
Table 5.
Reported QSAR studies used in neurodegenerative diseases drug discovery.
| Author (Year of Publication) | Diseases | QSAR Technique Performed | Chemical Scaffold Under Study | Target | Significance of Study | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Rezaei Makhuri et al. (2015) [61] | ALS | 3D-QSAR using CoMFA, CoMSIA and Auto GPA | A series of 47 N-(benzothiazolyl)-2-phenyl-acetamides | CK-1δ | The results of 3D-QSAR analyses revealed that hydrophobic and negatively charged groups at 6th position of benzothiazole ring and positively charged and bulky groups at ortho position of phenyl ring in N-(benzothiazolyl)-2-phenyl-acetamides were favorable for high bioactivity. | |||||
| Zhu et al. (2006) [164] |
PD, AD | 3D-QSAR using CoMFA and CoMSIA techniques | A set of 55 tripeptide aldehyde inhibitors | 20S proteasome | The contour maps corroborated with the structural features of the binding pocket of β5 subunit of 20S proteasome, which proposed that the built models could be applied to pre-screen compounds to expedite the development of lead-compounds with optimized pharmacokinetic properties. | |||||
| Fresqui et al. (2013) [165] |
anti-AD, antidepressant and anti-PD | 3D-QSAR | A set of 34 amphetamine derivatives (the R and S configurations of a series of MAO A inhibitors) | MAO A | Six descriptors, namely, CHELPG atomic charges C3, C4 and C5, electrophilicity, molecular surface area and logP were found to be significant, considering both the configurations. | |||||
| Bharate et al. (2013) [166] |
AD | Descriptor based QSAR and pharmacophore based QSAR studies | A series of meridianin analogs | Dyrk1A | This study revealed that Kier Chi4 path/cluster (molecular connectivity index), total lipole (measure of the lipophilic distribution in a 3D space), VAMP polarization (polarizability coordinate), Dp and logP play vital role in Dyrk1A inhibition. | |||||
| Tong et al. (1996) [167] |
AD | 3D-QSAR (CoMFA) study | A series of 1-benzyl-4-[2-(N- benzoylamino)ethyl] piperidine derivatives and of N-benzylpiperidine benzisoxazoles | AChE | i) Substitutions with bulky and/or lipophilic groups at the benzisoxazole and benzoyl moieties are important for the activity; ii) The oxygen in isoxazole ring, if replaced with less electronegative atom like nitrogen or sulfur, is found to diminish the potency; iii) The basicity of the nitrogen atom in N-piperidine ring is important in contributing to the activity; iv) Occupying the ortho position of the benzoyl moiety with steric bulk negatively affects the activity. |
|||||
| Ponmary et al. (2010) [168] |
PD | SW-MLR method | Compounds structurally similar to glycerol | Parkinson’s disease causing targets | The results demonstrated the high robustness and real predictive power of IC50 model. | |||||
| Jung et al.(2007) [169] | AD | (GA)-MLR and (SA)-MLR |
Tacrine derivatives (a set of 80 structurally heterogeneous compounds composed of 11H-indeno-[1,2-b]-quinolin-10-ylamine derivatives, thiopyranoquinolines, pyranoquinolines and benzonaphthyridines, tacrine-E2020 hybrids, bis-tacrine congeners, and tacrine-hurprine heterodimers) | AChE | The best equation was obtained from SA MLR with greater explanatory and prediction capability. The results suggested the important roles of hydrophobic and electrostatic interactions on increasing the structure’s AChE activity. | |||||
| Author (Year of Publication) | Diseases | QSAR Technique Performed | Chemical Scaffold Under Study | Target | Significance of Study | |||||
| Chen et al. (2012) [170] |
Neuroprotect-ive profile and a moderate Ca2+ channel blockade effect | 3D-QSAR models based on the flexible docking alignment using CoMFA and CoMSIA | multi-target-directed AChEIs of tacrine-nimodipine dihydropyridine | AChE | The results indicated that the IC50 can be improved by means of increasing the electronegativity and introducing small volume substituent at 3-position of the DHP (1,4-dihydropyridines) and hydrophobic like methoxy group was favorable to the 4-position of the benzene ring of DHP. | |||||
| Hoeglund et al. (2010) [171] | Cancer progression, MS, obesity, diabetes, AD, and chronic pain | Integrated ligand-based computational strategies (binary QSAR), medicinal chemistry, and experimental enzymatic assays | Analogues of the most potent hit (H2L 7905958, IC50 of 1.6 ± 0.4 μM) | Autotaxin (ATX) | Analogues of the lead compound were examined and four of the 30 indicated IC50 less than or equal to the lead. The most potent analog indicated an IC50 of 900 nM with respect to ATX-mediated FS-3 hydrolysis with a Ki of 700 nM, making this compound approximately 3-fold more potent than the lead. | |||||
| Recanatini et al. (1997) [172] | AD | Comparative 2D-QSAR studies | Three classes of AChEIs, for example, physostigmine analogs, 1,2,3,4-tetrahydroacridines (tacrine analogs) and benzylamines | AChE | i) Hydrophobicity plays a crucial role in both the physostigmine and the benzylamine-derived classes; ii) electronic effects are vital for the interactions shown by the variable portion of benzylamine derivatives; and iii) steric factors are also important. | |||||
| Jain and Jadhav (2013) [173] | AD | 2D-QSAR using MLR | Aminoimidazoles dataset | β-Secretase (BACE-1) | The study revealed that thermodynamic descriptors (MR, logP, van der Waals energy, polar surfacearea) and steric descriptors (Harary index, Randic index) play important role in β-secretase inhibition. | |||||
| Debord et al. (1997) [174] | AD | 2D-QSAR | Derivatives of 2-amino-4,6-dimethylpyridine, aryl (alkyl) carboxamides, thiocarbamides and amidrazones | AChE and BChE | The binding affinity was improved by the structural changes like: i) increase in molecular volume; ii) decrease in the energy of the LUMO; iii) insertion of a methylene group between the amide carbonyl and the aromatic ring; and iv) replacement of the amide oxygen by sulfur. | |||||
| Huang et al. (2013) [175] | AD | 3D-QSAR using Topomer CoMFA | 125 BACE-1 inhibitors |
BACE1 | Topomer search was used for VS in lead-like compounds present in ZINC databases and as a result, they successfully designed 30 new molecules with better activity than those present in the dataset. | |||||
| Hossain et al. (2013) [176] | AD | QSAR (3D-QSAR, HQSAR) and pharmacophore mapping studies | Structurally diverse BACE inhibitors | BACE | Both types of studies confirmed the importance of terminal meta-tolyl sulfonamide piperazine core and heterocyclic ring along with adjacent nucleophilic hydroxyl group and amide linkage. Finally, it was concluded from the findings that hydrogen bond donor and acceptor, hydrophobicity, electrostatic and steric properties of ligand are the important features for interaction with receptor cavity. | |||||
| Dessalew et al. (2007) [177] | cancer, chronic inflammation, bipolar disorders and AD | 3D-QSAR studies using CoMFA and CoMSIA | Novel class of pyrazolopyrimidine derivatives | GSK-3 | Based on the contour analysis, authors deduced that improvement in GSK-3β binding affinity can be achieved through conformationally restricted substitution at N1 position near region A and keeping the electronegative group to the central core in region B.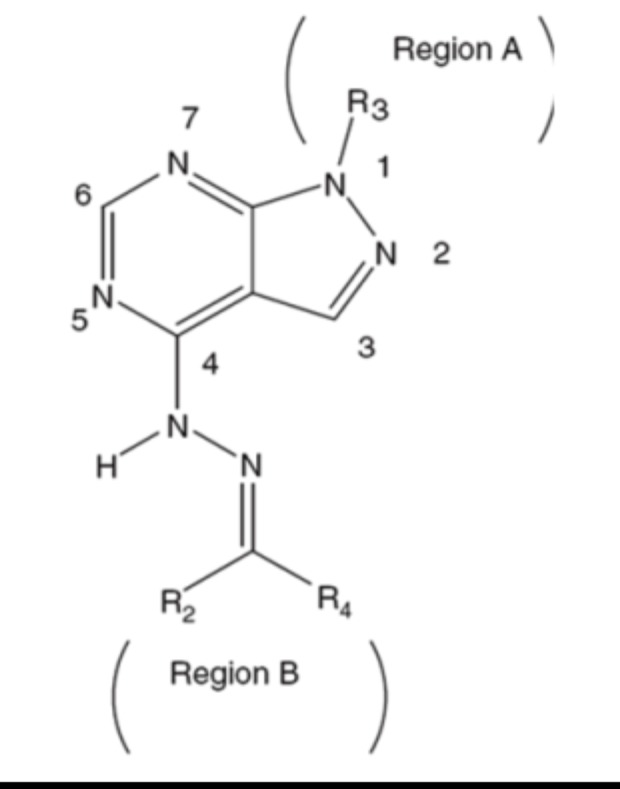
|
|||||
| Author (Year of Publication) | Diseases | QSAR Technique Performed | Chemical Scaffold Under Study | Target | Significance of Study | |||||
| Garcı´a et al. (2010) [178] | anti-AD, anti-parasitic, anti-fungi and antibacterial | Multi-target LDA | Heterogeneous structural GSK-3 inhibitors | GSK-3 | The reported LDA model was significant, since one can use a single equation to predict the results of heterogeneous series of organic compounds in 42 different experimental tests instead of developing and using 42 different QSAR models. | |||||
| Bhadoriya et al. (2014) [179] | AD | 3D-QSAR using kNN-MFA | A series of 34 fused 5,6-bicyclic heterocycles | g-secretase | The developed kNN-MFA model highlighted the importance of shape of the molecules, that is, hydrophobic and steric descriptors at the grid points His83 and Ser183, Ser227 for γ-secretase binding interaction. | |||||
| Barreca et al. (2003) [180] | Anticonvulsa-nts and neuroprotectants | A four-point Catalyst HIPHOP pharmacophore | 14 noncompetitive AMPA receptor antagonists | AMPA | This hypothesis, which consisted of two hydrophobic regions, one hydrogen bond acceptor and one aromatic region was employed to screen the Maybridge database and select eight compounds for testing of which six of these were found to be active in vivo as anticonvulsants. | |||||
| Valasani et al. (2013) [181] | AD | 2D-QSAR | Frentizole, benzothiazole-urea derivatives | ABAD | Based on QSAR studies of frentizole and benzothiazole-urea derivatives, authors designed and synthesized novel small drug molecules as benzothiazole-urea and frentizole phosphonate derivatives, which might have the capacity to cross the BBB and inhibit ABAD interaction. | |||||
| Kaur et al. (2000) [182] | AD | 2D-QSAR | Derivatives of physostigmine, tacrine, donepezil, huperzine A | AChE | It was concluded that all inhibitors were of hydrophobic nature as suggested by the presence of logP in the majority of QSAR models. Additionally, it was observed that all classes of inhibitors contained ionizable nitrogen. | |||||
| Zhou et al. (2015) [183] | AD | 3D-QSAR (CoMFA and CoMSIA), molecular docking, and MD | 60 tacrine derivatives | AChE | The contour maps for five fields obtained from the optimal 3D-QSAR models revealed that the steric and H-bond fields of these compounds were essential for their activities. Some key residues such as Tyr70, Trp84, Tyr121, Trp279, and Phe330 at the binding site of AChE were identified from molecular docking. | |||||
| Pourbasheer et al. (2015) [184] | PD | 3D-QSAR using CoMFA | A series of pyrimidines such as AA2AR antagonists | AA2AR | Based on the derived results some novel potent AA2AR antagonists have been designed and the proposed models were used to predict the AA2AR antagonist activity of newly designed compounds. | |||||
| Dinata et al. (2013) [185] | CJD | QSAR using LR, a statistical model of parabolic regression and multiple regression. | 2-aminothiazole derivatives | PrPSc | The results indicated that steric and lipophilic were the parameter most closely related to improve the biological activity of the compound 2-aminothiazole derivatives. | |||||
| Hajimahdi et al. (2016) [186] | AD, HD and PD | SW-MLR | A series of 53 potent 1,2-benzisothiazol-3-one derivatives | Caspase-3 (cysteine-dependent aspartyl-specific protease) | The results indicated that atomic masses, atomic Sanderson electronegativities, atomic van der Waals volumes and atom-centered fragments had a key role in regulating the caspase-3 inhibitory activity. | |||||