signaling to avoid overactive immune responses.31 The therapeutic approach of targeting GR is therefore widely used in medicine to treat inflammation, infectious and autoimmune diseases. Previous studies have shown that the modulation of GR signaling impacts on many different types of immune cells in both innate and adaptive immunity.32 Recently, we and others have found that GR signaling may play a role in modulating the physiology and function of MDSCs.15,33,34,35 In the present study, we have revealed that MDSCs isolated from both systemic inflammation-driven hepatic injury, especially in innate immune cell-mediated hepatic injury mice and in human patients with hepatitis, display significantly lower levels of GR compared to healthy controls. Treatment with the GR agonist dexamethasone (Dex) restored GR expression in MDSCs and ameliorated mortality and inflammatory insults in IMH, implying that GR signaling in MDSCs is a potential therapeutic target in treating innate immune cell-mediated hepatic injury.
Mechanistically, the upregulation of GR by Dex protects mice against immune-mediated hepatic injuries through potentiating MDSC suppressive activities, reducing their pro-inflammatory cytokines and increasing anti-inflammatory cytokine production. Conversely, the down-regulation of GR by RU-486 can significantly inhibit the immunosuppressive activities of MDSCs, increase pro-inflammatory cytokine production and reduce anti-inflammatory cytokine secretion, consequentially breaking immune tolerance in a chronically LPS-induced immune tolerance model. Previous studies suggested that ErK,36,37 JNK and p38MAPK signaling pathways are important downstream signaling nodes of GR in various cellular and pathological contexts.29,30 While we failed to reveal any significant changes in these signaling pathways, as indicated by a comparable level of p-ErK, p-p38, and p-JNK in MDSCs following the modulation of GR, we still cannot exclude the involvement of these pathways in fine-tuning GR signaling and impacting the function of MDSCs. Importantly, our current work implicates an important role of the GR-HIF1α signaling axis in regulating the glycolytic activity of MDSCs. GR signaling suppresses HIF1α and HIF1α-dependent glycolysis in MDSCs and thus promotes the immune suppressive activity of MDSCs in protecting against immunological hepatic injuries.
HIF1α is a transcription factor known to play an important role in regulating the innate immune function in response to hypoxia.38 Furthermore, HIF1α also responds to normoxia and plays a critical role in proinflammatory cytokine production.39,40 As a tumor grows, it rapidly outgrows its blood supply, leaving portions of the tumor with regions where the oxygen concentration is significantly lower than in healthy tissues.38 As such, hypoxia microenvironments in solid tumors stabilize HIF1α and, in turn, alter the function of MDSCs through selectively up-regulating an immune checkpoint molecule, programmed death 1 ligand, and redirecting MDSC differentiation toward tumor-associated macrophages.41,42 Recent studies also implicate their involvement in several other pathological contexts, including inflammatory injuries.8,9 In the present study, we showed that HIF1α is responsible for the GR-regulated functions of MDSCs in systemic inflammation-driven immune-mediated hepatic injury mouse models. These data suggested that GR-HIF1α signaling could be a potential therapeutic target in hepatic diseases that are driven by innate immune cell-mediated systemic inflammation.
To elicit rapid and robust immune responses under diverse metabolic and immune conditions, our immune system has evolved to coordinate nutrient metabolism and bioenergetic capacity with immune cell proliferation and activation. Such coordination is the result of the convergence of the signaling pathways that control metabolic activities and the signaling pathways that mediate immune functions. The inability to meet metabolic requirements would cause the imbalances of immune homeostasis. Heightened aerobic glycolysis has been implicated as a key metabolic feature in many pro-inflammatory immune cells, such as M1 macrophages, TLR-stimulated dendritic cells, and TH17.43,44,45,46 Myc and HIF1α, two key transcriptional factors, play an important role in regulating metabolic gene expression that reprograms cellular metabolism to support tumor malignant growth.47,48 Notably, HIF1α has also been implicated in regulating the glycolytic program in M1 macrophages, TH9 and TH17 cells and their pro-inflammatory functions.27,44,46,49 Consistent with the pro-inflammatory function of HIF1α in macrophages and TH17 cells, we found that genetic ablation of HIF1α or pharmacological inhibition of glycolysis suppresses the pro-inflammatory cytokine production in MDSCs while enhancing their anti-inflammatory activities. Similarly, the increased GR expression following Dex treatment not only leads to the downregulation of HIF1α and glycolytic activity but also significantly enhances the anti-inflammatory function of MDSCs and protects mice against innate immune cell-mediated hepatic injury. In conclusion, our current work implicates a critical role of the GR-HIF1α axis in regulating glycolysis and the function of MDSCs in innate immune cell-mediated hepatic injury. The pharmacological targeting of GR or MDSC metabolism may represent a promising therapeutic approach in treating human patients with systemic inflammation-driven hepatic injury, especially in innate immune cell-mediated hepatic injury.
Electronic supplementary material
Acknowledgements
This research is supported by grants from the National Natural Science Foundation for General Programs of China (31671524, 31171407 and 81273201, GL), the Key Basic Research Project of the Science and Technology Commission of Shanghai Municipality (12JC1400900, GL), the Innovation Program of Shanghai Municipal Education Commission (14ZZ009, GL), the Excellent Youth Foundation of Chinese Academy of Sciences (KSCX2-EW-Q-7, GL), R21AI117547, 1R01AI114581, V2014-001 from the V-foundation, and 128436-RSG-15-180-01-LIB from the American Cancer Society (RW).
Conflict of interest
The authors declare no conflict of interest.
Contributor Information
Yong Zhao, Email: zhaoy@ioz.ac.cn, Email: ruoning.wang@nationwidechildren.org, Email: liugw@bnu.edu.cn.
Ruoning Wang, Email: zhaoy@ioz.ac.cn, Email: ruoning.wang@nationwidechildren.org, Email: liugw@bnu.edu.cn.
Guangwei Liu, Email: zhaoy@ioz.ac.cn, Email: ruoning.wang@nationwidechildren.org, Email: liugw@bnu.edu.cn.
Electronic supplementary material
Supplementary Information for this article can be found on the Cellular & Molecular Immunology website 10.1038/cmi.2017.5
References
- 1.Bowen DG, Walker CM. Adaptive immune responses in acute and chronic hepatitis C virus infection. Nature. 2005;436:946–952. doi: 10.1038/nature04079. [DOI] [PubMed] [Google Scholar]
- 2.Das M, Sabio G, Jiang F, Rincon M, Flavell RA, Davis RJ. Induction of hepatitis by JNK-mediated expression of TNF-alpha. Cell. 2009;136:249–260. doi: 10.1016/j.cell.2008.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liberal R, Grant CR, Longhi MS, Mieli-Vergani G, Vergani D. Diagnostic criteria of autoimmune hepatitis. Autoimmun Rev. 2014;13:435–440. doi: 10.1016/j.autrev.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 4.Yang H, Bi Y, Xue L, Wang J, Lu Y, Zhang Z, et al. Multifaceted Modulation of SIRT1 in Cancer and Inflammation. Crit Rev Oncog. 2015;20:49–64. doi: 10.1615/CritRevOncog.2014012374. [DOI] [PubMed] [Google Scholar]
- 5.Zhao Y, Wu T, Shao S, Shi B. Phenotype, development, and biological function of myeloid-derived suppressor cells. Oncoimmunology. 2016;5:e1004983. doi: 10.1080/2162402X.2015.1004983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lechner MG, Liebertz DJ, Epstein AL. Characterization of cytokine-induced myeloid-derived suppressor cells from normal human peripheral blood mononuclear cells. J Immunol. 2010;185:2273–2284. doi: 10.4049/jimmunol.1000901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pallett LJ, Gill US, Quaglia A, Sinclair LV, Jover-Cobos M, Schurich A, et al. Metabolic regulation of hepatitis B immunopathology by myeloid-derived suppressor cells. Nat Med. 2015;21:591–600. doi: 10.1038/nm.3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xia S, Sha H, Yang L, Ji Y, Ostrand-Rosenberg S, Qi L. Gr-1+ CD11b+ myeloid-derived suppressor cells suppress inflammation and promote insulin sensitivity in obesity. J Biol Chem. 2011;286:23591–23599. doi: 10.1074/jbc.M111.237123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arocena AR, Onofrio LI, Pellegrini AV, Carrera Silva AE, Paroli A, Cano RC, et al. Myeloid-derived suppressor cells are key players in the resolution of inflammation during a model of acute infection. Eur J Immunol. 2014;44:184–194. doi: 10.1002/eji.201343606. [DOI] [PubMed] [Google Scholar]
- 10.van de Garde MD, Martinez FO, Melgert BN, Hylkema MN, Jonkers RE, Hamann J. Chronic exposure to glucocorticoids shapes gene expression and modulates innate and adaptive activation pathways in macrophages with distinct changes in leukocyte attraction. J Immunol. 2014;192:1196–1208. doi: 10.4049/jimmunol.1302138. [DOI] [PubMed] [Google Scholar]
- 11.Rauch A, Seitz S, Baschant U, Schilling AF, Illing A, Stride B, et al. Glucocorticoids suppress bone formation by attenuating osteoblast differentiation via the monomeric glucocorticoid receptor. Cell Metab. 2010;11:517–531. doi: 10.1016/j.cmet.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 12.De Bosscher K, Van Craenenbroeck K, Meijer OC, Haegeman G. Selective transrepression versus transactivation mechanisms by glucocorticoid receptor modulators in stress and immune systems. Eur J Pharmacol. 2008;583:290–302. doi: 10.1016/j.ejphar.2007.11.076. [DOI] [PubMed] [Google Scholar]
- 13.Yoo YM, Baek MG, Jung EM, Yang H, Choi KC, Yu FH, et al. Parathyroid hormone-related protein and glucocorticoid receptor beta are regulated by cortisol in the kidney of male mice. Life Sci. 2011;89:615–620. doi: 10.1016/j.lfs.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 14.Gong H, Jarzynka MJ, Cole TJ, Lee JH, Wada T, Zhang B, et al. Glucocorticoids antagonize estrogens by glucocorticoid receptor-mediated activation of estrogen sulfotransferase. Cancer Res. 2008;68:7386–7393. doi: 10.1158/0008-5472.CAN-08-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liao J, Wang X, Bi Y, Shen B, Shao K, Yang H, et al. Dexamethasone potentiates myeloid-derived suppressor cell function in prolonging allograft survival through nitric oxide. J Leukoc Biol. 2014;96:675–684. doi: 10.1189/jlb.2HI1113-611RR. [DOI] [PubMed] [Google Scholar]
- 16.Bala S, Tang A, Catalano D, Petrasek J, Taha O, Kodys K, et al. Induction of Bcl-3 by acute binge alcohol results in toll-like receptor 4/LPS tolerance. J Leukoc Biol. 2012;92:611–620. doi: 10.1189/jlb.0112050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aneja RK, Tsung A, Sjodin H, Gefter JV, Delude RL, Billiar TR, et al. Preconditioning with high mobility group box 1 (HMGB1) induces lipopolysaccharide (LPS) tolerance. J Leukoc Biol. 2008;84:1326–1334. doi: 10.1189/jlb.0108030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li C, Bi Y, Li Y, Yang H, Yu Q, Wang J, et al. Dendritic cell MST1 inhibits Th17 differentiation. Nat Commun. 2017;8:14275. doi: 10.1038/ncomms14275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schumann J, Prockl J, Kiemer AK, Vollmar AM, Bang R, Tiegs G. Silibinin protects mice from T cell-dependent liver injury. J Hepatol. 2003;39:333–340. doi: 10.1016/S0168-8278(03)00239-3. [DOI] [PubMed] [Google Scholar]
- 20.Liu G, Burns S, Huang G, Boyd K, Proia RL, Flavell RA, et al. The receptor S1P1 overrides regulatory T cell-mediated immune suppression through Akt-mTOR. Nat Immunol. 2009;10:769–777. doi: 10.1038/ni.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu G, Bi Y, Shen B, Yang H, Zhang Y, Wang X, et al. SIRT1 limits the function and fate of myeloid-derived suppressor cells in tumors by orchestrating HIF-1alpha-dependent glycolysis. Cancer Res. 2014;74:727–737. doi: 10.1158/0008-5472.CAN-13-2584. [DOI] [PubMed] [Google Scholar]
- 22.Wu T, Sun C, Chen Z, Zhen Y, Peng J, Qi Z, et al. Smad3-deficient CD11b(+)Gr1(+) myeloid-derived suppressor cells prevent allograft rejection via the nitric oxide pathway. J Immunol. 2012;189:4989–5000. doi: 10.4049/jimmunol.1200068. [DOI] [PubMed] [Google Scholar]
- 23.Murphey ED, Fang G, Varma TK, Sherwood ER. Improved bacterial clearance and decreased mortality can be induced by LPS tolerance and is not dependent upon IFN-gamma. Shock. 2007;27:289–295. doi: 10.1097/01.shk.0000245024.93740.28. [DOI] [PubMed] [Google Scholar]
- 24.Patenaude J, D'Elia M, Cote-Maurais G, Bernier J. LPS response and endotoxin tolerance in Flt-3L-induced bone marrow-derived dendritic cells. Cell Immunol. 2011;271:184–191. doi: 10.1016/j.cellimm.2011.06.020. [DOI] [PubMed] [Google Scholar]
- 25.Liu G, Bi Y, Wang R, Yang H, Zhang Y, Wang X, et al. Targeting S1P1 receptor protects against murine immunological hepatic injury through myeloid-derived suppressor cells. J Immunol. 2014;192:3068–3079. doi: 10.4049/jimmunol.1301193. [DOI] [PubMed] [Google Scholar]
- 26.Jang GR, Wrighton SA, Benet LZ. Identification of CYP3A4 as the principal enzyme catalyzing mifepristone (RU 486) oxidation in human liver microsomes. Biochem Pharmacol. 1996;52:753–761. doi: 10.1016/0006-2952(96)00357-7. [DOI] [PubMed] [Google Scholar]
- 27.Na YR, Gu GJ, Jung D, Kim YW, Na J, Woo JS, et al. GM-CSF Induces Inflammatory Macrophages by Regulating Glycolysis and Lipid Metabolism. J Immunol. 2016;197:4101–4109. doi: 10.4049/jimmunol.1600745. [DOI] [PubMed] [Google Scholar]
- 28.Bowden SD, Rowley G, Hinton JC, Thompson A. Glucose and glycolysis are required for the successful infection of macrophages and mice by Salmonella enterica serovar typhimurium. Infect Immun. 2009;77:3117–3126. doi: 10.1128/IAI.00093-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang R, Green DR. Metabolic checkpoints in activated T cells. Nat Immunol. 2012;13:907–915. doi: 10.1038/ni.2386. [DOI] [PubMed] [Google Scholar]
- 30.Kharwanlang B, Sharma R. Molecular interaction between the glucocorticoid receptor and MAPK signaling pathway: a novel link in modulating the anti-inflammatory role of glucocorticoids. Indian J Biochem Biophys. 2011;48:236–242. [PubMed] [Google Scholar]
- 31.Radu CG, Cheng D, Nijagal A, Riedinger M, McLaughlin J, Yang LV, et al. Normal immune development and glucocorticoid-induced thymocyte apoptosis in mice deficient for the T-cell death-associated gene 8 receptor. Mol Cell Biol. 2006;26:668–677. doi: 10.1128/MCB.26.2.668-677.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsutsui S, Vergote D, Shariat N, Warren K, Ferguson SS, Power C. Glucocorticoids regulate innate immunity in a model of multiple sclerosis: reciprocal interactions between the A1 adenosine receptor and beta-arrestin-1 in monocytoid cells. FASEB J. 2008;22:786–796. doi: 10.1096/fj.07-9002com. [DOI] [PubMed] [Google Scholar]
- 33.Varga G, Ehrchen J, Tsianakas A, Tenbrock K, Rattenholl A, Seeliger S, et al. Glucocorticoids induce an activated, anti-inflammatory monocyte subset in mice that resembles myeloid-derived suppressor cells. J Leukoc Biol. 2008;84:644–650. doi: 10.1189/jlb.1107768. [DOI] [PubMed] [Google Scholar]
- 34.Diao W, Jin F, Wang B, Zhang CY, Chen J, Zen K, et al. The protective role of myeloid-derived suppressor cells in concanavalin A-induced hepatic injury. Protein Cell. 2014;5:714–724. doi: 10.1007/s13238-014-0069-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Semple JW. Move over Tregs, MDSCs are here. Blood. 2016;127:1526–1528. doi: 10.1182/blood-2016-01-692160. [DOI] [PubMed] [Google Scholar]
- 36.Wang HC, Zentner MD, Deng HT, Kim KJ, Wu R, Yang PC, et al. Oxidative stress disrupts glucocorticoid hormone-dependent transcription of the amiloride-sensitive epithelial sodium channel alpha-subunit in lung epithelial cells through ERK-dependent and thioredoxin-sensitive pathways. J Biol Chem. 2000;275:8600–8609. doi: 10.1074/jbc.275.12.8600. [DOI] [PubMed] [Google Scholar]
- 37.Lv WM, Zhao Y, Yang G, Dong SY, Zhang GH, Zhang Y, et al. Role of Ras, ERK, and Akt in glucocorticoid-induced differentiation of embryonic rat somatotropes in vitro. Mol Cell Biochem. 2014;391:67–75. doi: 10.1007/s11010-014-1988-4. [DOI] [PubMed] [Google Scholar]
- 38.Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer. 2008;8:705–713. doi: 10.1038/nrc2468. [DOI] [PubMed] [Google Scholar]
- 39.Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–657. doi: 10.1016/S0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walmsley SR, Cadwallader KA, Chilvers ER. The role of HIF-1alpha in myeloid cell inflammation. Trends Immunol. 2005;26:434–439. doi: 10.1016/j.it.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 41.Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014;211:781–790. doi: 10.1084/jem.20131916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. 2010;207:2439–2453. doi: 10.1084/jem.20100587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Farooque A, Afrin F, Adhikari JS, Dwarakanath BS. Polarization of macrophages towards M1 phenotype by a combination of 2-deoxy-d-glucose and radiation: Implications for tumor therapy. Immunobiology. 2016;221:269–281. doi: 10.1016/j.imbio.2015.10.009. [DOI] [PubMed] [Google Scholar]
- 44.Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. 2011;146:772–784. doi: 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pearce EJ, Everts B. Dendritic cell metabolism. Nat Rev Immunol. 2015;15:18–29. doi: 10.1038/nri3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hedl M, Yan J, Abraham C. IRF5 and IRF5 disease-risk variants increase glycolysis and human M1 macrophage polarization by regulating proximal signaling and Akt2 activation. Cell Rep. 2016;16:2442–2455. doi: 10.1016/j.celrep.2016.07.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xue G, Yan HL, Zhang Y, Hao LQ, Zhu XT, Mei Q, et al. c-Myc-mediated repression of miR-15-16 in hypoxia is induced by increased HIF-2alpha and promotes tumor angiogenesis and metastasis by upregulating FGF2. Oncogene. 2015;34:1393–1406. doi: 10.1038/onc.2014.82. [DOI] [PubMed] [Google Scholar]
- 48.Gordan JD, Thompson CB, Simon MC. HIF and c-Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell. 2007;12:108–113. doi: 10.1016/j.ccr.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Y, Bi Y, Chen X, Li C, Li Y, Zhang Z, et al. Histone deacetylase SIRT1 negatively regulates the differentiation of interleukin-9-producing CD4(+) T cells. Immunity. 2016;44:1337–1349. doi: 10.1016/j.immuni.2016.05.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.