

Abstract
Vascular endothelial growth factor (VEGF) is a potent mitogen that regulates proliferation, migration, and tube formation of endothelial cells (EC). VEGF has recently become a target for severe retinopathy of prematurity (ROP) therapy. We tested the hypothesis that a specific VEGF isoform and/or receptor acts synergistically with insulin-like growth factor (IGF)-I to alter normal retinal microvascular EC angiogenesis and RNA interference can be used to reverse VEGF effects. We used small interfering RNA (SiRNA) transfection to target VEGF isoforms, IGFs, and their receptors in human retinal microvascular endothelial cells (HRECs). Media was collected at 24 and 48 hours post transfection for measurement of VEGF, sVEGFR-1 and IGF-1 levels; and HRECs were assessed for migration, tube formation, VEGF signaling genes, oxidative stress, and immune-reactivity. At 24 hours post transfection VEGF increased with VEGFR-2; sVEGFR-1 decreased with VEGF165, VEGFR-2, and IGF-1R; and IGF-I increased with VEGF189, VEGFR-1, IGF-2R, IGF+VEGF165, and IGF+VEGF121. IGF-I transfection with each VEGF isoform reduced sphere- forming and migration capacities with robust upregulation of caspase-9, COX-2, MAPK, PKC, and VEGF receptors. At 48 hours, the effects were reversed with a majority of genes downregulated, except with IGF-I and NP-1 transfection. Using RNA interference for targeted inhibition of VEGF isoforms in conjunction with IGF-I may be preferable for suppression of HREC overgrowth in vasoproliferative retinopathies such as ROP.
Keywords: Angiogenesis, insulin-like growth factor, retinal endothelial cells, small interfering RNAs, vascular endothelial growth factor
Introduction
Vascular endothelial growth factor (VEGF) is a potent endothelial mitogen and trophic factor that promotes proliferation, migration and tube formation leading to angiogenic growth of new blood vessels [1,2]. The VEGF family and its receptor system (VEGFR-1, VEGFR-2, VEGFR-3, and neuropilin or NP) are fundamental regulators in cell signaling of angiogenesis [2]. VEGF is the main growth factor involved in retinal vessel growth with a primary role in both phases of retinopathy of prematurity (ROP) [3-8]. To date, six isoforms of human VEGF have been identified: VEGF121 (VEGF120 in rodents), VEGF145, VEGF165 (VEGF164 in rodents), VEGF189 (VEGF188 in rodents), VEGF183, and VEGF206; they are produced by alternate splicing of the VEGF gene. VEGF121, VEGF165, and VEGF189 are the most abundant isoforms expressed by all retinal cells with VEGF165 mediating the most potent angiogenesis [9]. The lower isoform, VEGF121, lacks heparin binding, is not bound to the extracellular matrix (ECM), is freely diffused over considerable areas within tissues, and plays a major role as a long range attractant [10,11]. The higher isoforms VEGF189 is tightly heparin-bound, serves as a spatially restricted stimulatory cue, and can compensate for loss of the lower isoforms during development [11,12]. Mice expressing only VEGF120 die perinatally, with simplified vasculature whereas mice expressing only VEGF188 displayed abnormal vessel branching with excessively thin and disorganized branches, and mice expressing only VEGF164 were normal [13].
VEGF mediates its actions via different receptors, which are expressed on endothelial cells (ECs) and are required for angiogenesis. VEGF signaling through VEGFR-2 induces permeability, proliferation, migration, and EC survival [14]. VEGFR-1 has a high affinity for VEGF, however, its activity is 10-fold lower than that of VEGFR-2 and therefore only weakly stimulates EC proliferation [15]. The splice variant of VEGFR-1, soluble VEGFR-1 (sVEGFR-1), functions as a decoy or VEGF sink by limiting the action of VEGFR-2 and retaining VEGF in the plasma membrane. It also acts to guide emerging vessel sprouts away from the parent vessel [16]. VEGFR-3 is mainly expressed in lymph-ECs and regulates lymph angiogenesis [15]. Neuropilin (NP)-1 and -2 are co-expressed only with VEGFR-2 to increase its potency [14]. Furthermore, NPs have been implicated in regulation of vessel sprouting [16,17]. Multiple studies have shown that IGF-1 acts as a permissive agent in VEGF-mediated angiogenesis [18-20]. The relationship between VEGF isoforms, IGF-1, and their receptor in angiogenesis is still poorly understood, but appears to play a key role in the development of ROP [18,19].
Recently, there has been increased use of anti-VEGF therapy (intravitreal bevacizumab or ranibizumab) in extremely low birth weight (ELBW) neonates with severe ROP. This therapeutic intervention is highly invasive and results in many complications [21-25] including recurrence of severe ROP and late detachment, myopia, cytokine derangements and reduction of systemic VEGF for up to 2 months after intravitreal administration [26]. Because of the complexity of the VEGF gene and its interaction with other factors as well as its involvement in the vascular development of other organs, targeted therapy against specific isoforms is more desirable in order to prevent or mitigate side effects. In this study, we tested the hypothesis that a specific VEGF isoform and/or receptor acts synergistically with IGF-I to alter normal angiogenesis in human retinal endothelial cells (HRECs). To test our hypothesis, we evaluated the effects of targeted SiRNA transcription of VEGF isoforms, IGFs, and their receptors on cell viability; VEGF, sVEGFR-1, IGF-I production; sphere-forming and migration capacities; and VEGF signaling in HRECs.
Material and methods
Cells
HRECs (ACBRI-181) were purchased from Cell Systems (Kirkland, WA) in a flask of proliferating cells at 80% confluence (1.5×106 cells). The cells were allowed to acclimatize for 2-3 hours in an incubator at 37°C, 5% CO2, after which the transport medium was discarded and the cells were plated in P75 flasks in specialized medium warmed to 37°C and activated with culture boost containing growth factors, Bac-Off containing antibiotics, and 5% amphotericin B. The media was changed every 2 days until confluence. At 80% confluence, the cells were passaged. After 4 passages (24 flasks), the cells were seeded onto 24-well plates (4×104 cells in 0.5 mL media/well) coated with attachment factor, an ECM product that promotes cell attachment, and incubated at 37°C, 5% CO2, 100% humidity, as previously described [27,28].
Transfection
The cells were transfected 24 hours post seeding onto 24-well plates. Total VEGF, VEGF189, VEGF165, VEGF121, VEGFR-1, VEGFR-2, NP-1, NP-2, IGF-I, IGF-2, IGF-IR, IGF-2R and positive and negative control SiRNAs were purchased from Qiagen (Valencia, CA). Custom and Qiagen-ready SiRNAs were diluted to a concentration of 37.5 ng and 1.5 μL added to 100 μL culture medium without antibiotics to give a final SiRNA concentration of 5 nM after addition to the cells. Three μL HiPerfect transfection reagent (Qiagen) was added to the diluted SiRNA and mixed by vortexing. The mixture was incubated at room temperature for 10 minutes to allow formation of a transfection complex, which was added as one drop (20 µL) per well onto the cells. The cells with the transfection complexes were incubated under these growth conditions for 24 and 48 hours. A total of 19 groups were studied: 1) control (no SiRNA); 2) total VEGF; 3) VEGF189; 4) VEGF165; 5) VEGF121; 6) VEGFR-1; 7) VEGFR-2; 8) NP-1; 9) NP-2; 10) IGF-I; 11) IGF-2; 12) IGF-IR; 13) IGF-2R; 14) Total VEGF+IGF-I; 15) VEGF189+IGF-I; 16) VEGF165+IGF-I; 17) VEGF121+IGF-I; 18) positive control; and 19) negative control.
Sample collection
Media and cells were collected at 24 and 48 hours post transfection and frozen at -80°C prior to assay. In each group, two wells were pooled for a total of 12 media samples and 4 wells were pooled for a total of 3 cell samples. Cells from the remaining 12 wells were pooled, and re-plated for tube formation and migration assays.
SiRNA transfection optimization
To obtain the highest transfection efficiency and low non-specific effects, various concentrations of SiRNA and Lipofectamine RNAiMAX complexes were tested. Similarly, a transfection efficiency test was performed with varying concentrations of SiRNA and HiPerfect transfection reagent according to the manufacturer’s HiPerfect Transfection Reagent Handbook. A BLOCK-iT transfection optimization kit was purchased from ThermoFisher Scientific in order to determine the optimal amount of Oligo required to obtain a strong fluorescent signal according to the manufacturer’s protocol. High transfection efficiency was verified with fluorescent siRNA, fluorescent Block-IT. Validation of siRNA and functional assays were performed at 24, and 48 hours post-transfection.
Immunofluorescence
Cells were plated at the same time onto sterile 16-well culture slides (Fisher Scientific, Pittsburgh, PA) and exposed to similar conditions as described above for the 24-well plates. At the end of each experimental time, the slides were washed, fixed in 4% paraformaldehyde, permeabilized and incubated with primary antibodies (Santa Cruz Biotechnology, Dallas, TX); and Alexa Fluor fluorescent secondary antibodies (Life Technologies, Grand Island, NY). Cells were imaged at 20× magnification using an Olympus IX73 inverted microscope system and CellSens imaging software (Olympus, Center Valley, PA).
Lactate dehydrogenase assay
Cell viability was assessed using the in-vitro lactate dehydrogenase (LDH) activity kit purchased from Sigma-Aldrich (St. Louis, MO) according to the manufacturer’s protocol. The LDH assay is a simple and accurate method that yields reproducible results. The amount of LDH activity was used as an indicator of relative cell viability and membrane function integrity.
Assay of VEGF, sVEGFR-1 and IGF-I
VEGF, sVEGFR-1 and IGF-I levels in the media were assayed using commercially-available human sandwich immunoassay kits (R&D Systems, Minneapolis, MN, USA). The VEGF assay predominantly binds the monomeric VEGF165 but will also detect the VEGF121 isoform. The assay utilizes a monoclonal anti-VEGF, anti-sVEGFR-1, or anti-IGF-I detection antibody conjugated to horseradish peroxidase and color development with tetramethylbenzidine/hydrogen peroxide (TMB solution). All assays were performed according to the manufacturer’s protocol. The coefficient of variation from inter- and intra-assay precision assessment was less than 10% for all assays.
Migration capacity
Becton Dickinson (BD)-BioCoat Angiogenesis System-EC tube formation 96-well plates (BD Biosciences, Bedford, MA) were used for migration assays according to the manufacturer’s protocol. Cells from each group were plated at 2×104 in 50 μL media in each well. The plates were incubated for 16-18 hours at 37°C and 5% CO2 atmosphere after which the plates were labeled with BD calcein AM fluorescent dye (BD Biosciences). The plates were imaged and the number of cells that migrated to the bottom was determined.
Sphere-forming capacity
Cells from each group were resuspended in serum-free media containing 20 ng/mL epidermal growth factor and 20 ng/mL fibroblast growth factor (warmed to 37°C), and seeded onto 96 well plates. The plates were placed incubated under their growth conditions for 7 days and scored for the presence or absence of spheres.
Lipid peroxidation
Lipid peroxidation generally refers to the oxidative degradation of cellular lipids by reactive oxygen species. Lipid peroxidation plays a key role in pathological processes and it is often the cause of free radical-mediated damage in cells. We used the Image-iT lipid peroxidation assay (ThermoFisher Scientific, Waltham, MA, USA), a sensitive fluorescent reporter for lipid peroxidation. Upon oxidation in live cells, fluorescence shifts from red to green. Live cells were treated with Image-iT lipid peroxidation Sensor at a final concentration of 10 µM, incubated for 30 minutes at 37°C, washed with PBS, fixed in 4% PFA, washed and counter stained with DAPI. Cells were imaged at 20× magnification.
Real-time PCR
Cells from 4 wells per group were harvested for extraction of total RNA and real-time PCR arrays. Total RNA was extracted using RNA Pro solution (MP Bio, Solon, Ohio) using the FastPrep-24 instrument (MP Bio) according to the manufacturer’s protocol. Cleanup of the RNA was performed using RNEasy mini cleanup kits (Qiagen, Valencia, CA) followed by on-column treatment with DNase I (Qiagen). Reverse transcriptase was performed using a RT2 First Strand kit purchased from SABiosciences (Frederick, MD), a Qiagen company. The real-time PCR arrays were carried out in duplicate using the human VEGF Signaling PCR Arrays (SABiosciences/Qiagen) using a BioRad IQ5 real-time instrument (BioRad, Hercules, CA). Each PCR array plate consisted of a panel of 5 housekeeping genes to normalize the PCR array data; replicate genomic DNA controls to detect non-transcribed genomic DNA contamination with a high levels of sensitivity; replicate reverse transcription controls to test the efficiency of the RT2 first strand reaction; and replicate positive PCR controls to test the efficiency of the PCR reaction itself using a pre-dispensed artificial DNA sequence and the primer set that detects it. The replicate controls also test for inter-well and intra-plate consistency. Calculations were made by exporting the data into an Excel spreadsheet using the Qiagen PCR Array Data Analysis Excel Template and uploading the real-time amplification data into the SABiosciences RT2 Profiler PCR Array Data Analysis web portal.Quantitative PCR was based on the cycle threshold (Ct) value. A gene was considered not detectable if the Ct value was >35. The ΔCt for each gene was calculated as Ct (gene of interest) - Ct (housekeeping gene). Expression changes were quantified by fold regulation; and differences that were 10-fold greater than control were determined to be significant.
Statistical analysis
One-way analysis of variance was used to determine differences among the groups for normally-distributed data, and Kruskal-Wallis test was used for non-normally-distributed data following Bartlett’s test for equality of variances. Post hoc analysis was performed using the Dunnet’s tests for significance comparing all groups versus control. Significance was set at P<0.05 and data are reported as mean ± SEM. All analyses were two-tailed and performed using SPSS version 16.0 (IBM Analytics - Chicago IL).
Results
Cell viability
There was a significant increase in LDH activity, and implicitly cell death, with selective inhibition of VEGF and VEGF isoforms at 24 hr. (Figure 1A). At 48 hr. LDH activity increased with knockdown of IGF and combination IGF and VEGF isoforms (Figure 1B).
Figure 1.
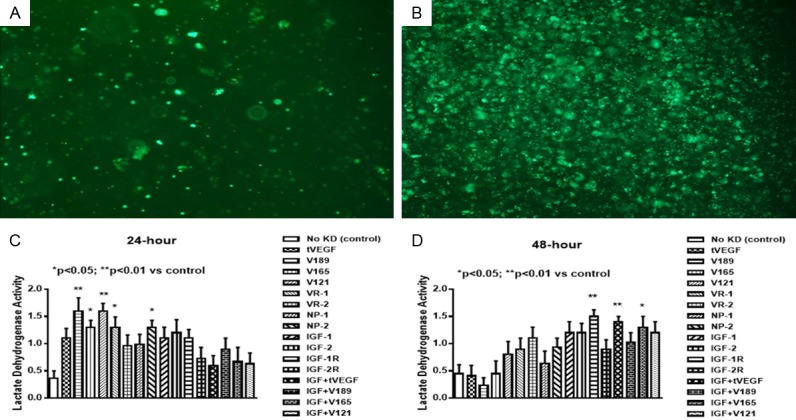
Uptake of VEGF SiRNA at 24 (A) and 48 (B) hours post transfection. Effect of selective SiRNA inhibition of VEGF isoforms, VEGF receptors, and/or IGF-I on lactate dehydrogenase activity, in the media of proliferating human retinal endothelial cells (HRECs) at 24 (C) and 48 (D) hours post transfection. Data are expressed as mean ± SEM (n=12 samples/group; *P<0.05, **P<0.01 vs. control). Images were captured at 4× magnification.
Effects on VEGF
VEGF levels were generally low in the media and were higher at 24 hr. (Figure 2A) compared to 48 hr. (Figure 2B). At 24 hr. VEGF levels were significantly higher with VEGFR-2 knockdown alone. At 48 hr. VEGF levels were higher with IGF-I knockdown suggesting a role for IGF in VEGF retention to the ECM. VEGF levels were lower with knockdown of VEGF121 in combination with IGF. This isoform lacks heparin-binding domains and is not bound to the ECM.
Figure 2.
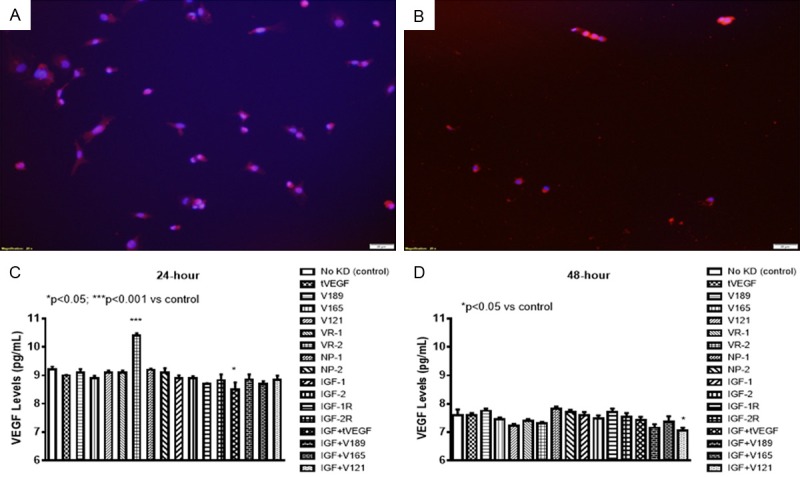
Expression of total VEGF in cells at 24 (A) and 48 (B) hours post transfection. Effect of selective SiRNA inhibition of VEGF isoforms, VEGF receptors, and/or IGF-I on VEGF levels in the media of proliferating human retinal endothelial cells (HRECs) at 24 (C) and 48 (D) hours transfection. Data are expressed as mean ± SEM (n=12 samples/group; *P<0.05, ***P<0.001 vs. control). Images were captured at 20× magnification.
Effects on sVEGFR-1
The levels of sVEGFR-1 (an endogenous negative regulator of VEGF), in the media were about 800-fold higher than that of VEGF. At 24 hr., knockdown of VEGF165, VEGFR-2, and IGF-1R caused a decrease in sVEGFR-1 (Figure 3A). In contrast, knockdown of NP-2 and combined IGF-I with VEGF189, or VEGF121 increased sVEGFR-1 levels. At 48 hr., sVEGFR-1 levels remained suppressed with IGF-1R knockdown (Figure 3B).
Figure 3.
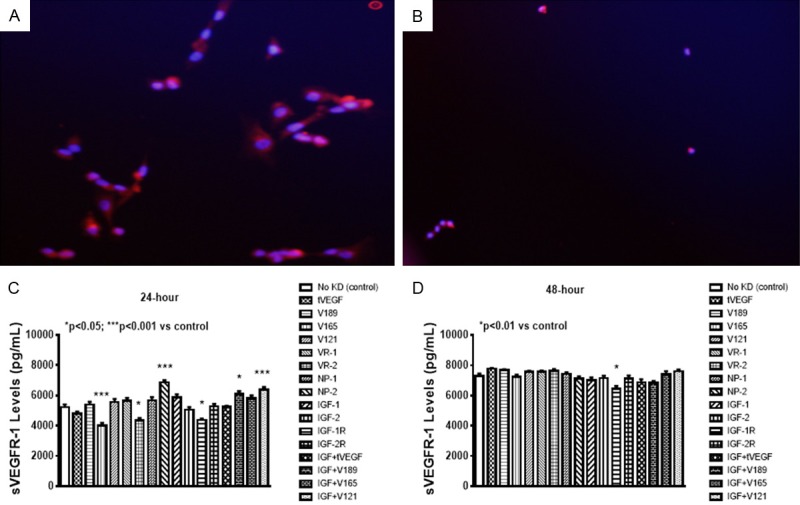
Expression of total VEGFR-1 in cells at 24 (A) and 48 (B) hours post transfection. Effect of selective SiRNA inhibition of VEGF isoforms, VEGF receptors, and/or IGF-I on sVEGFR-1 levels in the media of proliferating human retinal endothelial cells (HRECs) at 24 (C) and 48 (D) hours post transfection. Data are expressed as mean ± SEM (n=12 samples/group; *P<0.05, ***P<0.01 vs. control). Images were captured at 20× magnification.
Effects on IGF
IGF levels were increased at 24-hours with knockdown of VEGF189, VEGFR-2, and combination IGF-I with VEGF165 and VEGF121. IGF-I levels were dramatically decreased to almost zero with knockdown of the VEGF121 (Figure 4A). At 48, IGF levels were decreased with knockdown of VEGF isoforms and VEGFR-2, but remained elevated with IGF-I alone and combination with VEGF189, and VEGF165 (Figure 4B).
Figure 4.
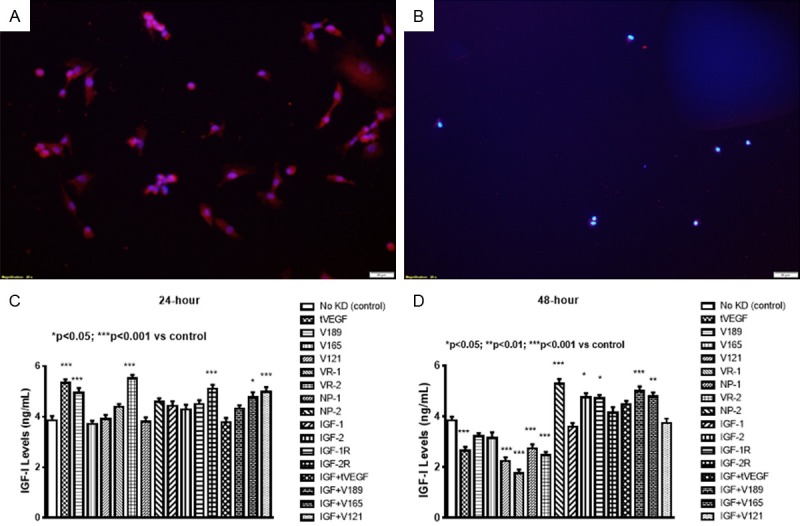
Expression of total IGF-I in cells at 24 (A) and 48 (B) hours post transfection. Effect of selective SiRNA inhibition of VEGF isoforms, VEGF receptors, and/or IGF-I on IGF-I levels in the media of proliferating human retinal endothelial cells (HRECs) at 24 (C) and 48 (D) hours post transfection. Data are expressed as mean ± SEM (n=12 samples/group; *P<0.05, **P<0.01, ***P<0.001 vs. control). Images were captured at 20× magnification.
Migration capacity
Figure 5 represents a single well showing EC migration using fluorescence staining. Migration capacity was determined by the number of cells that migrated to the bottom of the wells and presented as the mean number cells per well. Figure 5A shows more migration and appearance of tube formation. Figure 5B shows less migration. Decreased migration and tube formation capacities occurred with selective inhibition of VEGF189 isoform alone and in combination with IGF at 24 hr. (Figure 5C). However, at 48 hr. we noted a nonsignificant increase in migration capacity, when VEGF165, V121, and VEGFR-1 were knocked down (Figure 5D). Knockdown of the lower isoforms may increase the expression of the higher isoform, VEGF189 thus causing more migration.
Figure 5.
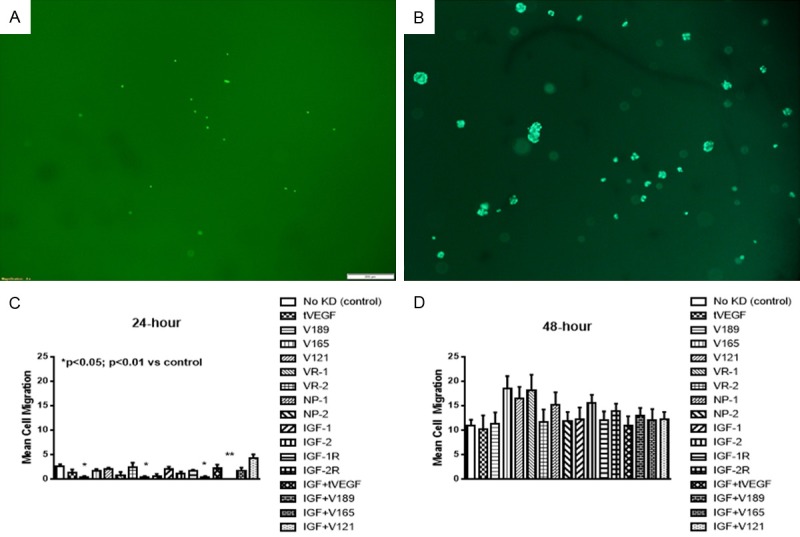
Representative sample of migration capacity of human retinal endothelial cells (HRECs) at 24 (A) and 48 (B) hours post VEGF transfection. Cells were stained with calcein AM fluorescent dye, the plates were imaged, and the number of cells that migrated to the bottom was determined at 24 (C) and 48 (D) hours. Data are expressed as mean ± SEM (n=4 samples/group; *P<0.05, **P<0.01 vs. control). Images were captured at 4× magnification.
Sphere-forming capacity
Figure 6 represents a single well showing EC sphere formation. Spheres were determined if the cells formed complete or almost complete circles. The number of circles were counted and presented as the mean number of spheres per well. Figure 6A shows three complete circles or spheres. Figure 6B shows no spheres and was counted as zero. Our data showed decreased sphere-forming capacity at 24 hr. with all knockdowns except IGF-I (Figure 6C) and this finding was consistent at 48 hr (Figure 6D).
Figure 6.
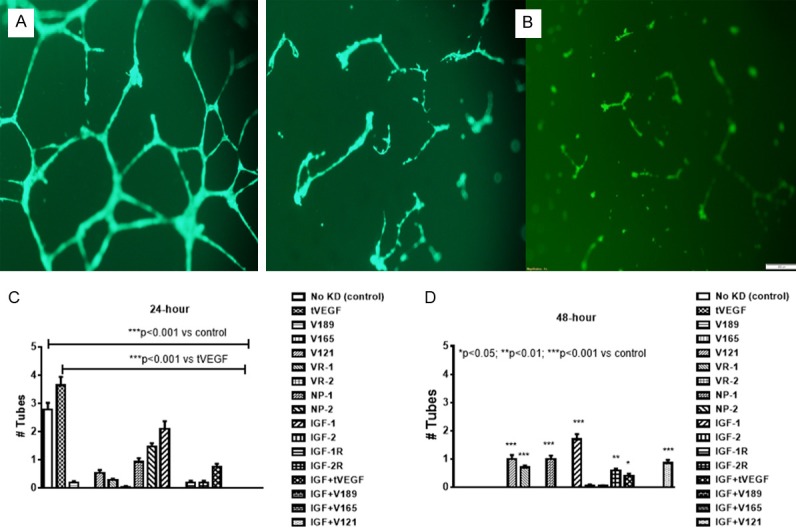
Representative sample of tube formation capacity of human retinal endothelial cells (HRECs) at 24 (A) and 48 (B) hours post transfection. Cells were resuspended in serum-free media and seeded onto 96 well plates. The plates were placed incubated under their growth conditions for 7 days and scored for the presence or absence of spheres at 24 (C) and 48 (D) hours. Data are expressed as mean ± SEM (n=96 samples/group; *P<0.05, **P<0.01, ***P<0.001 vs. control). Images were captured at 20× magnification.
Lipid peroxidation
Figure 7 represents cells assessed for lipid peroxidation using the Image-iT lipid peroxidation assay kit. Normal non-SiRNA exposed cells at 24 and 28 hours are represented in Figure 7A and 7B, respectively. These cells show no lipid peroxidation as evidenced by green color. In contrast, cells exposed to VEGF SiRNA showed increased green color at 24 hours (Figure 7C), an effect that intensified at 48 hours (Figure 7D).
Figure 7.
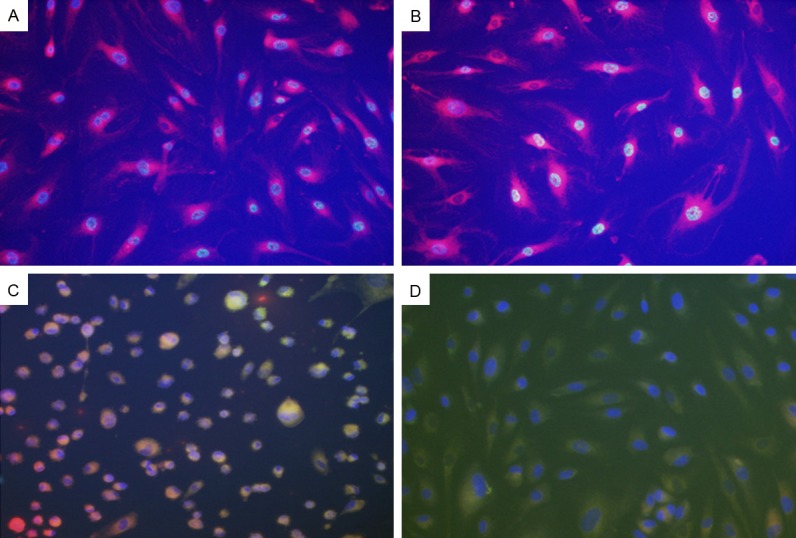
Representative sample of oxidative stress in of human retinal endothelial cells (HRECs) at 24 (C) and 48 (D) hours post VEGF transfection. Panels (A and B) represent control non-transfected cells at 24 and 48 hours, respectively. Oxidative stress was determined using the Image-It lipid peroxidation assay. Images show intense green staining in the SiRNA exposed cells suggesting lipid peroxidation. Images were captured at 20× magnification.
VEGF signaling
At 24 hours, most genes were robustly upregulated with VEGF189+IGF-I, VEGF165+IGF-I and VEGF121+IGF-I (Table 1). SiRNA of the total VEGF gene combined with IGF-I moderately upregulated affected only MAPK-3, PKCβ, and PKCγ. These same genes were upregulated with SiRNA of all genes except total VEGF and VEGF189. At 48 hours, most genes were downregulated, particularly MAPK-3, PKCβ, and PKCγ which were mostly affected by VEGF189 and VEGF165. In contrast, NP-1, IGF-I and VEGF189+IGF-I caused upregulation of most genes although the effect was less robust than at 24 hours (Table 2).
Table 1.
Gene expression of angiogenic growth factors in human retinal endothelial cells in response to 24-hour SiRNA exposure
| Genes of Interest | Total VEGF | VEGF189 | VEGF165 | VEGF121 | VEGFR-1 | VEGFR-2 | NP-1 | NP-2 | IGF-I | IGF-2 | IGF-IR | IGF-2R | Total VEGF+IGF-I | VEGF189+IGF-I | VEGF165+IGF-I | VEGF121+IGF-I |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Casp 9 | 2.4 | 3.2 | 8.8 | 1.6 | 1.8 | 1.3 | 3.5 | 1.7 | -2.0 | 2.2 | 1.2 | 7.4 | -1.1 | 17.5* | 76.4 | 74.5* |
| Cav 1 | 1.0 | -1.2 | -1.3 | -1.1 | -1.4 | -1.6 | -2.2 | -1.2 | -1.5 | -1.2 | -1.7 | -2.1 | -1.8 | -1.4 | -1.1 | -1.6 |
| COX-2 | 1.2 | 1.0 | -2.0 | 1.4 | 1.5 | 1.6 | -1.3 | 2.0 | -4.7 | 1.5 | 2.5 | 1.6 | 1.8 | 3.8 | 16.5* | 16.1* |
| eNOS | -1.0 | 1.1 | 1.1 | 2.4 | -3.0 | 1.2 | 1.2 | -1.3 | 1.2 | 1.3 | 2.3 | -1.0 | 1.0 | 1.6 | 6.9 | 6.7 |
| HIF1α | -1.1 | 1.1 | 1.1 | 1.3 | 1.2 | -1.3 | 1.1 | 1.2 | -1.2 | -1.0 | 1.2 | -2.5 | -1.1 | -1.7 | 2.6 | 2.5 |
| MAPK 1 | -1.0 | 1.1 | 1.2 | 1.6 | -2.0 | -1.1 | -1.0 | -1.2 | -1.2 | 1.0 | -1.1 | -1.7 | -1.1 | -1.2 | 2.8 | 2.7 |
| MAPK 3 | 1.1 | 2.1 | 21.3* | 4.2 | 11.1 | 2.5 | 26.0* | 13.3* | 5.3 | 2.5 | 18.3* | 74.5* | 13.5* | 122.4* | 2553* | 553.7* |
| NP-1 | 1.1 | 1.4 | 2.1 | 1.7 | 1.4 | 1.2 | -3.0 | 1.2 | -1.4 | 1.1 | 1.4 | 4.1 | 1.2 | 4.6 | 69.7 | 45.6* |
| NP-2 | 1.5 | 1.2 | 1.2 | 1.5 | -1.0 | -1.0 | 1.2 | -1.3 | 1.2 | -1.1 | 1.7 | -4.3 | 1.3 | -3.1 | 1.4 | 1.4 |
| PDGF | 1.2 | 2.2 | 5.0 | 3.2 | -1.3 | -1.3 | 1.6 | -1.3 | 1.0 | -1.2 | 1.3 | 3.3 | 1.9 | 7.8 | 44.6 | 33.1* |
| PKCα | -1.1 | 1.6 | -1.0 | 2.0 | 2.0 | -1.1 | 2.0 | 1.5 | 1.22 | -1.0 | 1.2 | 2.1 | 1.1 | 3.4 | 14.9 | 14.5* |
| PKCβ | 1.4 | 3.0 | 35.2* | 4.9 | 12.8* | 5.2 | 41.8* | 11.7* | 20.3* | 4.2 | 23.8* | 108.7* | 37.8* | 299.9* | 1691* | 3852* |
| PKCγ | -1.4 | 1.8 | 28.6* | 4.8 | 19.4* | 4.1 | 38.5* | 18.3* | 5.4 | -1.1 | 13.5* | 81.2* | 9.8 | 191.3* | 835.5* | 814.2* |
| VEGF A | 1.2 | 1.3 | -1.4 | -1.1 | 1.6 | 1.3 | 1.8 | 2.6 | -3.9 | -1.0 | 2.0 | 4.0 | -2.2 | 9.1 | 39.8* | 38.8* |
| VEGF B | -1.0 | 1.1 | -1.5 | 1.2 | 1.4 | 1.2 | -1.2 | -3.0 | 1.5 | 1.2 | 1.2 | -2.8 | -3.0 | -1.2 | 3.6 | 3.5 |
| VEGF C | -1.2 | -1.6 | 1.4 | 1.5 | 1.2 | 1.0 | 1.9 | -1.3 | -1.5 | 1.2 | 1.7 | 5.0 | 1.8 | 7.2 | 31.6* | 30.8* |
| VEGF D | -1.3 | 1.5 | 19.9* | 2.6 | 9.2 | 2.4 | 23.0 | 11.0* | 3.0 | 1.1 | 7.5 | 36.8* | 7.1 | 110.4* | 311.4* | 401.4* |
| VEGFR-1 | 1.2 | 1.0 | -3.0 | 1.1 | -4.3 | -2.2 | -1.0 | -1.5 | -1.3 | -1.6 | -1.2 | -1.0 | -8.5 | 2.3 | 10.0* | 9.8 |
| VEGFR-2 | 1.0 | 1.0 | 1.7 | 1.3 | -1.3 | -2.0 | 1.0 | 1.1 | -1.1 | -1.8 | 1.0 | 3.0 | -2.2 | 3.3 | 8.8 | 7.2 |
| VEGFR-3 | 1.1 | 1.7 | 1.2 | 1.7 | -1.5 | -2.3 | 1.5 | 4.7 | 1.5 | 1.0 | 2.7 | 2.8 | -1.6 | 6.6 | 188.2* | 27.9* |
Data are fold change from control (no knockdown). All data were corrected using 5 different housekeeping genes. Genes are selected from a profile of 84 genes. Genes of interest are: Casp 9, caspase 9; Cav 1, caveolin-1; COX-2, cyclooxygenase-2; eNOS, endothelial cell nitric oxide synthase; HIF1a, hypoxia inducible factor 1a; MAPK, mitogen-activated protein kinase; NP, neuropilin; PDGF, platelet-derived growth factor; PKC, protein kinase C; VEGF, vascular endothelial growth factor; VEGFR, VEGF receptor.
p<0.05 versus control.
Table 2.
Gene expression of angiogenic growth factors in human retinal endothelial cells in response to 48-hour SiRNA exposure
| Genes of Interest | Total VEGF | VEGF189 | VEGF165 | VEGF121 | VEGFR-1 | VEGFR-2 | NP-1 | NP-2 | IGF-I | IGF-2 | IGF-IR | IGF-2R | Total VEGF+IGF-I | VEGF189+IGF-I | VEGF165+IGF-I | VEGF121+IGF-I |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Casp 9 | -1.2 | -1.2 | -5.0 | -3.5 | -2.3 | 3.4 | 11.9* | -1.6 | 17.4* | -1.3 | -6.7 | 2.6 | -2.1 | 8.0 | 2.5 | -2.7 |
| Cav 1 | 1.3 | 1.4 | 1.8 | 1.0 | 1.8 | 1.3 | -1.7 | -1.4 | -1.2 | -1.5 | 1.2 | 1.4 | -1.1 | -1.1 | 1.0 | -1.3 |
| COX-2 | 1.8 | 1.1 | -1.7 | -2.7 | -3.9 | 3.1 | 11.9* | -1.6 | 17.4* | -1.3 | 1.0 | 1.8 | -2.1 | 8.0 | 2.5 | 1.3 |
| eNOS | -1.6 | 1.2 | 2.0 | -2.4 | -2.2 | 2.2 | 8.5 | 1.7 | 12.4* | -1.8 | -1.3 | 1.3 | 1.0 | 5.7 | 1.8 | 1.6 |
| HIF1α | -3.3 | -1.4 | -1.4 | -2.5 | -2.4 | -2.5 | 1.2 | -1.5 | 1.7 | -1.4 | 1.8 | -5.4 | -2.0 | -1.3 | -1.6 | -1.6 |
| MAPK 1 | -3.7 | -2.0 | -2.7 | -2.0 | -1.7 | -2.4 | -1.1 | -2.0 | 1.4 | -3.0 | -2.0 | -2.0 | -2.7 | -1.6 | -1.3 | -2.6 |
| MAPK 3 | 2.0 | -21.0* | -26.9* | -4.7 | -4.3 | 9.5 | 21.8* | -1.3 | 1.4 | -1.2 | -10.4* | 1.5 | -2.0 | 7.0 | 10.6* | -2.1 |
| NP-1 | -1.3 | -1.1 | -1.5 | -1.8 | -1.8 | 1.7 | 20.8* | -2.4 | 1.1 | 1.7 | -1.7 | 2.3 | 1.2 | 14.1* | 5.0 | 1.0 |
| NP-2 | -1.0 | 4.6 | 4.0 | 1.0 | 1.4 | 2.5 | 3.3 | -1.0 | 5.6 | 2.2 | 4.8 | -1.6 | 1.1 | 2.2 | -1.4 | 5.7 |
| PDGF | -1.5 | -2.0 | -1.2 | -1.7 | -1.4 | 4.0 | 8.8 | -2.2 | 12.9* | 1.5 | -2.7 | 1.3 | -3.0 | 5.9 | 4.5 | -2.5 |
| PKCα | 1.3 | -1.5 | -1.2 | -1.8 | 1.6 | 2.3 | 8.8 | 1.7 | 12.9* | 1.1 | -1.2 | 1.3 | -2.3 | 8.9 | 3.2 | -2.2 |
| PKCβ | 1.5 | -54.7* | -17.9* | -9.1 | -2.4 | 1.4 | 6.8 | -1.1 | 44.5* | 2.5 | -24.9* | -1.3 | -4.9 | 15.0* | 6.3 | -2.2 |
| PKCγ | -1.2 | -23.6* | -5.1 | -6.2 | -4.0 | 3.1 | 11.9* | -1.6 | 17.4* | -1.3 | -10.8* | 1.8 | -2.1 | 8.0 | 2.5 | -2.7 |
| VEGF A | -1.2 | -3.0 | -5.1 | -6.2 | -4.0 | 3.1 | 11.9* | -1.6 | 17.4* | -1.3 | -1.6 | 1.8 | -2.1 | 8.0 | 2.5 | -2.7 |
| VEGF B | -9.0 | -2.0 | -1.6 | -1.4 | -1.3 | -2.7 | 1.4 | -3.0 | 2.1 | 1.7 | -1.4 | -2.4 | -1.9 | -1.0 | -1.0 | 1.0 |
| VEGF C | -1.2 | -5.0 | -2.5 | -1.4 | -4.0 | 3.0 | 11.6* | -1.6 | 17.0* | -1.3 | -2.2 | 1.8 | -2.2 | 7.9 | 2.5 | -1.2 |
| VEGF D | -1.1 | -14.2* | -3.9 | -5.2 | -3.0 | 3.4 | 14.7* | -1.0 | 29.4* | 1.4 | -7.0 | 2.5 | -1.5 | 10.8* | 2.5 | -2.0 |
| VEGFR-1 | -1.2 | 1.4 | 1.2 | -5.0 | 1.0 | 3.1 | 11.9* | -1.1 | 17.4* | -1.3 | -1.2 | 1.8 | 1.1 | 8.0 | 2.5 | -1.3 |
| VEGFR-2 | -3.8 | -1.2 | -2.7 | -9.0 | -2.0 | 3.7 | 6.2 | 1.1 | 12.6* | -1.2 | -4.2 | -2.0 | -3.0 | 6.5 | 5.2 | -2.6 |
| VEGFR-3 | -1.2 | -1.4 | 1.2 | -5.1 | -4.0 | 3.1 | 12.0* | -1.6 | 17.4* | -1.3 | -1.4 | 1.8 | -2.1 | 8.0 | 2.9 | -2.7 |
Data are fold change from control (no knockdown). All data were corrected using 5 different housekeeping genes. Genes are selected from a profile of 84 genes. Genes of interest are as described in Table 1.
p<0.05 versus control.
Discussion
In this study we present data showing a significant inhibition of sphere-forming capacity with selective VEGF isoforms knockdown. The most profound effect occurred with VEGF189 and VEGF165. This demonstrates that inhibition of selective VEGF isoforms may be an effective targeting strategy for the control of abnormal/hypervascularization in the development of diseases such as ROP. We further showed that proliferating HREC under normoxia (or normal conditions) produce significant amounts of sVEGFR-1 as compared with total VEGF production, suggesting that sVEGFR-1 is an important by-product of proliferating retinal ECs. This finding corroborates those of Chappell et al. [29] who demonstrated that sVEGFR-1 is necessary for local sprout guidance and pushing of emerging sprouts away from the parent vessels. High levels of sVEGFR-1 may neutralize the VEGF gradient which could account for the low levels observed. As a corollary, low VEGF levels in the media may be due to retention of higher isoforms to their membrane receptors. We also showed a differential effect among VEGF isoforms alone and in combination with IGF-I with respect to the ability of HRECs to form spheres. The levels of VEGF were not appreciably affected by selective knockdown of individual isoforms but was significantly increased with VEGFR-2 and inhibited with tVEGF+IGF-I at 24-hr. Together, these findings lead us to speculate that sVEGFR-1 may be a more important regulator of angiogenesis in proliferating HRECs in vitro.
Few studies have examined the role of individual VEGF isoforms and its receptors in HREC angiogenesis [16,30,31] despite the development and use of anti-VEGF medications with unknown long-term side effects. The findings we present in this paper suggest the possibility of minimizing side effects by developing targeted therapies to selectively inhibit at least one of the VEGF isoforms. It is known that VEGF isoforms differ by the presence or absence of sequences encoded by exon 6 and 7 [32]. VEGF189 is tightly bound to the ECM while VEGF165 is partially bound and VEGF121 is freely diffusible. The relative solubility of VEGF isoforms influences their specific bioactivities [33]. Our data show that manipulation of VEGF121 has little influence on HREC production of growth factors or their migration and tube forming capacities, except for decreasing IGF-I levels at 48 hr. Our findings suggest that unlike VEGF165 and VEGF189, VEGF121 may not be vital for normal angiogenesis of HRECs.
The observation that sVEGFR-1 level was predominantly expressed in the media of proliferating HRECs may be linked to its role in cell migration, a process that is essential in the initiation of angiogenesis. The trigger for guided vessel network patterning is provided by several signaling pathways [26,34-36]. It seems plausible that newly proliferating HRECs require initiation of migration in order for the template of new vessels to be laid down as the first step of angiogenesis. Therefore, the levels of sVEGFR-1 are upregulated at this stage as an essential step in the process of normal physiological angiogenesis. Relatively low VEGF levels in the media surrounding proliferating HRECs may be important for curtailing extrinsic vessel patterning by overwhelming production of sVEGFR-1 to regulate local sprout guidance [37]. This process of VEGF regulation may be required to achieve normal angiogenesis. Our findings emphasize and support the notion that sVEGFR-1 is more important than VEGF for initiation of angiogenesis.
IGF is known as a permissive factor for VEGF activity, and in settings were IGF level is low, angiogenesis is abnormal [18-20]. The increase of IGF determines increase in VEGF with secondary over-vascularization (premature infants with low IGF have worse ROP). We have shown that both VEGF and sVEGFR-1 production is decreased by manipulation of IGF-I and its receptor. These findings concur with previous reports demonstrating that normal IGF levels are necessary for normal angiogenesis.
VEGF interacts with two high affinity tyrosine kinase receptors on endothelial cells, VEGFR-1 and VEGFR-2, which have differential roles in angiogenesis [9]. The pathway activation of VEGFR-2 by VEGF in cells lacking VEGFR-1 results in a mitogenic response, while activation of VEGFR-1 does not induce cell proliferation. Though VEGF signaling through VEGFR-2 is well understood, the role of VEGFR-1 is less clear; however, genetic loss of VEGFR-1 leads to vessels dysmorphogenesis and overgrowth [38], a key finding in ROP. The manipulation of VEGFR-2 and IGF had a significant impact on VEGF expression. VEGFR-2 inhibition resulted in an initial increase in VEGF expression at 24 hours, with the effect disappearing by 48 h. This observation may be due to an initial release of VEGF from ECM by decreasing VEGFR-2 binding ability. In contrast, manipulation of IGF or its receptor determined a decrease of VEGF. Interestingly, these cells preserved their ability to differentiate and form spheres, further implying a lesser role of VEGF than sVEGFR-1 in proliferating cells.
It was interesting to note that knockdown of selective isoforms plus IGF-I induced the upregulation of apoptosis gene, caspase-9, suggesting that IGF-I is important for VEGF signaling and HREC viability. MAPK3 and PKC were upregulated by knockdown of most genes. MAPKs play an important role in inflammatory conditions of the retina [39] and PKC is involved in vascular permeability and loss of tight junction proteins in retinal vessels [40]. Their upregulation with VEGF165, NP, IGFR and VEGF+IGF-I knockdown demonstrate the importance of these genes in EC integrity and may suggest alternative pathways for targeted therapies in vasoproliferative retinopathies. These observations strongly imply that the primary response of HRECs to knockdown of VEGF, its receptors and/or IGF-I is apoptosis. Despite the clinical importance of this study, one limitation is that the experiments were conducted under normoxic conditions, and therefore the effects do not accurately represent hyperoxic and/or hypoxic conditions that are known to contribute to ROP [41]. In addition, the temporary nature of selective gene silencing should also be considered when results are interpreted at 24 or 48 hours. Collecting data at shorter intervals to show a true longitudinal time course of effects is warranted and would have significant clinical value.
In light of current attempts at treating pathologic angiogenic conditions such as ROP with complete VEGF inhibition, our work may be relevant in three ways. First, our results indicate that whole inhibition may be counter-productive, in as much as it may lead to an actual increase in sVEGFR-1 and IGF, and not impede sphere formation. Second, targeted inhibition is likely to be preferable because it could minimize side effects associated with complete VEGF inhibition, and our results show that such selective inhibition can be effective in suppressing sphere formation. Finally, by identifying the particular isoforms that are most effective in suppressing sphere formation, future research on the side-effects of VEGF suppression therapy could be easier to pursue as they need to focus only on the subset of isoforms/receptors we identified as most effective in suppression. Targeted obliteration of VEGF189 rather than non-selective VEGF inhibition may be a more desirable therapy for ROP to prevent unwanted side effects. IGF-I is a key factor regulating VEGF signaling and should be considered when anti-VEGF medication is contemplated.
Acknowledgements
This work was made possible through a grant from Memorial Medical Center Foundation, Long Beach, CA 90806, and the Eunice Kennedy Shriver National Institute of Child Health & Human Development Grant # U54HD071594.
Disclosure of conflict of interest
None.
References
- 1.Herbert SP, Stainier DY. Molecular control of endothelial cell behavior during blood vessel morphogenesis. Nat Rev. 2011;12:551–564. doi: 10.1038/nrm3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ferrara N. Vascular endothelial growth factor: molecular and biological aspects. Curr Top Microb Immuno. 1999;237:1–30. doi: 10.1007/978-3-642-59953-8_1. [DOI] [PubMed] [Google Scholar]
- 3.Penn JS, Madan A, Caldwell RB. Vascular endothelial growth factor in eye disease. Prog Retin Eye Res. 2008;27:331–71. doi: 10.1016/j.preteyeres.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pierce EA, Foley ED, Smith LE. Regulation of vascular endothelial growth factor by oxygen in a model of ROP. Arch Ophthalmol. 1996;114:1219–1228. doi: 10.1001/archopht.1996.01100140419009. [DOI] [PubMed] [Google Scholar]
- 5.Heidary G, Löfqvist C, Mantagose L, et al. Retinopathyof prematurity. Clinical insights from molecular studies. Neoreviews. 2009;10:e550. [Google Scholar]
- 6.Quinn GE, Gilbert C, Darlow BA, Zin A. Retinopathy of prematurity: an epidemic in the making. Chin Med J (Engl) 2010;123:2929–2937. [PubMed] [Google Scholar]
- 7.Romagnoli C. Risk factors and growth factors in ROP. Early Hum Dev. 2009;85:S79–S82. doi: 10.1016/j.earlhumdev.2009.08.026. [DOI] [PubMed] [Google Scholar]
- 8.Chen J, Smith LE. Retinopathy of prematurity. Angiogenesis. 2007;10:133–140. doi: 10.1007/s10456-007-9066-0. [DOI] [PubMed] [Google Scholar]
- 9.Robinson CJ, Stringer SE. The splice variants of VEGF and their receptors. J Cell Sci. 2001;114:853–865. doi: 10.1242/jcs.114.5.853. [DOI] [PubMed] [Google Scholar]
- 10.Ruhrberg C. Growing and shaping the vascular tree: multiple roles for VEGF. BioEssays. 2003;25:1052–1060. doi: 10.1002/bies.10351. [DOI] [PubMed] [Google Scholar]
- 11.Gerhardt H. VEGF and endothelial guidance in angiogenic sprouting. Organogenesis. 2008;4:241–246. doi: 10.4161/org.4.4.7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ruhrberg C, Gerhardt H, Golding M, Watson R, Ioannidou S, Fujisawa H, Betsholtz C, Shima DT. Spatially restricted patterning cues provided by heparin-binding VEGF-A control blood vessel branching morphogenesis. Genes Dev. 2002;16:2684–98. doi: 10.1101/gad.242002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stalmans I, Ng YS, Rohan R, Fruttiger M, Bouché A, Yuce A, Fujisawa H, Hermans B, Shani M, Jansen S, Hicklin D, Anderson DJ, Gardiner T, Hammes HP, Moons L, Dewerchin M, Collen D, Carmeliet P, D’Amore PA. Arteriolar and venular patterning in retinas of mice selectively expressing VEGF isoforms. J Clin Invest. 2002;109:327–336. doi: 10.1172/JCI14362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999;13:9–22. [PubMed] [Google Scholar]
- 15.Shibuya M. Vascular endothelial growth factor-dependent and -independent regulation of angiogenesis. BMB Rep. 2008;41:278–286. doi: 10.5483/bmbrep.2008.41.4.278. [DOI] [PubMed] [Google Scholar]
- 16.Pan Q, Chathery Y, Wu Y, Rathore N, Tong RK, Peale F, Bagri A, Tessier-Lavigne M, Koch AW, Watts RJ. Neuropilin-1 binds to VEGF121 and regulates endothelial cell migration and sprouting. J Biol Chem. 2007;282:24049–24056. doi: 10.1074/jbc.M703554200. [DOI] [PubMed] [Google Scholar]
- 17.Whitaker GB, Limberg BJ, Rosenbaum JS. Vascular endothelial growth factor receptor -2 and Neuroplilin-1 form a receptor complex that is responsible for differential signaling potency of VEGF 165 and VEGF 121. J Biol Chem. 2001;276:25520–25531. doi: 10.1074/jbc.M102315200. [DOI] [PubMed] [Google Scholar]
- 18.Hellstrom A, Carlsson B, Niklasson A, Segnestam K, Boguszewski M, de Lacerda L, Savage M, Svensson E, Smith L, Weinberger D, Albertsson Wikland K, Laron Z. IGF-1 is critical for normal vascularization of the human retina. J Clinic Endocrinol Metab. 2002;87:3413–3416. doi: 10.1210/jcem.87.7.8629. [DOI] [PubMed] [Google Scholar]
- 19.Hellstrom A, Perruzzi C, Ju M, Engstrom E, Hard AL, Liu JL, Albertsson-Wikland K, Carlsson B, Niklasson A, Sjodell L, LeRoith D, Senger DR, Smith LE. Low IGF-1 suppresses VEGF-survival signaling in retinal endothelial cells: direct correlation with clinical ROP. Proc Natl Acad Sci U S A. 2001;98:5804–5808. doi: 10.1073/pnas.101113998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith LE, Shen W, Perruzzi C, Soker S, Kinose F, Xu X, Robinson G, Driver S, Bischoff J, Zhang B, Schaeffer JM, Senger DR. Regulation of vascular endothelial growth factor-dependent retinal neovascularization by insulin-like growth factor 1 receptor. Nat Med. 1999;5:1390–1395. doi: 10.1038/70963. [DOI] [PubMed] [Google Scholar]
- 21.Jang SY, Choi KS, Lee SJ. Delayed-onset retinal detachment after an intravitreal injection of ranibizumab for zone 1 plus retinopathy of prematurity. J AAPOS. 2010;14:457–459. doi: 10.1016/j.jaapos.2010.05.011. [DOI] [PubMed] [Google Scholar]
- 22.Hård AL, Hellström A. On safety, pharmacokinetics and dosage of bevacizumab in ROP treatment-a review. Acta Paediatr. 2011;100:1523–1527. doi: 10.1111/j.1651-2227.2011.02445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Honda S, Hirabayashi H, Tsukahara Y, Negi A. Acute contraction of the proliferative membrane after an intravitreal injection of bevacizumab for advanced retinopathy of prematurity. Graefes Arch Clin Exp Ophthalmol. 2008;246:1061–1063. doi: 10.1007/s00417-008-0786-7. [DOI] [PubMed] [Google Scholar]
- 24.Wu WC, Lai CC, Chen KJ, Chen TL, Wang NK, Hwang YS, Yeung L, Li LM. Long term tolerability and serum concentrations of Bevacizumab (Avastin) when injected in newborn rabbit eyes. Invest Ophthalmol Vis Sci. 2011;51:3701–3708. doi: 10.1167/iovs.09-4425. [DOI] [PubMed] [Google Scholar]
- 25.Atchaneeyasakul LO, Trinavarat A. Choroidal rupture after adjuvant intravitreal injection of bevacizumab for aggressive posterior retinopathy of prematurity. J Perinatol. 2010;30:497–499. doi: 10.1038/jp.2009.166. [DOI] [PubMed] [Google Scholar]
- 26.Hartnett ME. The role of cytokines and treatment algorithms in retinopathy of prematurity. Curr Opin Ophthalm. 2017;28:282–288. doi: 10.1097/ICU.0000000000000360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beharry KD, Cai CL, Valencia GB, Lazzaro D, Valencia AM, Salomone F, Aranda JV. Human retinal endothelial cells and astrocytes cultured on 3-D scaffolds for ocular drug discovery and development. Prostaglandins Other Lipid Mediat. 2017;134:93–107. doi: 10.1016/j.prostaglandins.2017.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quan M, Cai CL, Valencia GB, Aranda JV, Beharry KD. MnTBAP or catalase is more protective against oxidative stress in human retinal endothelial cells exposed to intermittent hypoxia than their co-administration (EUK-134) React Oxyg Species. 2017;3:47–65. doi: 10.20455/ros.2017.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chapell JC, Taylor SM, Ferrara N, Bautch VL. Local guidance of emerging vessel sprouts requires soluble Flt-1. Dev Cell. 2009;17:377–386. doi: 10.1016/j.devcel.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stewart EA, Samaranayake GJ, Browning AC, Hopkinson A, Amoaku WM. Comparison of choroidal and retinal endothelial cells: characteristics and response to VEGF isoforms and anti-VEGF treatments. Exp Eye Res. 2011;93:761–766. doi: 10.1016/j.exer.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 31.Wilting J, Birkenhäger R, Eichmann A, Kurz H, Martiny-Baron G, Marmé D, McCarthy JE, Christ B, Weich HA. VEGF121 induces proliferation of vascular endothelial cells and expression of flk-1 without affecting lymphatic vessels of the chorioallantoic membrane. Dev Biol. 1996;176:76–85. doi: 10.1006/dbio.1996.9993. [DOI] [PubMed] [Google Scholar]
- 32.Tischer E, Mitchell R, Hartman T, Silva M, Gospodarowicz D, Fiddes JC, Abraham JA. The human gene for VEGF. Multiple protein forms are encoded through alternative exon splicing. J Biol Chem. 1991;266:11947–11954. [PubMed] [Google Scholar]
- 33.Takahashi H, Shibuya M. The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under pathological conditions. Clin Sci (London) 2005;109:227–241. doi: 10.1042/CS20040370. [DOI] [PubMed] [Google Scholar]
- 34.Eichman A, Yuan L, Moyon D, le Noble F, Pardanaud L, Breant C. Vascular development: from precursors cells to branched arterial and venous networks. Int J Dev Biol. 2005;49:259–267. doi: 10.1387/ijdb.041941ae. [DOI] [PubMed] [Google Scholar]
- 35.Holerfield MT, Hughes CC. Crosstalk between vascular endothelial growth factor-beta in vascular morphogenesis. Circ Res. 2008;102:637–652. doi: 10.1161/CIRCRESAHA.107.167171. [DOI] [PubMed] [Google Scholar]
- 36.Kappas NC, Zeng G, Chappell J, Kearney JB, Hazarika S, Kallianos KG, Patterson C, Annex BH, Bautch VL. The VEGF receptor Flt-1 spatially modulates Flk-1 signaling and blood vessels branching. J Cell Biol. 2008;181:847–858. doi: 10.1083/jcb.200709114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A, Jeltsch M, Mitchell C, Alitalo K, Shima D, Betsholtz C. VEGF guides angiogenic sprouting utilizing tip cell filopodia. J Cell Biol. 2003;161:1163–1177. doi: 10.1083/jcb.200302047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roberts DM, Kearney JB, Johnson JH, Rosenberg MP, Kumar R, Bautch VL. The vascular endothelial growth factor (VEGF) receptor Flt-1(VEGFR-1) modulates Flk (VEGFR-2) signaling during blood vessels formation. Am J Pathol. 2004;164:2511–2524. doi: 10.1016/S0002-9440(10)63711-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Du Y, Tang J, Li G, Berti-Mattera L. Effects of p38 MAPK inhibition on early stages of diabetic retinopathy and sensory nerve function. Invest Ophthalmol Vis Sci. 2010;51:2158–2164. doi: 10.1167/iovs.09-3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim JH, Kim JH, Jun HO, Yu YS, Kim KW. Inhibition of protein kinase C delta attenuates blood-retinal barrier breakdown in diabetic retinopathy. Am J Pathol. 2010;176:1517–1524. doi: 10.2353/ajpath.2010.090398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith LE. Through the eyes of a child: understanding retinopathy through ROP the Friedenwald lecture. Invest Ophthalmol Vis Sci. 2008;49:5177–82. doi: 10.1167/iovs.08-2584. [DOI] [PubMed] [Google Scholar]