

There exist many different ion channel modulators that inhibit channel function, but few that increase it. Liin et al. tested 18,000 compounds by high-throughput electrophysiology and found several channel openers with a biaryl-sulfonamide motif that acts on the voltage-sensing machinery.
Abstract
Voltage-gated ion channels are key molecules for the generation of cellular electrical excitability. Many pharmaceutical drugs target these channels by blocking their ion-conducting pore, but in many cases, channel-opening compounds would be more beneficial. Here, to search for new channel-opening compounds, we screen 18,000 compounds with high-throughput patch-clamp technology and find several potassium-channel openers that share a distinct biaryl-sulfonamide motif. Our data suggest that the negatively charged variants of these compounds bind to the top of the voltage-sensor domain, between transmembrane segments 3 and 4, to open the channel. Although we show here that biaryl-sulfonamide compounds open a potassium channel, they have also been reported to block sodium and calcium channels. However, because they inactivate voltage-gated sodium channels by promoting activation of one voltage sensor, we suggest that, despite different effects on the channel gates, the biaryl-sulfonamide motif is a general ion-channel activator motif. Because these compounds block action potential–generating sodium and calcium channels and open an action potential–dampening potassium channel, they should have a high propensity to reduce excitability. This opens up the possibility to build new excitability-reducing pharmaceutical drugs from the biaryl-sulfonamide scaffold.
Introduction
Many types of disease caused by increased electrical excitability, such as epilepsy, cardiac arrhythmia, and pain, are sometimes caused by ion-channel mutations and can, in some cases, be treated by ion-channel modulators (Catterall, 2014; Zamponi et al., 2015). The majority of existing pharmaceutical drugs that function as ion-channel modulators block the ion-conducting pore and thus decrease the channel current (Hille, 1977; Ragsdale et al., 1994). It is mechanically simple for a compound to block a channel by plugging the pore and thereby preventing ion flux (Hille, 1977; Ragsdale et al., 1994; Zhou et al., 2001). In contrast, a few drugs and drug-like compounds increase the current (Wuttke et al., 2005; Peretz et al., 2010; Li et al., 2013; Ottosson et al., 2015; Salari et al., 2018). However, to increase a current, another mechanism must be used to keep the channel open. For instance, retigabine, an antiepileptic drug that opens the voltage-gated potassium channel KV7.2, binds near the gate that opens and closes the channel and bends the gate open (Wuttke et al., 2005). Alternatively, some compounds act on the channel’s voltage-sensing mechanism to activate the voltage sensor of the channel’s voltage-sensor domains (VSD) and thereby indirectly pull the gate open.
VSD-acting compounds can enter the VSD of KV7-type channels from the extracellular solution and bind at the center, between the four transmembrane segments, S1–S4, to keep the voltage sensor S4 in an activated, up, state (Peretz et al., 2010; Li et al., 2013). Alternatively, hydrophobic and negatively charged polyunsaturated fatty acids (PUFAs) or resin acids act from the lipid-facing part of the VSD of KV1- or KV7-type channels to electrostatically attract the positively charged voltage sensor S4 to activate and open the channel (Börjesson et al., 2008, 2010; Börjesson and Elinder, 2011; Ottosson et al., 2014, 2015, 2017; Liin et al., 2015, 2016a; Yazdi et al., 2016; Elinder and Liin, 2017; Salari et al., 2018; Silverå Ejneby et al., 2018). To specifically control excitability in different cell types, new types of ion-channel modulators are needed; new modulators based on new scaffolds can potentially be developed into either openers or closers and can potentially be designed to act on specific channels.
To search for new ion-channel modulator scaffolds, we performed an electrophysiological screen of almost 18,000 small-molecule compounds from a diverse range of compound libraries. We found that sulfonamides, a sulfonyl group connected to a nitrogen (Fig. 1 A), formed a family of KV-channel–opening compounds. The minimal requirement for the active compounds was a biaryl-sulfonamide motif (e.g., Fig. 1 B, which we will refer to as the short biaryl-sulfonamide motif). Our data suggest that these molecules bind to the VSD of the voltage-gated Shaker KV channel to keep the voltage sensor S4 in an activated, up, state, and the pore gate open.
Figure 1.

High-throughput screen of ion-channel modulators. (A) General sulfonamide motif. (B) Example of a biaryl sulfonamide motif. (C) The voltage protocol used in in the high-throughput screen together with a representative effect of an ion-channel opening compound on the current. (D) Summary of data from almost 18,000 compounds tested on the 3R Shaker KV channel expressed in CHO cells. Ion currents were recorded by IonWorks (see Materials and methods) and measured at the end of a 150-ms-long voltage-clamp step to +10 mV. Holding voltage, −80 mV. The data points are aligned in potency order. The gray area indicates no effect, the dashed and dotted lines define clear openers (243 compounds, 1.3% of all tested compounds) or clear blockers (140 compounds, 0.8% of all tested compounds; see text for a detailed description).
Materials and methods
Shaker KV channels
The Shaker H4 channel (accession no. NM_167595.3 in the Bluescript II KS[+] plasmid; Kamb et al., 1987), with removed N-type inactivation (ShH4IR; Hoshi et al., 1990), referred to as the WT Shaker KV channel, was used throughout this work. In addition, a modified, so-called 3R Shaker KV channel was used, in which two introduced positively charged arginines (M356R and A359R), in addition to one native arginine (R362), made the channel more sensitive to PUFAs and resin acids (Ottosson et al., 2014). The 3R Shaker KV channel was also expressed in a CHO-K1 stable cell line (Ottosson et al., 2015). To measure gating currents, we used the W434F mutation in the WT Shaker KV channel background (Perozo et al., 1993). Point mutations in the Shaker KV channel were introduced using the QuikChange Site-Directed Mutagenesis kit (Stratagene) and verified by sequencing, as previously described (Börjesson and Elinder, 2011).
Chemical libraries
Compounds were selected from several chemical libraries: Acids (Vitas-M Laboratory); Consortium set (Specs)*; Drug Like (Enamine)*; Hit Finder (Maybridge)*; Ion Channel Ligand and Nuclear Receptor Ligand (Enzo Life Sciences); Known Drugs (Prestwick Chemical); LCBKI Primary Screening Set (Chemical Biology Consortium Sweden)*; Natural derivatives (TimTec); NIH Clinical Collection (NIH); Peptidomimetics (ChemDiv Inc.)*; and Tocriscreen Plus (Tocris Bioscience). Apart from LCBKI, provided by the Chemical Biology Consortium Sweden, and the NIH clinical collection, provided by the Libraries Roadmap Initiative, all compound libraries were commercially available. Whenever a selection was made (*), a chemist was instructed to search for small, hydrophobic, and partially charged compounds.
Cell culture and preparation for high-throughput screen
CHO-K1 cells were grown to semiconfluence (maximum 75%) in T-150 Costar flasks (Corning) at 37°C (final 12–15 h before experiment at 30°C) in a humidified environment (5% CO2), in DMEM/F12+ Glutamax, supplemented with 10% FBS, G418 sulfate, and 1% MEM nonessential amino acids. Just before use, the cells were washed twice with prewarmed Mg- and Ca-free PBS (Invitrogen) and detached with 3 ml TrypLE (Gibco) for 4 min at 30°C. Cells were then detached from the bottom of the flask by gentle tapping, and 7 ml (Mg- and Ca-containing) Dulbecco’s PBS (D-PBS) was added and aspirated into a centrifuge tube before centrifugation (50 g for 3 min). The resulting supernatant was discarded, and the cell pellet was gently resuspended in 5 ml D-PBS. A 0.1-ml aliquot of cell suspension was removed, and the cell concentration was determined using a Burker chamber. After the cells had been counted, the cell suspension was adjusted with PBS to reach a cell concentration of 1.3 million/ml and finally added to the IonWorks cell boat.
Compound handling for high-throughput screening
Assay-ready plates were prepared at the High Throughput Center at Karolinska Institutet. All compounds were dissolved in 100% DMSO to a final stock concentration of 10 mM. 100 nl of the 10-mM DMSO solutions of compounds and controls were transferred to low-volume 384-well polypropylene Greiner plates by acoustic dispensing (Echo 550; Labcyte). For single-concentration measurements, compounds were placed in columns 3–22, and DMSO controls and a known modulator were placed in columns 1, 2, 23, and 24. The assay-ready plates were heat-sealed using a thermal microplate sealer (PlateLoc; Agilent) and stored overnight at 4°C. On the day of the experiment, the compound plates were diluted by adding 30 µl PBS to each well and mixed using a Biomek FxP Lab Automation workstation (Beckman Coulter), giving a compound plate concentration of 30.3 µM (0.33% DMSO; three times their final test concentration). Before the experiment, all plates were centrifuged (1,000 g for 2 min) to remove air bubbles. In one library, two compounds were tested per well, and if the double compound effect was significant, the two compounds were retested separately.
Automated planar patch-clamp electrophysiology for high-throughput screening
Recordings were performed in CHO-K1 cells using the PPC mode of the IonWorks Quattro automated patch-clamp system (Molecular Devices). The operation protocol for this system has been previously described (Schroeder et al., 2003; Bridgland-Taylor et al., 2006; Harmer et al., 2008). The intracellular solution contained (in mM) 100 potassium-glutamate, 40 KCl, 3.2 MgCl2, 3 EGTA, and 5 HEPES, pH 7.25–7.3 with KOH, and the extracellular solution was D-PBS (Gibco) and contained (in mM) 138 NaCl, 2.7 KCl, 1.5 KH2PO4, 8 Na2HPO4, 0.9 CaCl2, 0.5 MgCl2, and 5.5 glucose. The tested compounds were added to the extracellular solution. To establish the whole-cell perforated patch configuration, 25 mg/ml amphotericin B was added to the intracellular solution, resulting in a final concentration of 150 µg/ml. After attaining the whole-cell configuration, the voltage protocol was applied once to obtain a control recording (prescan) and again after compound application (post-scan) to measure the potentiation of potassium currents. During the compound incubation (∼2 min), that is, between the pre- and post-scans, there was no clamping of the membrane potential. Before the test voltage protocol, the cells were held at ‒80 mV for 20 s. Currents were evoked by a three-pulse voltage train (Fig. 1 C): pulse 1, a 150-ms square-shaped step to +10 mV; pulse 2, a 150-ms square-shaped step to +30 mV; and pulse 3, a 200-ms linear ramp from −80 mV to +50 mV directly followed by a 100-ms step to +50 mV. The cells were kept at ‒80 mV for 300 ms between each step. Wells with a seal resistance of <20 MΩ or with a current <0.05 nA were not included in the analysis. The current signal was sampled at 10 kHz. All recordings were made at room temperature (∼21°C). To minimize compound well carryover, we included a wash step in the IonWorks protocol with ethanol and PBS in between the addition of compounds. Moreover, in between the experiments, we also ran an IonWorks cleaning protocol with ethanol and PBS.
Preparation and handling of Xenopus laevis oocytes
All animal experiments were approved by the local Animal Care and Use Committee and followed international guidelines. Generally, 50 nl WT or mutated Shaker KV channel cRNA (50 pg) were injected into each oocyte using a Nanoject injector (Drummond Scientific). X. laevis surgery, oocyte isolation, and oocyte storage after RNA injection followed our previously described protocol (Börjesson et al., 2010). Injected oocytes were kept at 8°C for 1–6 d before electrophysiological recordings were performed. All chemicals were supplied by Sigma-Aldrich if not stated otherwise.
Manual electrophysiology
All manual electrophysiological recordings were performed 1–6 d after cRNA injection by the two-electrode voltage clamp technique performed at room temperature (20–23°C), using a CA-1B amplifier (Dagan). Signals were lowpass-filtered at 5 kHz and digitized by a Digidata 1440A converter (Molecular Devices). The amplifier’s leak compensation was used, and Clampex 10.5 software (Molecular Devices) was used to obtain data and create voltage protocols. The extracellular control solution (1K) contained (in mM) 88 NaCl, 1 KCl, 15 HEPES, 0.4 CaCl2, and 0.8 MgCl2, pH adjusted to 7.4 by NaOH (reaching a sodium concentration of ∼100 mM). Compounds were dissolved to 100 mM in 99.5% EtOH and stored at ‒20°C. Compounds were diluted in the extracellular control solution to the desired test concentration immediately before experiments. To measure closing kinetics, we used a high potassium–containing solution (100K) in which NaCl in the 1K control solution was replaced with KCl and pH set with KOH, yielding a potassium concentration of ∼100 mM.
The extracellular control solution was added to the bath with a gravity-driven perfusion system. Sulfonamides were added to the bath using a Mini-Peristaltic Pump II (Harvard Apparatus). Glass microelectrodes were pulled from borosilicate glass, filled with 3 M KCl, and had a resistance of 0.5–2 MΩ. The holding voltage was set to ‒80 mV. Currents were evoked by 100-ms-long square-like steps ranging from −80 up to +50 mV (WT) and +70 mV (3R) in steps of 5 mV. The pulses occurred at a frequency of 0.2 Hz. To study gating currents, the linear membrane capacitance was compensated for by the use of the amplifier’s capacitance compensation for a 10-ms voltage pulse between 0 and ‒10 mV.
Analysis of electrophysiological data
The manual electrophysiological data were processed with Clampfit 10.4 (Molecular Devices) and GraphPad Prism 5 (GraphPad Software).
Analysis of automated planar patch clamp electrophysiology
Modulation of the KV-mediated current was assessed by dividing the post-scan steady-state KV current (for the last 10 ms of each pulse) by the respective prescan KV current for each well. For the quantitative analysis presented in this paper, we used the data from +10 mV. In control solution (at +10 mV) during the experiment, there was a consistent rundown of the relative current by −20 ± 7% (mean ± SD). Therefore, all experimental data (post-scan/prescan) between 0.6 and 1.0 (mean ± 3 SD) were regarded as without effect.
Analysis of X. laevis oocyte recordings
Conductance G(V) was calculated as
| (1) |
where IK is the mean current from the steady-state phase at the end of each 100-ms pulse, V is the membrane voltage, and Vrev is the reversal potential for K+ (set to ‒80 mV). To analyze gating currents, a leakage current correction was performed offline. This was achieved by using Clampfit software to eliminate the steady-state leakage current at the end of the ON pulse or the OFF pulse, after the current had reached steady state. The gating charge was calculated as the area under the curve from 1.5 ms after onset of the OFF pulse (to exclude the remaining capacitive current) to 100 ms after onset of the OFF pulse. These data were fitted to a Boltzmann-inspired equation:
| (2) |
where A is the amplitude of the curve, V is the absolute membrane voltage, V1/2 is the midpoint, s is the slope (Börjesson et al., 2008), and n is an exponent. For most of the ion-current analyses, n was set to 4 to allow a good fit to the steep foot of the G(V) curve. For the gating current analyses, n was set to 1. The compound-induced G(V) shift (ΔVG(V)) was determined at the 10% level of the maximum conductance in control solution (Börjesson et al., 2008).
Data for concentration- and pH-dependent G(V) shifts (ΔVG(V)) in oocytes were fitted to a concentration–response equation:
| (3) |
where ΔVG(V),max is the maximal shift, c1/2 is half-maximal effective concentration (or pKa value in the pH experiments), and c is the concentration.
Calculation of chemical properties
Marvin was used to draw chemical structures (Marvin 16.12.9, 2016, ChemAxon). The logarithm of the acid dissociation constants, pKa, for the ionic forms of the compounds were calculated using the Marvin Calculation plugin. The pH range for calculating microspecies distribution (%) was set to 0–14 and temperature to 298°K, and the pKa was obtained from the global mass and charge conservation law (Macro mode). The ionic strength was considered to be 0.1 M. The octanol-water partitioning coefficient, log P, for uncharged compounds was calculated using the Marvin Calculation plugin. The consensus model in ChemAxon was used for calculations, and electrolyte concentrations were set to 0.1 M for Cl−, Na+, and K+.
Statistical analysis
Mean values are expressed as mean ± SEM. One-way ANOVAs, followed by Dunnett’s multiple comparison test, were used to compare the effects of sulfonamides on each mutant to the corresponding effects on the WT Shaker KV channel. Two-tailed t tests were used to compare the effects of sulfonamides on the WT Shaker KV channel to a hypothetical value. P < 0.05 was considered significant for all tests.
Molecular docking
The RosettaLigand docking (Lemmon and Meiler, 2012) software (v2016.16) was used to dock the Cmpd 5 and Cmpd 6 ligands to the Shaker KV channel VSD. A homology model for the structure of the Shaker KV channel was constructed using HHpred (Hildebrand et al., 2009) and Modeller 9v13 (Sali and Blundell, 1993) with the KV1.2–KV2.1 paddle chimera channel (PDB ID: 2R9R) as template. Alignment for the homology modeling is provided as a supplemental file. Ligand conformers were constructed using OMEGA from OpenEye Scientific (Hawkins et al., 2010), with default parameters resulting in 201 and 30 conformers for Cmpd 5 and Cmpd 6, respectively. The ligand was placed in an approximate binding pocket suggested by a recent study on NaV1.7 channels (Ahuja et al., 2015); before each docking, the ligand was randomly rotated and translated between 0 and 2 Å uniformly. The docking protocol samples all the different conformers of the ligand, allowing the side-chains and backbone of the protein receptor close to the ligand (<6 Å) to adapt to the ligand. In total, 49,277 and 42,004 docking poses were generated for Cmpd 5 and Cmpd 6, respectively. In addition, 40,000 docking poses were generated for Cmpd 6 in the deprotonated state. The 5% lowest-energy docking poses were ranked by the interface delta energy, defined as the total energy of the complex with ligand bound minus the total energy of the complex and ligand alone.
Online supplemental material
Files, scripts, and instructions on how to reproduce the docking are provided in the datasets, as well as a pymol session of the best scoring docking poses.
Results
A high-throughput screen suggests biaryl sulfonamides as K-channel openers
To search for compounds modulating a voltage-gated K channel, we screened almost 18,000 compounds on a specially designed Shaker KV channel expressed in Chinese hamster ovary (CHO) cells (Ottosson et al., 2015). This channel was developed to be sensitive to negatively charged hydrophobic compounds that electrostatically activate the voltage sensor and thereby open the ion channel. Two extra (positively charged) arginines were introduced at the top of the voltage sensor S4 (M356R/A359R, called the 3R channel because there are three critical arginines in positions 356, 359, and 362; Ottosson et al. [2014]). The rationale for using this channel was to identify compounds for further investigation and refinement. However, this mutant does not exclude the detection of compounds acting on other sites via other mechanisms. The compounds were selected from several compound libraries (see Materials and methods), and the selection criteria were partly hydrophobic compounds with a negative charge. The compounds’ effects (at a concentration of 10 µM) were explored using a high-throughput electrophysiological patch-clamp technique (IonWorks). As a readout, we used the steady-state current in compound solution relative control solution, measured during the last 10 ms of a 150-ms voltage step to +10 mV (from a holding voltage of −80 mV; pulse 1 in Fig. 1 C).
The current in the compound solution relative to the current in the control solution for all tested compounds is shown in Fig. 1 D. Note that the number of experiments is slightly lower than 18,000 because some compounds were tested two at a time; if the effect was significant, both compounds were rerun individually. Because of the experimental rundown during the experiment (see Materials and methods), all data between 0.6 and 1.0 was regarded as without effect (shaded area in Fig. 1 D). Almost 20% of the compounds caused a significant current reduction (<0.6), and 6.5% of all data showed a significant current increase (>1.0), but it should be noted that the actual fraction is slightly lower because this data also include reruns and confirmatory runs. To reduce the number of false hits, we considered only compounds with a relative current of 1.2 as clear K-channel openers (dashed line in Fig. 1 D) and 0.4 as clear K-channel blockers (dotted line in Fig. 1 D). In the screen, 243 compounds (1.3% of all tested compounds) were classified as clear K-channel openers, and 140 as clear blockers (0.8% of all tested compounds). 26 of the clear openers (11% of the hits; Table 1) were sulfonamides characterized by the motif in Fig. 1 A, whereas none of the clear blockers had this motif.
Table 1. Sulfonamides identified in the high-throughput screen.
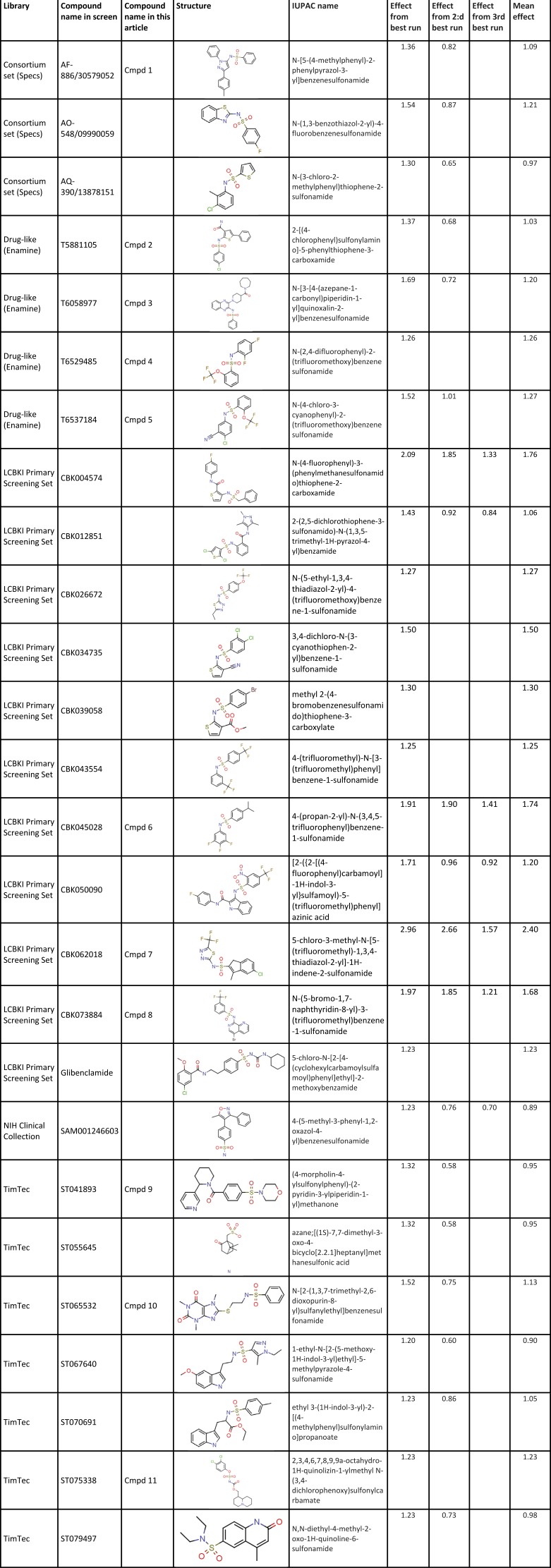 |
The sulfonamide family was selected for closer analysis. Most of the sulfonamide compounds (19 of 26) were tested in repeated screens, and seven of them in triplicate. Libraries, names, structures, and data from the screen of identified sulfonamides are listed in Table 1. Most of the compounds have one ring structure at each end of the sulfonamide motif. One compound has one atom between the two rings (the nitrogen trapped in the ring), 16 have two atoms (O and N), one has three atoms, three have four atoms, one has five atoms, one has six atoms, and three have a free end with just one ring connected to the sulfonamide motif. Thus, 16 of the 26 compounds have a characteristic short biaryl-sulfonamide motif with just the two-atom sulfonamide motif between the two rings (Fig. 1 B). Interestingly, of the ten best compounds (mean channel-opening effect of >1.25; Table 1), nine had a biaryl motif and one had just an additional atom in between. In the less potent group (mean channel-opening effect <1.25), 7 of 16 compounds had a short biaryl-sulfonamide motif, whereas 9 had other types of sulfonamide structures. Thus, the short biaryl-sulfonamide motif could be the key to the channel-opening effect. However, it should be noted that there was variability between the different runs, and some of the compounds were run only once.
A short biaryl-sulfonamide motif affects the voltage dependence of the WT Shaker KV channel
In the screen described above, compounds were tested on the 3R Shaker KV channel in an attempt to find compounds electrostatically acting on the outer end of S4, as has been described for PUFAs and resin acids (Börjesson and Elinder, 2011; Ottosson et al., 2014, 2015, 2017; Liin et al., 2015; Yazdi et al., 2016; Silverå Ejneby et al., 2018). In the next step, we explored 11 selected sulfonamide compounds on the WT Shaker KV channel expressed in X. laevis oocytes at 10 and 100 µM at pH 7.4. The compound selection was designed to choose diverse types of compounds, with respect to both molecular structures and effects in the screen.
Cmpd 5 at 100 µM clearly increased the WT Shaker KV channel current at −30 mV (Fig. 2 A). The effect reached saturation within 2 min. After washout, the effect recovered to control level within ∼5 min. This time course of the effect was typical for the compounds, and we made sure to reach saturation in all experiments. At −35 mV, 100 µM of Cmpd 5 increased the current fivefold (Fig. 2 B) by shifting the conductance-versus-voltage curve, G(V), by −10.8 ± 0.6 mV (n = 4), with only minor effects on the maximum conductance (Fig. 2 C). Other typical examples of the effect of compounds on the G(V) curve are shown in Fig. 2 (D–F). Of the 11 tested compounds, 5 displayed clear effects of approximately equal magnitude (blue symbols in Fig. 2 G). One compound had small but significant effects at 100 µM (black symbols in Fig. 2 G). Five of the compounds displayed no significant effects on the WT Shaker KV channel (red symbols in Fig. 2 G). The reason for this lack of effect, despite effects in the primary screen, could be (a) the differing expression system (X. laevis oocytes vs. CHO cells), (b) differing channels (WT vs. 3R channel), or (c) false-positive effects in the initial screen. However, there is a clear pattern on the WT Shaker KV channel that resembles the pattern from the initial screen on the 3R Shaker KV channel; all effective compounds share the short biaryl-sulfonamide motif (Fig. 1 B), whereas all compounds lacking this motif (Cmpds 9–11) had no effect. This suggests that the WT Shaker KV channel has a distinct binding site for the short biaryl-sulfonamide motif. To explore the mechanism of effective sulfonamides further, we selected two of the active compounds for a more detailed characterization. Cmpd 5 (Fig. 2 H) had the largest effect at 100 µM, and Cmpd 6 (Fig. 2 H) had the largest effect at 10 µM.
Figure 2.

Short biaryl sulfonamides activate the WT Shaker KV channel expressed in X. laevis oocytes. (A) Representative plot of the steady-state current amplitude at −30 mV during wash-in and wash-out of a sulfonamide compound (100 µM of Cmpd 5). Holding voltage, −80 mV; pulse length, 100 ms; pulse frequency, 0.1 Hz. (B) Representative current traces in control solution (black) and after application of 100 µM Cmpd 5 (red). Holding voltage, −80 mV; test-step voltage, −35 mV. (C) G(V) curve for the cell shown in B. The arrow denotes the shift in voltage dependence (ΔVG(V) = −10.8 mV) measured at 10% of the maximum conductance in control (dotted line; see Materials and methods for a detailed description). The continuous curves are best fitted to Eq. 2; the exponent n was fixed to 4; the slope s was constrained to be equal for the two sets of data; and V1/2 was shifted −11.7 mV. (D–F) Representative effects on the G(V) curve for Cmpds 4, 6, and 7. The shifts are −0.8, −13.2, and −1.2 mV, respectively, measured by Eq. 2. (G) Summary of the dose-dependent effect of 11 selected sulfonamides on ΔVG(V) of the WT Shaker KV channel. n = 3–6. Mean ± SEM. The compounds are color coded depending on their effect on ΔVG(V): red for no significant effect, black for significant but small effect, and blue for significant prominent effect. The effect of 100 µM Cmpd 2 could not be determined, as application of the compound resulted in massive cell leakage. A two-tailed t test was used to compare effects to a hypothetical value of 0. *, P < 0.05; **, P < 0.01. The remaining data points had P > 0.05. (H) Molecular structures of the two compounds selected for further investigation.
To further test the hypothesis that a short biaryl-sulfonamide motif is required to open the Shaker KV channel, we tested the antidiabetic drug glibenclamide (also called glyburide), which closes the ATP-sensitive K channel, depolarizes pancreatic β cells, and increases the release of insulin (Ashcroft and Ashcroft, 1990). This drug was in fact one of the 26 compounds identified in the original screen (Table 1), but because of its elongated biaryl-sulfonamide motif (Fig. 3 A), we did not predict it to open the WT Shaker KV channel. Consistent with this hypothesis, 30 µM of the drug had no effect on the Shaker KV channel (ΔVG(V) = −0.3 ± 0.2 mV, n = 3; Fig. 3, A and B). To test if it is the two aromatic rings in combination with the amine group and not the sulfonic group that opens the channel, we tested another ion-channel modulating drug in clinical use, the antiepileptic drug retigabine, which opens the voltage-gated KV7.2/7.3 M-channel and dampens excitability (Main et al., 2000; Rundfeldt and Netzer, 2000; Wickenden et al., 2000; Wuttke et al., 2005). Retigabine, at 30 µM, had no effect on the WT Shaker KV channel (ΔVG(V) = 0.0 ± 0.0 mV, n = 3; Fig. 3, C and D).
Figure 3.

An elongated biaryl sulfonamide or a biaryl amine lack effect on the WT Shaker KV channel. (A) Representative current traces of the WT Shaker KV channel in control solution and after application of 30 µM glibenclamide. Holding voltage, −80 mV; test-step voltage, −35 mV. (B) G(V) curve for the cell shown in A. (C) Representative current traces of the WT Shaker KV channel in control solution and after application of 30 µM retigabine. Holding voltage, −80 mV; test-step voltage, −35 mV. (D) G(V) curve for the cell shown in C.
Selective effect on the closing kinetics compared with opening kinetics
Cmpd 5 had no effect on the channel-opening kinetics at +60 mV (Fig. 4 A); the opening time constant in compound solution relative control solution was 0.95 ± 0.02 (n = 5; ns). In contrast, Cmpd 5 clearly slowed down the channel-closing kinetics at −60 mV (Fig. 4 B) by a factor of 4.0 ± 0.7 (n = 4; P < 0.05). The selective effect on channel closing by Cmpd 5 is shown in Fig. 4 C. Cmpd 6 had a very similar profile to Cmpd 5. Cmpd 6 had no effect on the channel-opening kinetics at +60 mV (Fig. 4 D); the opening time constant in compound solution relative control solution was 1.17 ± 0.11 (n = 5; ns). In contrast, Cmpd 6 clearly slowed down the channel-closing kinetics at −60 mV (Fig. 4 E) by a factor of 6.2 ± 1.5 (n = 4; P < 0.05). The selective effect on channel closing by Cmpd 6 is shown in Fig. 4 F.
Figure 4.

Effects on the opening and closing kinetics of the WT Shaker KV channel. (A) Effect of 100 µM Cmpd 5 on the opening at +60 mV. Holding voltage, −80 mV; extracellular solution, 100K; black curve, control solution; red curve, compound solution. Single exponential curves (dashed lines) were fitted from 1.5 to 30 ms. τ(control) = 1.01 ms, τ(Cmpd 5) = 0.88 ms. (B) Effect of 100 µM Cmpd 5 on the closing at −60 mV. Prepulse voltage = +60 mV for 100 ms. Extracellular solution, 100K; black curve,control solution; red curve, compound solution. Tail currents were normalized to the current at the end of the activation pulse. Single exponential curves (dashed lines) were fitted to the tail currents after removal of capacitive spikes. τ(control) = 1.88 ms, τ(Cmpd 5) = 10.8 ms. (C) Effect of 100 µM Cmpd 5 on time constants at different voltages. Time constants for channel opening (as in A but 1K solution, open symbols). Time constants for channel closing (as in B, closed symbols). Black symbols, control solution; red symbols, compound solution. (D) Same as in A but Cmpd 6. τ(control) = 0.89 ms, τ(Cmpd 6) = 1.20 ms. (E) Same as in B but Cmpd 6. τ(control) = 1.22 ms, τ(Cmpd 6) = 10.6 ms. (F) Same as in C but Cmpd 6.
To test if the compound also affected the voltage sensor movement (directly or indirectly), we measured gating currents. The W434F Shaker KV channel mutant does not conduct ions, so isolated gating currents can easily be measured (Perozo et al., 1993). The ON-gating currents, generated when the oocyte is pulsed from a negative holding voltage of −80 mV to more positive voltages, showed a characteristic amplitude and voltage dependence. Cmpd 5 had no effect on the ON-gating currents (Fig. 5 A). The small effects during the first milliseconds are because of alterations in the capacitance compensations during the experiment. In contrast, Cmpd 5 clearly slowed down the OFF-gating currents at −80 mV (from different prepulse voltages; Fig. 5 B) by a factor of 4.8 ± 0.1 (n = 3; P = 0.013). The steady-state gating-charge movement at different voltages, Q(V), was measured by integrating the OFF-gating current (as shown in Fig. 5 B) and plotted versus the prepulse voltage (Fig. 5 C). Cmpd 5 shifted the Q(V) curve by −7.6 mV ± 1.1 mV (n = 3, P = 0.022) but did not affect the maximum gating charge (−3 ± 2%, n = 3, ns). Cmpd 6 had similar effects to Cmpd 5. The ON-gating currents were not affected (Fig. 5 D). The OFF-gating currents were clearly slowed down (Fig. 5 E; by a factor of 2.7 ± 0.2 at −80 mV; n = 7; P < 0.0001). The Q(V) curve (Fig. 5 F) was shifted by −3.4 mV ± 0.4 mV (n = 7, P = 0.0003), but the maximum gating charge was not affected (+1 ± 4%, n = 4, ns).
Figure 5.

Effects on the gating currents of the W434F Shaker KV channel. (A) Effect of 100 µM Cmpd 5 on the ON-gating currents at denoted voltages. Holding voltage, −80 mV; extracellular solution, 1K; black curves, control solution; red curves, compound solution. (B) Effect of 100 µM Cmpd 5 on the OFF-gating currents at −80 mV, after 500-ms prepulses between −100 and +10 mV in steps of 5 mV. Extracellular solution, 1K; black curves, control solution; red curves, compound solution. (C) Effect of 100 µM Cmpd 5 on gating charge versus voltage, Q(V), curve. Extracellular solution, 1K. The gating charges were calculated as the area under the curve in B from 1.5 to 100 ms. Black symbols and curves, control solution; red symbols and curves, compound solution. Data were fitted to Eq. 2 (with exponent n = 1). ΔV1/2 = −8.3 mV. (D) Same as in A but Cmpd 6. (E) Same as in B but Cmpd 6. (F) Same as in C but Cmpd 6. ΔV1/2 = −3.8 mV. (G–I) Effects of 100 µM Cmpd 5 and 6 on the activation time constant (G), the deactivation time constant (H), and the steady-state curves (I). Channel activation is measured as the opening in Fig. 4 (A and D) and as the single exponential relaxation phase (as recorded in Fig. 5 A at 0 mV). Channel closing is measured as the closing in Fig. 4 (B and E) and as the single exponential relaxation phase at −80 mV after a prepulse to 0 mV as recorded in Fig. 5 (B and E). Shifts of G(V) (from Fig. 2 C), and Q(V) as measured in Fig. 5 (C and F). Mean ± SEM. Number of experiments indicated in the figure. A two-tailed t test was used to compare effects to a hypothetical value of 1 in G and H and 0 in I. ns, P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
In summary, neither Cmpd 5 nor Cmpd 6 had any effects on the ion-channel opening kinetics or the ON-gating charge movements (Fig. 5 G). Both Cmpds 5 and 6 clearly slowed down channel closure and return of gating charges (Fig. 5 H). Both Cmpds 5 and 6 clearly shifted G(V) and Q(V) in a negative direction along the voltage axis. Altogether, this suggests that the short biaryl-sulfonamide compounds trap the ion channel in an open state with the voltage sensor in an activated state.
The negative charge of the compounds or a deprotonated binding site is probably required for the effect
Cmpd 5 dose-dependently increased the current of the WT Shaker KV channel by shifting the G(V) curve in a negative direction along the voltage axis (Fig. 6 A). As shown by Eq. 3 in Materials and methods, the maximum shift (ΔVG(V),max) was −14.8 mV, and the concentration causing 50% shift (c1/2) was 35 µM. The nitrogen of the compound is protonable, and the molecule is expected to be uncharged at low pH and negatively charged at high pH. The calculated pKa value is 6.85, and an alteration in extracellular pH is expected to alter the charge of the compound around neutral pH if the compound experiences a watery surrounding. To test whether the charge is important for the G(V) shift, we therefore studied the effect of different pHs. Cmpd 5 had no effect at pH 5.0 or 6. At pH 7.4 the effect peaked, but surprisingly, it decreased at higher pH (Fig. 6 B). We do not know the reason for this decreased effect at high pH, but the data are consistent with the idea that the negative charge of the compound is important for the effect. Like Cmpd 5, Cmpd 6 dose-dependently increased the current of the WT Shaker KV channel by shifting the G(V) curve in a negative direction along the voltage axis (Fig. 6 C). At a concentration of 30 µM, the compound showed a saturated voltage shift. The maximum shift (ΔVG(V),max) was −10.0 mV, and the concentration causing 50% shift (c1/2) was 9.4 µM. The calculated pKa value was 7.69, and the experimentally obtained pKa value was 6.7 (Fig. 6 D; Eq. 3), which suggests that the molecule (or the binding site) is almost fully deprotonated at a neutral pH value. To summarize, short biaryl sulfonamides bound to the channel caused G(V) shifts up to ‒15 mV, 10–30 µM of the compounds caused 50% of the maximum shift, and the negatively charged form of the compound (or a deprotonated binding site) is possibly required for these effects.
Figure 6.

Characterization of the effects of Cmpd 5 and Cmpd 6 on the WT Shaker KV channel. (A) Concentration–response curve for Cmpd 5–induced shift of the G(V). Mean ± SEM (n = 2–4). Best fit of Eq. 3: ΔVG(V),max = −14.8 ± 1.5 mV; c1/2 = 35 ± 9 µM. (B) pH dependence of Cmpd 5–induced ΔVG(V) (30 µM). Mean ± SEM (n = 3). Data points connected with line. (C) Concentration–response curve for Cmpd 6–induced shift of the G(V). Mean ± SEM (n = 3–6). Best fit of Eq. 3: ΔVG(V),max = −10.0 ± 0.9 mV; c1/2 = 9.4 ± 3.2 µM. (D) pH dependence of Cmpd 6–induced ΔVG(V) (30 µM). Mean ± SEM (n = 3–5). Best fit of Eq. 3: ΔVG(V),max = −8.5 ± 0.9 mV, apparent pKa = 6.7.
The biaryl sulfonamides are suggested to bind to the top of the VSD
Where is the site of action for the short biaryl-sulfonamide compounds? Negatively charged PUFAs and resin acids bind in the lipid bilayer, close to the gating charges of the VSD, and, from this position, they electrostatically attract the gating charges to pull the channel open (Börjesson et al., 2008; Börjesson and Elinder, 2011; Ottosson et al., 2014, 2015, 2017; Liin et al., 2015, 2016b; Yazdi et al., 2016; Silverå Ejneby et al., 2018). Because a negative charge is also probably required for the short biaryl sulfonamides to open the channel, we hypothesized that they share a binding site and act via the same mechanism as PUFAs and resin acids. A characteristic feature for compounds acting via this site and mechanism is that the introduction of two positive charges at the extracellular end of the voltage sensor S4 (facing the lipid bilayer, i.e., the 3R, or M356R/A359R, channel) increases the drug-induced G(V) shift by a factor of ∼3 (Ottosson et al., 2014, 2015). However, the 3R mutation (M356R/A359R/R362R; Table 2) did not alter the shift for either Cmpd 5 or Cmpd 6 (Fig. 7, A–C), thus refuting this hypothesis.
Table 2. Summary of biophysical properties of Shaker KV channel mutants.
| Construct | V½ (mV) | s (mV) | n |
|---|---|---|---|
| WT | ‒23.3 ± 1.1 | 6.6 ± 0.2 | 15 |
| M356R/A359R/R362R (=3R) | +24.2 ± 0.9 | 9.3 ± 0.5 | 7 |
| Y323C | ‒5.7 ± 1.8 | 9.6 ± 1.1 | 8 |
| F324C | ‒9.5 ± 1.4 | 13.4 ± 0.6 | 10 |
| T326C | ‒10.0 ± 2.4 | 13.1 ± 0.9 | 9 |
| Y323C/F324C/T326C | +17.9 ± 2.5 | 13.1 ± 0.4 | 7 |
| R365C | +19.2 ± 1.0 | 8.9 ± 0.6 | 7 |
V½ and s were determined using Eq. 2 in Materials and methods raised to the power of 1. Data shown as mean ± SEM.
Figure 7.

Mutational effect on Cmpd 6– and Cmpd 5–induced G(V) shifts. (A, B, D, and E) Representative effect of 30 µM Cmpd 5 on the G(V) curve of four different WT or mutated Shaker KV channels. Data fitted to Eq. 2 with the exponent n = 4. ΔVG(V) = −8.6 mV (A), −9.4 mV (B), −3.0 mV (D), and −0.4 mV (E). (C) Summary of data from all mutations. n = 4–6; mean ± SEM. One-way ANOVA followed by Dunnett’s multiple comparison test to compare the effect of Cmpd 5 or Cmpd 6 on each mutant to the corresponding effect on the WT Shaker KV channel. *, P < 0.05; ***, P < 0.001; ns, P > 0.05. Dashed lines denote WT data. (F and G) Dose–response curves for Cmpds 5 and 6 on WT (closed symbols, data from Fig. 6) and the triple mutant Y323C/F324C/T326C (open symbols; mean ± SEM, n = 3–5). Best fit of Eq. 3 with ΔVG(V),max constrained to be equal for WT and the triple mutant. (F) ΔVG(V),max = −14.3 ± 2.3 mV; c1/2(WT) = 32 ± 14 µM; c1/2(Y323C/F324C/T326C) = 276 ± 96 µM. (G) ΔVG(V),max = −10.0 ± 1.1 mV; c1/2(WT) = 9.4 ± 1.8 µM; c1/2(Y323C/F324C/T326C) = 141 ± 52 µM.
Instead, we hypothesized that the compounds bind to the top of the VSD in the pocket formed at the center between the transmembrane segments S1, S2, S3, and S4 of the VSD. This site accommodates other types of ion-channel modulators (Peretz et al., 2010; Li et al., 2013; Ahuja et al., 2015). To test whether the biaryl sulfonamides could bind to this site, we performed in silico docking of Cmpd 5 and 6. Both molecules bound with favorable binding energies (Fig. 8, B and D), with an energy gap similar to that found for known binders of similar size (Meiler and Baker, 2006). Cmpd 5 had a well-defined binding pose, with all of the top 10 binding poses more or less identical, differing only slightly in their binding energies (Fig. 8 B). In this binding pose, Cmpd 5 is interacting with residues Y323, F324, and T326 at the extracellular end of S3 and the gating charges R365, R368, and R371 of S4 (Fig. 8 A). The Cmpd 6 binding pose formed two distinct clusters, one with the difluorophenyl ring pointing outward and interacting with F324 or P273 (Cluster 1), and one with the difluorophenyl ring pointing inward with the fluorines interacting with the gating charges R365 and R368 (Cluster 2); in both clusters, the benzene group was interacting with Y323 (Fig. 8 C). Because Cmpd 6 is more active at high pH and its nitrogen is most likely deprotonated at neutral pH (Fig. 6 D), dockings with a deprotonated Cmpd 6 were also performed. These dockings generated similar binding poses as with the neutral nitrogen, protonated state, but with slightly more poses in cluster 1 compared with cluster 2 among the top 10. The deprotonated Cmpd 6 also exhibits more favorable binding energies compared with the protonated Cmpd 6 (Fig. 8 D), in agreement with the higher activity of Cmpd 6 at higher pH (Fig. 6 D).
Figure 8.

Molecular docking of Cmpd 5 and Cmpd 6. (A) Top 10 ligand poses for Cmpd 5. Note that they are virtually identical and superimpose on top of each other. (B) Rank versus binding energy measured in Rosetta Energy Units for Cmpd 5. (C) Top 10 ligand poses for Cmpd 6 and two representative clusters. (D) Rank versus binding energy measured in Rosetta Energy Units for Cmpd 6 and with a deprotonated nitrogen (Cmpd 6-H).
To verify the suggested binding poses of Cmpd 5 and Cmpd 6, we mutated the residues suggested to be important for the binding. Table 2 shows the electrophysiological characteristics of these mutations. The Cmpd 5–induced G(V) shift was decreased by ∼50% by the Y323C, F324C, and T326C mutations (Fig. 7, C and D). The Cmpd 6–induced G(V) shift was also reduced by ∼50% by the Y323C mutation (Fig. 7 C). Both the Cmpd 5– and Cmpd 6–induced G(V) shifts were completely abolished when all three residues were mutated at the same time, Y323C/F324C/T326C (Fig. 7, C and E), thus supporting the hypothesis that these residues are important for the Cmpd 5 and 6 effects. To test if the triple mutation Y323C/F324C/T326C affected the affinity to a binding pocket, we also measured the effect at different concentrations. Data are consistent with a decreased affinity by a factor of 9 for Cmpd 5 (Fig. 7 F) and by a factor of 15 for Cmpd 6 (Fig. 7 G).
To test if the (probably) negatively charged biaryl sulfonamide electrostatically attracts the positive gating charges in the gating canal to pull S4 into an up position and keep the channel open, we tested the effect of neutralization of a single gating charge, R365C. However, this single mutation was not enough to prevent the G(V)-shifting effect of the compounds (Fig. 7 C).
Discussion
In the present study, we have identified the short biaryl-sulfonamide motif as a common motif for several openers of the Shaker KV channel. No sulfonamide compounds with longer (or shorter) motifs between the two rings opened the Shaker KV channel. The pH experiments suggested that the negatively charged form of the compound is the active variant to facilitate opening of the channel (or alternatively that the compound is more likely to bind to a deprotonated form of the binding site). Our data suggest that the biaryl sulfonamides bind to the extracellular central pocket of the Shaker KV channel VSD. The bound compounds keep the channel in an open state, most likely by keeping S4 in an activated up state, preventing downward movement.
This central VSD site is different from what has been found for PUFAs and resin acids, but the chemical properties of the compounds are also significantly different. However, in contrast with the proposed binding site for the biaryl sulfonamides, in the extracellular water-facing crevice of the VSD, these compounds bind at the interface between the lipid bilayer and the VSD. These different binding sites are reflected in the different chemical properties of the compounds. The lipid-facing PUFAs and resin acids have LogP values between 5.5 and 6.5 (Ottosson et al., 2017), whereas the supposedly water-facing biaryl sulfonamides have lower LogP values (Cmpd 5, LogP = 4.35; Cmpd 6, LogP = 4.13). Similarly, there is a large difference between calculated and experimentally obtained pKa values. PUFAs and resin acids have calculated pKa values ∼3.5–4.5, but their functional pKa values are ∼3 pH-steps higher (Ottosson et al., 2017), reflecting a binding site in an environment different from water. In contrast, the biaryl-sulfonamide compounds studied in the present investigation have pKa values around 7, similar to their functional pKa values, reflecting a binding site in a water environment.
The negatively charged variant of PUFAs and resin acids open both the WT Shaker KV channel and the 3R Shaker KV channel, but the 3R channel to a larger extent, and the positively charged residues at the top of S4 are critical for the effect of the compounds (Börjesson et al., 2008, 2010; Börjesson and Elinder, 2011; Ottosson et al., 2014, 2015, 2017; Silverå Ejneby et al., 2018). The G(V)-shifting effect caused by the biaryl sulfonamide compound bound to its site is not sensitive to positively charged residues added to the extracellular end of S4, as tested using the 3R Shaker KV channel. This reinforces the view that the biaryl sulfonamide and resin acid/PUFA sites are different from each other. In the molecular docking, the charge of the biaryl-sulfonamide compound electrostatically interacts with residue R365 (the second arginine) of S4. However, when we neutralized R365 to a cysteine (R365C), the effect of the biaryl sulfonamides was not reduced. One possible explanation is that the biaryl-sulfonamide compound instead interacts with R368 or R371 when R365 is neutralized. Both R368 and R371 pass the hydrophobic barrier and are exposed on the extracellular side, when the channel switches from a resting state to the activated (open) state (Tao et al., 2010; Henrion et al., 2012).
The binding site proposed in this investigation, centrally in the VSD between S3 and S4, is close, but not identical, to the site found for other channel-opening compounds that bind within the VSD. NH29 opens the Kv7.2 channel by binding in the space between S2 and S4 (Peretz et al., 2010), and ztz240 and other compounds open the KV7.2 channel by binding deeper into the VSD (Li et al., 2013). Thus, the VSDs of KV channels seem to have a cleft at the top, which can harbor different small-molecule compounds to keep the channel activated.
The sulfonamide motif, a sulfonyl group connected to a nitrogen (Fig. 1 A), is widespread in medical drugs and covers a wide range of therapeutic uses. The motif even led to the naming of sulfa antibiotics, which preceded penicillin. Other sulfonamide compounds are anticonvulsant (e.g., sultiame), antihypertensive (e.g., thiazide diuretics), or antidiabetic (e.g., glibenclamide). The sulfonamide compounds have diverse sites and mechanisms of action. Many sulfonamides act by inhibiting different types of enzymes (Winum et al., 2006), but some block ion channels; for instance, the antidiabetic drug glibenclamide blocks the ATP-sensitive potassium channel in pancreatic β cells (Ashcroft and Ashcroft, 1990). Different types of aryl sulfonamides, including aminopiperidine sulfonamides, have been reported to block different types of voltage-gated calcium and sodium channels: CaV2.2 (Shao et al., 2012), CaV3.2 (Jarvis et al., 2014), and NaV1.7 (Focken et al., 2016). However, to our knowledge, there are no sulfonamide pharmaceutical drugs on the market acting on voltage-gated ion channels.
Recently, it was shown that some sulfonamides bind to the VSD of domain IV in a voltage-gated Na channel to lock the channel in a nonconducting state (McCormack et al., 2013; Ahuja et al., 2015). However, the voltage sensor S4 was locked in an activated up state, but the channel was probably inactivated and thus nonconducting. Thus, short biaryl sulfonamides are able to open K channels and close Na channels by similar mechanisms. This suggests that short biaryl sulfonamides can potentially reduce neuronal excitability by targeting multiple ion channels (a potentially powerful strategy (Tigerholm et al., 2012; Salari et al., 2018). It also suggests that short biaryl sulfonamides can potentially be designed to specifically open or close different types of voltage-gated ion channels.
Supplementary Material
Acknowledgments
We thank Gunnar Nordvall for chemistry advice and compound selection, Anders B. Eriksson (SciLifeLab) for help with compound handling, Andreas Nolting for construction of the cell line, Anders Fridberger for comments on the text, and Alexis Reisch for linguistic advice. We thank the Chemical Biology Consortium Sweden at the Science for Life Laboratory for providing the chemical libraries tested in the screen. The NIH Clinical Collection was provided through the NIH Molecular Libraries Roadmap Initiative. The LCBKI primary screening set was provided by CBCS. Part of this work was assisted by Karolinska High Throughput Center (KHTC), a core facility at Karolinska Institutet with affiliation to Science for Life Laboratory (www.scilifelab.se/facilities/khtc/).
This work was supported by grants from the Swedish Research Council (2016-02615), the Swedish Heart-Lung Foundation (20150672), the Swedish Brain Foundation (2016-0326), and the Swedish Society for Medical Research.
The authors declare no competing financial interests.
Author contributions: F. Elinder conceived the study. S.I. Liin, P-E. Lund, J.E. Larsson, J. Brask, and F. Elinder designed experiments and analyzed data. S.I. Liin and J.E. Larsson performed manual electrophysiological experiments. P-E. Lund performed automated electrophysiological experiments. B. Wallner performed molecular docking simulations. S.I. Liin, P-E. Lund, J.E. Larsson, J. Brask, B. Wallner, and F. Elinder wrote the paper.
Kenton J. Swartz served as editor.
References
- Ahuja S., Mukund S., Deng L., Khakh K., Chang E., Ho H., Shriver S., Young C., Lin S., Johnson J.P. Jr., et al. 2015. Structural basis of Nav1.7 inhibition by an isoform-selective small-molecule antagonist. Science. 350:aac5464 10.1126/science.aac5464 [DOI] [PubMed] [Google Scholar]
- Ashcroft S.J., and Ashcroft F.M.. 1990. Properties and functions of ATP-sensitive K-channels. Cell. Signal. 2:197–214. 10.1016/0898-6568(90)90048-F [DOI] [PubMed] [Google Scholar]
- Börjesson S.I., and Elinder F.. 2011. An electrostatic potassium channel opener targeting the final voltage sensor transition. J. Gen. Physiol. 137:563–577. 10.1085/jgp.201110599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Börjesson S.I., Hammarström S., and Elinder F.. 2008. Lipoelectric modification of ion channel voltage gating by polyunsaturated fatty acids. Biophys. J. 95:2242–2253. 10.1529/biophysj.108.130757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Börjesson S.I., Parkkari T., Hammarström S., and Elinder F.. 2010. Electrostatic tuning of cellular excitability. Biophys. J. 98:396–403. 10.1016/j.bpj.2009.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridgland-Taylor M.H., Hargreaves A.C., Easter A., Orme A., Henthorn D.C., Ding M., Davis A.M., Small B.G., Heapy C.G., Abi-Gerges N., et al. 2006. Optimisation and validation of a medium-throughput electrophysiology-based hERG assay using IonWorks HT. J. Pharmacol. Toxicol. Methods. 54:189–199. 10.1016/j.vascn.2006.02.003 [DOI] [PubMed] [Google Scholar]
- Catterall W.A. 2014. Sodium channels, inherited epilepsy, and antiepileptic drugs. Annu. Rev. Pharmacol. Toxicol. 54:317–338. 10.1146/annurev-pharmtox-011112-140232 [DOI] [PubMed] [Google Scholar]
- Elinder F., and Liin S.I.. 2017. Actions and mechanisms of polyunsaturated fatty acids on voltage-gated ion channels. Front. Physiol. 8:43 10.3389/fphys.2017.00043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Focken T., Liu S., Chahal N., Dauphinais M., Grimwood M.E., Chowdhury S., Hemeon I., Bichler P., Bogucki D., Waldbrook M., et al. 2016. Discovery of aryl sulfonamides as isoform-selective inhibitors of NaV1.7 with efficacy in rodent pain models. ACS Med. Chem. Lett. 7:277–282. 10.1021/acsmedchemlett.5b00447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmer A.R., Abi-Gerges N., Easter A., Woods A., Lawrence C.L., Small B.G., Valentin J.P., and Pollard C.E.. 2008. Optimisation and validation of a medium-throughput electrophysiology-based hNav1.5 assay using IonWorks. J. Pharmacol. Toxicol. Methods. 57:30–41. 10.1016/j.vascn.2007.09.002 [DOI] [PubMed] [Google Scholar]
- Hawkins P.C., Skillman A.G., Warren G.L., Ellingson B.A., and Stahl M.T.. 2010. Conformer generation with OMEGA: Algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 50:572–584. 10.1021/ci100031x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrion U., Renhorn J., Börjesson S.I., Nelson E.M., Schwaiger C.S., Bjelkmar P., Wallner B., Lindahl E., and Elinder F.. 2012. Tracking a complete voltage-sensor cycle with metal-ion bridges. Proc. Natl. Acad. Sci. USA. 109:8552–8557. 10.1073/pnas.1116938109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand A., Remmert M., Biegert A., and Söding J.. 2009. Fast and accurate automatic structure prediction with HHpred. Proteins. 77(S9, Suppl 9):128–132. 10.1002/prot.22499 [DOI] [PubMed] [Google Scholar]
- Hille B. 1977. Local anesthetics: Hydrophilic and hydrophobic pathways for the drug-receptor reaction. J. Gen. Physiol. 69:497–515. 10.1085/jgp.69.4.497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi T., Zagotta W.N., and Aldrich R.W.. 1990. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 250:533–538. 10.1126/science.2122519 [DOI] [PubMed] [Google Scholar]
- Jarvis M.F., Scott V.E., McGaraughty S., Chu K.L., Xu J., Niforatos W., Milicic I., Joshi S., Zhang Q., and Xia Z.. 2014. A peripherally acting, selective T-type calcium channel blocker, ABT-639, effectively reduces nociceptive and neuropathic pain in rats. Biochem. Pharmacol. 89:536–544. 10.1016/j.bcp.2014.03.015 [DOI] [PubMed] [Google Scholar]
- Kamb A., Iverson L.E., and Tanouye M.A.. 1987. Molecular characterization of Shaker, a Drosophila gene that encodes a potassium channel. Cell. 50:405–413. 10.1016/0092-8674(87)90494-6 [DOI] [PubMed] [Google Scholar]
- Lemmon G., and Meiler J.. 2012. Rosetta Ligand docking with flexible XML protocols. Methods Mol. Biol. 819:143–155. 10.1007/978-1-61779-465-0_10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P., Chen Z., Xu H., Sun H., Li H., Liu H., Yang H., Gao Z., Jiang H., and Li M.. 2013. The gating charge pathway of an epilepsy-associated potassium channel accommodates chemical ligands. Cell Res. 23:1106–1118. 10.1038/cr.2013.82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liin S.I., Silverå Ejneby M., Barro-Soria R., Skarsfeldt M.A., Larsson J.E., Starck Härlin F., Parkkari T., Bentzen B.H., Schmitt N., Larsson H.P., and Elinder F.. 2015. Polyunsaturated fatty acid analogs act antiarrhythmically on the cardiac IKs channel. Proc. Natl. Acad. Sci. USA. 112:5714–5719. 10.1073/pnas.1503488112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liin S.I., Karlsson U., Bentzen B.H., Schmitt N., and Elinder F.. 2016a Polyunsaturated fatty acids are potent openers of human M-channels expressed in Xenopus laevis oocytes. Acta Physiol. (Oxf.). 218:28–37. [DOI] [PubMed] [Google Scholar]
- Liin S.I., Larsson J.E., Barro-Soria R., Bentzen B.H., and Larsson H.P.. 2016b Fatty acid analogue N-arachidonoyl taurine restores function of IKs channels with diverse long QT mutations. eLife. 5:e20272 10.7554/eLife.20272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Main M.J., Cryan J.E., Dupere J.R., Cox B., Clare J.J., and Burbidge S.A.. 2000. Modulation of KCNQ2/3 potassium channels by the novel anticonvulsant retigabine. Mol. Pharmacol. 58:253–262. 10.1124/mol.58.2.253 [DOI] [PubMed] [Google Scholar]
- McCormack K., Santos S., Chapman M.L., Krafte D.S., Marron B.E., West C.W., Krambis M.J., Antonio B.M., Zellmer S.G., Printzenhoff D., et al. 2013. Voltage sensor interaction site for selective small molecule inhibitors of voltage-gated sodium channels. Proc. Natl. Acad. Sci. USA. 110:E2724–E2732. 10.1073/pnas.1220844110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiler J., and Baker D.. 2006. ROSETTALIGAND: protein-small molecule docking with full side-chain flexibility. Proteins. 65:538–548. 10.1002/prot.21086 [DOI] [PubMed] [Google Scholar]
- Ottosson N.E., Liin S.I., and Elinder F.. 2014. Drug-induced ion channel opening tuned by the voltage sensor charge profile. J. Gen. Physiol. 143:173–182. 10.1085/jgp.201311087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottosson N.E., Wu X., Nolting A., Karlsson U., Lund P.E., Ruda K., Svensson S., Konradsson P., and Elinder F.. 2015. Resin-acid derivatives as potent electrostatic openers of voltage-gated K channels and suppressors of neuronal excitability. Sci. Rep. 5:13278 10.1038/srep13278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottosson N.E., Silverå Ejneby M., Wu X., Yazdi S., Konradsson P., Lindahl E., and Elinder F.. 2017. A drug pocket at the lipid bilayer-potassium channel interface. Sci. Adv. 3:e1701099 10.1126/sciadv.1701099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peretz A., Pell L., Gofman Y., Haitin Y., Shamgar L., Patrich E., Kornilov P., Gourgy-Hacohen O., Ben-Tal N., and Attali B.. 2010. Targeting the voltage sensor of Kv7.2 voltage-gated K+ channels with a new gating-modifier. Proc. Natl. Acad. Sci. USA. 107:15637–15642. 10.1073/pnas.0911294107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perozo E., MacKinnon R., Bezanilla F., and Stefani E.. 1993. Gating currents from a nonconducting mutant reveal open-closed conformations in Shaker K+ channels. Neuron. 11:353–358. 10.1016/0896-6273(93)90190-3 [DOI] [PubMed] [Google Scholar]
- Ragsdale D.S., McPhee J.C., Scheuer T., and Catterall W.A.. 1994. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science. 265:1724–1728. 10.1126/science.8085162 [DOI] [PubMed] [Google Scholar]
- Rundfeldt C., and Netzer R.. 2000. The novel anticonvulsant retigabine activates M-currents in Chinese hamster ovary-cells tranfected with human KCNQ2/3 subunits. Neurosci. Lett. 282:73–76. 10.1016/S0304-3940(00)00866-1 [DOI] [PubMed] [Google Scholar]
- Salari S., Silverå Ejneby M., Brask J., and Elinder F.. 2018. Isopimaric acid: A multi-targeting ion channel modulator reducing excitability and arrhythmicity in a spontaneously beating mouse atrial cell line. Acta Physiol. (Oxf.). 222:e12895 10.1111/apha.12895 [DOI] [PubMed] [Google Scholar]
- Sali A., and Blundell T.L.. 1993. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234:779–815. 10.1006/jmbi.1993.1626 [DOI] [PubMed] [Google Scholar]
- Schroeder K., Neagle B., Trezise D.J., and Worley J.. 2003. Ionworks HT: A new high-throughput electrophysiology measurement platform. J. Biomol. Screen. 8:50–64. 10.1177/1087057102239667 [DOI] [PubMed] [Google Scholar]
- Shao P.P., Ye F., Chakravarty P.K., Varughese D.J., Herrington J.B., Dai G., Bugianesi R.M., Haedo R.J., Swensen A.M., Warren V.A., et al. 2012. Aminopiperidine sulfonamide Cav2.2 channel inhibitors for the treatment of chronic pain. J. Med. Chem. 55:9847–9855. 10.1021/jm301056k [DOI] [PubMed] [Google Scholar]
- Silverå Ejneby M., Wu X., Ottosson N.E., Münger E.P., Lundström I., Konradsson P., and Elinder F.. 2018. Atom-by-atom tuning of the electrostatic potassium-channel modulator dehydroabietic acid. J. Gen. Physiol. 150:731–750. 10.1085/jgp.201711965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao X., Lee A., Limapichat W., Dougherty D.A., and MacKinnon R.. 2010. A gating charge transfer center in voltage sensors. Science. 328:67–73. 10.1126/science.1185954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tigerholm J., Börjesson S.I., Lundberg L., Elinder F., and Fransén E.. 2012. Dampening of hyperexcitability in CA1 pyramidal neurons by polyunsaturated fatty acids acting on voltage-gated ion channels. PLoS One. 7:e44388 10.1371/journal.pone.0044388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickenden A.D., Yu W., Zou A., Jegla T., and Wagoner P.K.. 2000. Retigabine, a novel anti-convulsant, enhances activation of KCNQ2/Q3 potassium channels. Mol. Pharmacol. 58:591–600. 10.1124/mol.58.3.591 [DOI] [PubMed] [Google Scholar]
- Winum J.Y., Scozzafava A., Montero J.L., and Supuran C.T.. 2006. Therapeutic potential of sulfamides as enzyme inhibitors. Med. Res. Rev. 26:767–792. 10.1002/med.20068 [DOI] [PubMed] [Google Scholar]
- Wuttke T.V., Seebohm G., Bail S., Maljevic S., and Lerche H.. 2005. The new anticonvulsant retigabine favors voltage-dependent opening of the Kv7.2 (KCNQ2) channel by binding to its activation gate. Mol. Pharmacol. 67:1009–1017. 10.1124/mol.104.010793 [DOI] [PubMed] [Google Scholar]
- Yazdi S., Stein M., Elinder F., Andersson M., and Lindahl E.. 2016. The molecular basis of polyunsaturated fatty acid interactions with the shaker voltage-gated potassium channel. PLOS Comput. Biol. 12:e1004704 10.1371/journal.pcbi.1004704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi G.W., Striessnig J., Koschak A., and Dolphin A.C.. 2015. The physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol. Rev. 67:821–870. 10.1124/pr.114.009654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M., Morais-Cabral J.H., Mann S., and MacKinnon R.. 2001. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature. 411:657–661. 10.1038/35079500 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.