

Abstract
Neuronal processes contain mRNAs and membrane structures, and some forms of synaptic plasticity seem to require protein synthesis in dendrites of hippocampal neurons. To quantitate dendritic protein synthesis, we used multiphoton microscopy of green fluorescent protein synthesized in living isolated dendrites. Transfection of dendrites with mRNA encoding green fluorescent protein resulted in fluorescence that exponentially increased on stimulation with a glutamate receptor agonist; a reaction attenuated by the translation inhibitors anisomycin and emetine. Comparable experiments on whole neurons revealed that (RS)-3,5-dihydroxy-phenylglycine 0.5 H2O (DHPG)-stimulated fluorescence was linear in cell bodies relative to the exponential increase seen in dendrites. Detailed spatial analysis of the subdendritic distribution of fluorescence revealed “hotspots,” sites of dendritic translation that were spatially stable. However, detailed temporal analysis of these hotspots revealed heterogeneous rates of translation. A double-label protocol counterstaining for ribosomes indicated that sites of “fastest” translation correlated with increased ribosome density, consistent with ribosome subunit assembly for initiation, the first step of translation. We propose that dendrites have specific sites specialized for fast translation.
Protein synthesis can occur in neuronal dendrites, relatively long distances from neuronal cell bodies (1–4). Specific populations of dendritic mRNAs have been identified, some of which are actively transported from cell bodies to the most distal regions of dendrites when neurons are stimulated (1, 3, 5–10). Dendrites also contain ribosomes (6, 11–13) and membranous structures (13, 14) that have been extensively characterized. Furthermore, application of exogenous mRNAs to isolated dendrites has clearly demonstrated that dendritic protein synthesis can occur independently of cell bodies (2, 3).
Our laboratory has developed a means to measure protein synthesis in living dendrites whose cell bodies have been removed. We have transfected isolated dendrites with mRNA encoding green fluorescent protein (GFP; ref. 15) and measured GFP fluorescence as this mRNA is translated into protein (16). Fluorescence-based measurements of translation are reported to be more sensitive than alternatives, such as radioactivity incorporation (17), proportional to the concentration of mRNA (see Fig. 4; ref. 18), and are protein-specific. However, detailed measurements of fluorescence in living cells have until recently been restricted to few time points as a result of excessive phototoxicity and bleaching suffered when using conventional microscopy [for example, work by Aakalu et al. (4)]. We have overcome this obstacle by using multiphoton laser scanning light microscopy (MPLSM; ref. 19), microscopy that excites GFP fluorescence by using two or more photons of long wavelength (at subpicosecond pulses) rather than the more-damaging short wavelength photons used in confocal microscopy. We were therefore able to measure rates of GFP fluorescence from GFP mRNA in situ over time courses of tens of minutes and have discovered an interesting heterogeneity between sites of translation within isolated dendrites.
Figure 4.

DHPG-stimulated synthesis of GFP protein at specific sites along isolated dendrites. (A) Enlarged images of dendrite from black box in Fig. 1B during time course [(5-min intervals) Bar = 50 μm.] showing example hotspots analyzed in B and C. (B) Profile plots through reoriented images of dendrite displayed in A, as examples of hotspots analyzed in D. (Inset) Line of the profile plot across hotspots. Fluorescence range 0–255: 0 = black, 255 = saturated. (C) Examples of hotspots analyzed in D. (Inset) ROIs (white circles) were applied to fluorescent hotspots, and the mean fluorescence intensity per pixel in each ROI was measured across time. (Graph) Time course of hotspot fluorescence as a proportion of that at t0 (——) with exponential curves applied to hotspots 1 and 5 (- - - -). (D) Histogram of time constants for exponential curves applied to fluorescence time courses for ROIs (such as those in C) around 134 hotspots displayed in Fig. 1.
Materials and Methods
DNA encoding GFP was amplified with PfuTurbo polymerase (Stratagene, no. 600252-51) by PCR from pEGFP-N1 (CLONTECH, no. 6085-1), using primers TCAGATCCGC TAGCGCTACC and GCAGTGAAAA AAATGCTTTA. The PCR product was ligated into pCR II-TOPO vector (Invitrogen, no. 45-0640) and was sequenced to ensure the absence of PCR-generated mutations. Messenger RNA was synthesized in vitro from the sp6 promoter by using mMESSAGE mMACHINE (Ambion, Austin, TX; no. 1340). Immediately before transfection, ≈1 μmol of GFP mRNA was bound to 5 μl of the lipofection agent Geneporter (Gene Therapy Systems, San Diego; no. T210007) for 30 min at room temperature and incubated on ice for up to 1 h. Primary cultured hippocampal neurons were prepared from embryonic day 18 rats and grown at low density on CELLocate coverslips (Eppendorf, no. 5245 953.005) embedded into 3-mm dishes by MatTek, Ashland, MA (no. P35G-1.5-7-C-grid). Four days later, the neurons were washed in physiological salt solution (155 mM NaCl/3 mM KCl/1 mM MgCl2/3 mM CaCl2/10 mM Hepes/1 mM glucose) before cell bodies were removed from a specific region of the grid with a glass micropipette, controlled by an Olympus (New Hyde Park, NY) micromanipulator. Cell bodies were removed within a 300 μm radius of the analyzed field of view to ensure that all dendrites were truly isolated dendrites. Images were captured with a Sony (Tokyo) 3CCD DKC 5000 digital photo camera mounted on an Olympus IX70 microscope (×20 dry lens). By using a fresh micropipette, mRNA/Geneporter was sprayed onto the “isolated dendrites” and incubated at 37°C with 5% CO2 for 1 h. Coverslips were washed thoroughly and incubated at 37°C. For Fig. 2, 10 μg/ml of anisomycin or 1 μg/ml of emetine was applied directly after transfection.
Figure 2.

Translation inhibitors anisomycin and emetine attenuated DHPG-stimulated fluorescence in isolated dendrites. (A) After transfection with GFP mRNA, isolated dendrites were incubated with 10 μg/ml of anisomycin (Δ; triangles) or control DMSO carrier (⋅) and stimulated with DHPG at t0, as in Fig. 1. (B) The same experiment with 1 μg/ml of emetine (⊗; circles) and control ethanol carrier (⋅). n = 4. At t10, fluorescence in inhibitor-treated dendrites was significantly less than controls (t test, P < 0.05).
Images of the isolated dendrites were acquired every 2.5 min by using a Bio-Rad MPLSM mounted on a Nikon Eclipse TE300 inverted microscope, using a ×40/0.1 oil lens [differential interference contrast microscopy (DIC) H ∞/0.17 WD 0.16]. Cells were incubated at 37°C with a heated stage. GFP was excited with a 140-fs pulse of 800 nm of light, and images were acquired with the Exponential function (F = 5 and n = 20) as opposed to the Kalman function or the Accumulate function. At t0, the isolated dendrites were stimulated by addition of 40 μl of stock (RS)-3,5-dihydroxy-phenylglycine 0.5 H2O (DHPG; Tocris Neuramin, Bristol, U.K.; no. 0342) to 2 ml of physiological salt solution directly above the gridded coverslip. We believe this method of application created a “burst” of DHPG that diffused across the Petri dish to a final concentration of 20 μM.
Images were analyzed with National Institutes of Health IMAGE 1.61/68K software. Regions of interest (ROIs) were drawn around dendrites or individual hotspots, and the mean fluorescence intensity per pixel (± SEM) was measured across time, relative to t0 (20, 21). Care was taken to minimize “background pixels” within ROIs, and background fluorescence was subtracted from the mean fluorescence. Exponential curves were applied by using WaveMetrics (Lake Oswego, OR) igor pro (Figs. 1E, 2 A and B, 3E, and 4C), with the time constant taken as 1/K2. Data were displayed by using Adobe Systems (Mountain View, CA) photoshop and Microsoft powerpoint.
Figure 1.

Transfection of isolated dendrites with GFP mRNA resulted in fluorescence that increased exponentially when the dendrites were stimulated with DHPG. (A and B) Phase images of 4-day-old primary hippocampal neurons grown on gridded coverslips. Arrows indicate positions of cell bodies removed with a micropipette. (B) Bold black lines highlight positions of dendrites in image A that were transfected with GFP mRNA. Black box indicates site of example dendrite shown in detail in Fig. 4A. [Square = 175 μm2.] (C) Fluorescence images acquired by MPLSM every 5 min, before (first 4 images) and after addition of DHPG to a final concentration of 20 μM. (C 15) Arrows indicate sites of removed cell bodies from A. [Bar = 50 μm.] (D) Mean fluorescence per pixel in ROIs around 7 dendrites (observed within the field of view in C) over a time course of tens of minutes (arbitrary units normalized to t0, n = 3). t0 was significantly different from t5 (t test, P < 0.001). (E) Mean fluorescence in isolated dendrites from 6 experiments, 63 dendrites in total, including those in D (DHPG added at t0). Arrowhead = 7 min 14 s. t15 was significantly greater than t−2.5 (t test, P < 0.05).
Figure 3.

Transfection of intact hippocampal neurons with GFP mRNA resulted in fluorescence in cell bodies as well as dendrites. (A) Transmission image of cultured hippocampal neurons transfected with GFP mRNA by lipofection. Dashed box indicates region shown in B–D. Solid box indicates dendrite shown in F. (B) MPLSM fluorescence image of neurons within dashed box in A. [Bar = 100 μm.] (C) Fluorescence image of neurons in B after stimulation with 20 μM DHPG for 27 min 30 s. (D) Fluorescence image of neurons in B after stimulation with 20 μM DHPG for 37 min 30 s. Fluorescence appears in cell bodies as well as dendrites. (E) Mean fluorescence per pixel in ROIs around dendrites (⋅) and cell bodies (Δ) over a time course of tens of minutes (arbitrary units normalized to t0, n = 3). Small capital letters B, C, and D on the graph depict time points for images B–D. Translation was exponential in dendrites and linear in cell bodies. (F) Enlarged fluorescent images of example dendrite (solid box in A). [Bar = 10 μm.] Numbers indicate minutes of stimulation with 20 μM DHPG. Fluorescent puncta were spatially stable (highlighted by vertical dashed gray lines) over a time course of tens of minutes.
For anti-ribosomal costaining, various anti-ribosomal mAbs [generously provided by T.-A. Sato (Riken Institute, Japan)] were screened for dendritic fluorescence patterns. After dendrite transfection with GFP mRNA and stimulation with DHPG, dendrites were fixed with 4% paraformaldehyde (Electron Microscopy Sciences, Fort Washington, PA), first in physiological solution for 7 min, then in PBS (0.17 mM sodium dihydrogen orthophosphate/1.76 mM disodium hydrogen orthophosphate/150 mM sodium chloride/2.6 mM potassium chloride, pH 7.4) for 9 min. Then they were permeabilized in 0.3% Triton X-100/PBS for 3 min, blocked in 2.5% normal goat serum (Jackson ImmunoResearch)/PBS, and incubated with 1 in 100 monoclonal anti-ribosomal Ab at 4°C for 3 days and 20°C for 2 h. PBS (1 ml) was added and incubated at 4°C overnight and 20°C for 3 h. Dendrites were washed, incubated with 1 in 100 donkey anti-mouse Texas red (Jackson ImmunoResearch) for 60 min 20°C, washed again, mounted in vectorshield mountant, and sealed with rubber cement (Best-Test). For Fig. 5E, 1 μM phalloidin-tetramethylrhodamine B isothiocyanate (TRITC; Sigma, no. P1951) was used instead of anti-ribosomal Ab, and coverslips were incubated in PBS alone during the secondary Ab incubation, in parallel with those coverslips labeled for ribosomes. Double-label confocal images were collected sequentially (20 Kalmans) with a Bio-Rad Radiance 2000 confocal microscope (argon/krypton laser) and merged for display by using confocal assistant. Data displayed in Fig. 6 were collected with ROIs in National Institutes of Health image software and analyzed with igor pro as described previously.
Figure 5.

GFP hotspots colocalized with anti-ribosomal fluorescence. (A) Transmission image of isolated dendrites with black lines highlighting some larger dendrites. Black circles indicate sites of cell bodies before they were removed, and arrows show sites of example hotspots shown in B and C. One grid = 175 μm2. (B) MPLSM fluorescence images of dendrites in A after transfection with GFP mRNA and stimulation with DHPG for 7 min 30 s and 25 min. Gray circles indicate sites of cell bodies before they were removed, and arrows show example hotspots corresponding to those in C. (C) Confocal image of fluorescent dendrites from B that were fixed and counterstained with monoclonal anti-ribosomal Ab (red). Some red puncta appeared devoid of green fluorescence (arrowheads), but all green hotspots contained some red anti-ribosomal fluorescence (examples shown by arrows). [Bar = 100 μm.] (D) Example of an intact neuron stained under the same procedure as isolated dendrites in B and C. (Upper) Transmission image of neuron on gridded coverslip. One grid = 175 μm2. (Lower) Confocal image of green GFP fluorescence and red anti-ribosomal fluorescence. Intact dendrites showed the same fluorescence pattern as isolated dendrites in C. (E) Example of an isolated dendrite counterstained with tetramethylrhodamine B isothiocyanate (TRITC)-phalloidin (indicative of F-actin) under the same procedure as isolated dendrites in B and C (i.e., fluorescence pattern comparison using a protein not associated with translation). (Top) Transmission image of neuron on gridded coverslip before removal of cell body. (Middle) Transmission image of isolated dendrite after removal of the cell body. One grid = 175 μm2. (Bottom) Confocal image of green GFP fluorescence and red TRITC-phalloidin fluorescence. Arrow shows sever-point where cell body was removed. Many GFP hotspots did not contain red fluorescence; the pattern of fluorescence was different from the anti-ribosomal staining in C and D.
Figure 6.
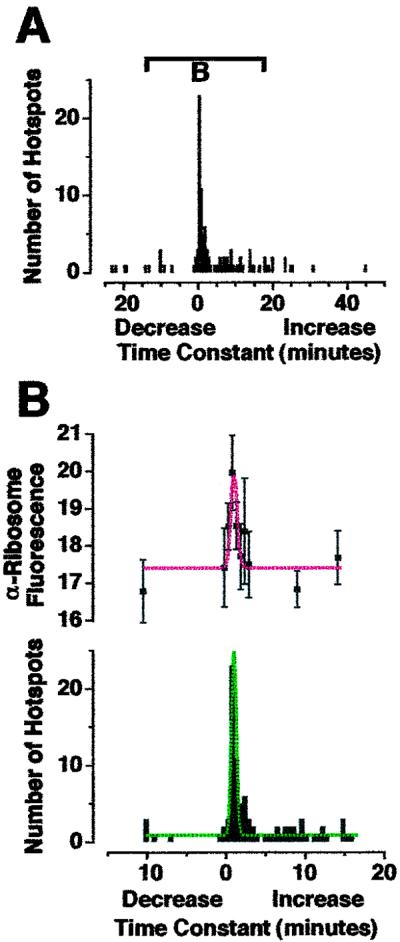
Greater anti-ribosomal fluorescence was associated with GFP hotspots that exhibited “fast” translation. (A) Histogram of time constants for all GFP hotpots within isolated dendrites shown in Fig. 5B (as calculated in Fig. 4C). Bar indicates region of histogram shown in B. (B) Mean intensity per pixel red anti-ribosomal fluorescence (Upper) found in GFP hotspots whose time constants are expressed in the histogram (Lower). Fluorescence range 0–255: 0 = black, 255 = saturated. Red fluorescence found in GFP hotspots with time constants between 30 s and 1 min was significantly greater than in hotspots with time constants between 3 min and 3 min 30 s (P < 0.05). Dashed lines are Gaussian curves. Greater ribosomal density at sites of fast translation is consistent with efficient recruitment of 40 S and 60 S ribosomal subunits as part of initiation, the first step of translation.
Results
Hippocampal neurons were cultured on gridded coverslips (from embryonic day 18 rats, for up to 5 days) and the resultant processes that extended from neuronal cell bodies were termed “dendrites” [according to definitions discussed by Sargent (22) and by immunostaining criteria published by Crino and Eberwine (3)]. Cell bodies were removed with a micropipette (Fig. 1 A and B), and remaining isolated dendrites were transfected with mRNA encoding GFP. After about 90 min, GFP fluorescence appeared in isolated dendrites (Fig. 1C −15–0). Previous studies on dendrites (observed by using immunohistochemistry on fixed cells) indicated that neuromodulators, such as neurotrophins (3) and the metabotropic glutamate receptor (mGluR) agonist DHPG (2, 23, 24), increase protein synthesis. To test the responsiveness of GFP synthesis to stimulation, we added DHPG to isolated dendrites and visualized a significant increase in fluorescence within minutes (Fig. 1C 0–30 and D). Quantification of 6 experiments (63 dendrites) showed that this reaction was exponential with completion by 15 min and a time constant of 7 min 14 s (Fig. 1E). Because these dendrites do not have cell bodies, this was strong evidence for dendritic protein synthesis. However, it was plausible that DHPG was not stimulating translation of GFP, but accelerating posttranslational processing of GFP, such as folding or chromophore formation. To address this possibility, we repeated these experiments on isolated dendrites in the presence of two translation inhibitors: anisomycin, which blocks peptide bond formation, and emetine, which blocks ribosome translocation along mRNA (25). DHPG-stimulated increases in GFP fluorescence were attenuated by both anisomycin (Fig. 2A) and emetine (Fig. 2B). Therefore, we were confident that we were observing stimulated dendritic translation (as well as posttranslational processing).
To check that DHPG-stimulated translation was not the result of mechanical damage caused by removal of cell bodies, these experiments were repeated on intact hippocampal neurons (Fig. 3A). After transfection with GFP mRNA, fluorescence appeared in both cell bodies and dendrites (Fig. 3B). On stimulation with DHPG, this fluorescence increased within tens of minutes (Fig. 3 C and D), as in isolated dendrites (Fig. 1). However, quantification revealed that fluorescence in the cell bodies increased linearly, whereas that in the dendrites increased exponentially (as in Fig. 1 D and E). This dendritic translation could be interpreted as “fast dendritic translation.” Furthermore, the GFP fluorescence was not evenly distributed within dendrites but appeared punctate along their length (Fig. 3F). Fluorescent “hotspots” appeared to be spatially stable over the time course of stimulation (tens of minutes; Figs. 1C, 3F, and 4A). We analyzed this pattern of fluorescence in detail and found that fluorescence intensity within these hotspots varied over time (Fig. 4B). Some puncta increased in intensity within 5 min of DHPG application (e.g., Fig. 4C 2–5), whereas others increased slowly (e.g., Fig. 4C 6) and some decreased (e.g., Fig. 4C 1). Histogram analysis of 134 puncta within 1 experiment revealed that the majority of hotspots showed a fast increase in fluorescence on stimulation with DHPG, whereas a minority showed a slow increase and some a decrease (Fig. 4D; same result observed in 5 additional experiments). Overall, these results demonstrate that rates of translation were heterogeneous within dendrites.
To confirm that the sites displaying fast increases in fluorescence were true sites of translation, we repeated these experiments, fixed the dendrites, and counterstained by using anti-ribosomal Abs (Fig. 5 A–C). A punctate pattern of anti-ribosomal fluorescence was observed along dendrites, consistent with that seen in previous studies (12, 13). Double-label imposition of red and green fluorescence revealed some pure-red puncta (Fig. 5 arrowheads). However, all green hotspots appeared to have some level of red anti-ribosomal fluorescence (appearing yellow in Fig. 5C; same results observed in 3 additional experiments). This pattern implies that all GFP hotpots were associated with ribosomes, but not all ribosomes associated with GFP mRNA. The same double-label pattern was observed in dendrites of intact neurons (Fig. 5D). A different double-label pattern was observed for F-actin (phalloidin-tetramethylrhodamine B isothiocyanate staining; Fig. 5E) [even though the pattern of fluorescence was consistent with those observed in previous studies; for example, Furukawa et al. (26)], confirming the specificity of the staining procedure. We then calculated the time constant for each GFP hotspot displayed in Fig. 5C (Fig. 6A) and measured the levels of red fluorescence within each green hotspot (Fig. 6B). There was a positive correlation between the hotspots with time constants between 30 s and 1 min and the mean intensity per pixel anti-ribosomal fluorescence. Because anti-ribosomal fluorescence could be considered indicative of ribosome density, these data are consistent with the classical understanding of “initiation,” when mRNA, amino acids, and the ribosomal subunits 60 S and 40 S combine to form a ribosome.
Discussion
We have presented, to our knowledge, the first detailed analysis of rates of synthesis for a specific protein in a living cellular compartment. We used GFP, which autocatalytically forms a chromophore by means of cyclization and oxidation. Experiments performed at 22°C indicate that oxidation is the rate-limiting step [discussed by Piston et al. (27)], requiring 90 min to 4 h (28). However, refolding of denatured GFP only takes tens of seconds (29). We show that application of DHPG results in an increase of GFP fluorescence that can be relatively fast at 37°C (time constant 7 min 14 s; Fig. 1E). Because this increase was attenuated by two distinct inhibitors of translation, we conclude that this increase in fluorescence is a representation of translation. We have also noted that the time scale of 90 min to 4 h for GFP oxidation was measured at 22°C by using wild-type GFP expressed in bacteria (28), whereas we performed our experiments in rat neurons at 37°C by using enhanced GFP (S65T mutant; CLONTECH). There are probably at least two reasons why our results differ from the bacterial study. (i) Because many chemical reactions occur faster at higher temperatures it is reasonable to think that GFP oxidation can occur faster at 37°C. (ii) Bacterial lysates may contain different levels of oxidants and antioxidants than the cytoplasm of rat hippocampal neurons. Because the folding of GFP is the rate-limiting step for GFP fluorescence, the time constant of 7 min 14 s is an estimate of GFP translation plus folding. It is therefore likely that our measurements of GFP fluorescence are maximal estimates of dendritic translation.
In addition to temporal analysis, the pattern of GFP fluorescence accurately represents sites of translation along dendrites, because counterstaining with an anti-ribosomal Ab revealed an exact colocalization of GFP hotspots with ribosomal subunits (Fig. 5C). GFP hotspots remained spatially stable over time courses of tens of minutes (Figs. 3F and 4A), appearing slightly diffuse at later time points (Fig. 4A 15–25), as though GFP (a cytoplasmic protein) was traveling away from the sites of translation. We did not observe movement of fluorescent hotspots along dendrites as whole units (Figs. 3F and 4A). This finding is interesting in light of observations made by Kosik and colleagues (6), who propose that “granules” of mRNA and translation machinery move from cell bodies to dendrites as translationally competent units. Because Kosik et al. labeled mRNA in the cell body, it is plausible that mobile Kosik granules are newly synthesized particles moving into dendrites to replenish spatially stable translation machinery.
Our measurements of GFP fluorescence have revealed two other important characteristics of translation in hippocampal neurons. First, GFP translation was exponential in dendrites but linear in cell bodies (Figs. 1D and 3E). Second, not all of the subdendritic sites of translation exhibited low time constants (Figs. 4D and 6A); they were heterogeneous. Considering the first observation (translation in dendrites vs. cell bodies), it is possible that resources necessary for translation (such as free amino acids) became depleted in dendrites but not cell bodies. This possibility is unlikely because dendritic hotspots farthest from their (removed) cell bodies did not necessarily reach completion any faster than more proximal hotspots (Figs. 1C, 3 B–D, 4A, and 5B). Because dendrites are pseudocylindrical, they will have a smaller cytoplasmic volume relative to surface membrane, compared with pseudoellipsoid cell bodies. It is therefore likely that reagents for translation (such as amino acids) are at greater concentrations within dendrite cytoplasm relative to the surface area of plasma membrane containing mGluRs. Also, secondary messenger systems may move over smaller distances from mGluRs at the plasma membrane to ribosomes in the dendroplasm. Finally, mGluR1 and mGluR5 (sites of DHPG action) are concentrated at perisynaptic sites along dendrites (30–32). These factors might contribute to greater levels of stimulation at sites of high surface area to volume ratio, thus driving translation to completion faster in dendrites than in cell bodies. Dendrite structure might therefore be optimal for fast translation. However, the exponential nature of translation in dendrites implies a synergistic interaction between translational regulators (secondary messengers, proteins and RNAs) that is not present in cell bodies (where translation is linear), thus faster dendritic translation may not simply be a structural phenomenon. Because dendrite functions include synaptic plasticity and some forms of synaptic plasticity seem to require protein synthesis, our results indicate that dendrites might be specialized for fast synthesis and processing of proteins necessary for synaptic plasticity. Many studies indicate that rapid dendritic protein synthesis is necessary for long-term changes in synaptic efficacy. Kang and Schuman (33) have suggested that neurotrophin-induced plasticity in hippocampal slices requires general protein synthesis within 30 min for increased synaptic efficacy over a time course of hours. In a similar study, Huber et al. (34) reported that general dendritic protein synthesis is required for maintenance of long-term depression within 10 min of DHPG stimulation. One protein that is abundant in hippocampal dendrites and may be required for synaptic plasticity (in its phosphorylated form) is α-CamKII (35). Estimates of α-CamKII levels in hippocampal slices indicate that dendritic α-CamKII synthesis is required within 5 min of tetanic stimulation (36), and in vitro synaptoneurosome preparations show increased levels of α-CamKII within minutes of stimulation with N-methyl-d-aspartate (NMDA; ref. 37). The time constant of 7 min 14 s that we measured for dendritic GFP synthesis is consistent with these measurements of protein synthesis-dependent synaptic plasticity.
With regard to our second observation, subdendritic sites of translation display heterogenous rate constants (Figs. 4 and 6), one can speculate as to spatial characteristics conducive to fast translation. An obvious question would be whether sites of fast translation are positioned at dendritic spines and synaptic connections. However, few of our translation hotspots colocalized with F-actin (Fig. 5E), which is enriched in dendritic spines (38). Furthermore, a study by Aakalu et al. (4) showed that myristylated GFP did not colocalize with the postsynaptic protein PSD-95 or the presynaptic protein synapsin. The determining requirements for sites of fast translation are therefore open to investigation.
So what is the functional purpose for fast translation at specific sites along dendrites? When we counterstained dendrites with anti-ribosomal Abs, we found a positive correlation between the rate of translation and the amount of anti-ribosomal fluorescence (Fig. 6B). According to well established literature (39), translation occurs in three basic steps: initiation (40 S and 60 S ribosomal subunits associate with mRNA and amino acids), elongation (peptide bonds are formed between amino acids and the ribosome translocates along the mRNA), and termination (mRNA, peptide, 40 S, and 60 S dissociate). One interpretation of the data displayed in Fig. 6 is that microenvironments within dendrites might be specialized for efficient recruitment of ribosomal subunits, thereby regulating initiation, the first step of translation. A study on rat neocortex by Khludova (11) showed increased ribosome numbers correlated with thickness of postsynaptic densities when the neocortex was stimulated, indicating that ribosomal clustering might be important for synaptic plasticity. Also, many studies have shown that ribosomal clustering beneath dendritic spines is important during development (40–45), presumably for maximal dendrite outgrowth and synaptic connectivity. Spatial regulation of translation could result in newly synthesized proteins (such as receptors) at specific distances from stimulated membranes within time windows necessary for synapse formation during development and for synaptic plasticity in mature neurons.
Acknowledgments
We greatly appreciate the efforts of Margie Maronsky who prepared the hippocampal cultures. We thank Irina Chernysh for technical assistance with MPLSM and James Sanzo for assistance with both multiphoton and confocal microscopes. We thank Taka-aki Sato (Riken Institute, Japan) for providing monoclonal anti-ribosomal Abs and Kevin Myashiro for screening anti-ribosomal fluorescence. We thank Gersham Dent, Eve Marder, and Yukihiko Sugimoto for helpful discussions. This work was funded in part by National Institutes of Health Grant A69900 and National Institute of Mental Health Grant 58561.
Abbreviations
- GFP
green fluorescent protein
- DHPG
(RS)-3,5-dihydroxy-phenylglycine 0.5 H2O
- MPLSM
multiphoton laser scanning light microscopy/microscope
- ROI
region of interest
- mGluR
metabotropic glutamate receptor
References
- 1.Torre E R, Steward O. J Neurosci. 1996;16:5967–5978. doi: 10.1523/JNEUROSCI.16-19-05967.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Estee Kacharmina J, Job C, Crino P, Eberwine J. Proc Natl Acad Sci USA. 2000;97:11545–11550. doi: 10.1073/pnas.97.21.11545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crino P B, Eberwine J. Neuron. 1996;17:1173–1187. doi: 10.1016/s0896-6273(00)80248-2. [DOI] [PubMed] [Google Scholar]
- 4.Aakalu G, Smith W B, Nguyen N, Jiang C, Schuman E M. Neuron. 2001;30:489–502. doi: 10.1016/s0896-6273(01)00295-1. [DOI] [PubMed] [Google Scholar]
- 5.Steward O. Neuron. 1997;18:9–12. doi: 10.1016/s0896-6273(01)80041-6. [DOI] [PubMed] [Google Scholar]
- 6.Rook M S, Lu M, Kosik K S. J Neurosci. 2000;20:6385–6393. doi: 10.1523/JNEUROSCI.20-17-06385.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miyashiro K, Dichter M, Eberwine J. Proc Natl Acad Sci USA. 1994;91:10800–10804. doi: 10.1073/pnas.91.23.10800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wallace C S, Lyford G L, Worley P F, Steward O. J Neurosci. 1998;18:26–35. doi: 10.1523/JNEUROSCI.18-01-00026.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roberts L A, Large C H, Higgins M J, Stone T W, O'Shaughnessy C T, Morris B J. Brain Res Mol Brain Res. 1998;56:38–44. doi: 10.1016/s0169-328x(98)00026-6. [DOI] [PubMed] [Google Scholar]
- 10.Steward O, Wallace C S, Lyford G L, Worley P F. Neuron. 1998;21:741–751. doi: 10.1016/s0896-6273(00)80591-7. [DOI] [PubMed] [Google Scholar]
- 11.Khludova G G. Neurosci Behav Physiol. 1999;29:175–180. doi: 10.1007/BF02465323. [DOI] [PubMed] [Google Scholar]
- 12.Tiedge H, Brosius J. J Neurosci. 1996;16:7171–7181. doi: 10.1523/JNEUROSCI.16-22-07171.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gardiol A, Racca C, Triller A. J Neurosci. 1999;19:168–179. doi: 10.1523/JNEUROSCI.19-01-00168.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tarrant S B, Routtenberg A. Tissue Cell. 1977;9:461–473. doi: 10.1016/0040-8166(77)90006-4. [DOI] [PubMed] [Google Scholar]
- 15.Sullivan K F, Kay S A, editors. Methods in Cell Biology: Green Fluorescent Protein. New York: Academic; 1999. [Google Scholar]
- 16.Eberwine J H, Miyashiro K, Estee Kacharmina J, Job C A. Proc Natl Acad Sci USA. 2001;98:7080–7085. doi: 10.1073/pnas.121146698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith C B. Neurochem Res. 1991;16:1037–1046. doi: 10.1007/BF00965848. [DOI] [PubMed] [Google Scholar]
- 18.Berestovskaya N G, Shaloiko L A, Gorokhovatsky A Y, Bondar V S, Vysotski E S, Maximov J E, von Doehren H, Alakhov Y B. Anal Biochem. 1999;268:72–78. doi: 10.1006/abio.1998.3051. [DOI] [PubMed] [Google Scholar]
- 19.Denk W, Strickler J H, Webb W W. Science. 1990;248:73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- 20.Lagnado L, Gomis A, Job C. Neuron. 1996;17:957–967. doi: 10.1016/s0896-6273(00)80226-3. [DOI] [PubMed] [Google Scholar]
- 21.Job C, Lagnado L. J Cell Biol. 1998;143:1661–1672. doi: 10.1083/jcb.143.6.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sargent P B. Trends Neurosci. 1989;12:203–205. doi: 10.1016/0166-2236(89)90121-5. [DOI] [PubMed] [Google Scholar]
- 23.Weiler I J, Greenough W T. Proc Natl Acad Sci USA. 1993;90:7168–7171. doi: 10.1073/pnas.90.15.7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weiler I J, Irwin S A, Klintsova A Y, Spencer C M, Brazelton A D, Miyashiro K, Comery T A, Patel B, Eberwine J, Greenough W T. Proc Natl Acad Sci USA. 1997;94:5395–5400. doi: 10.1073/pnas.94.10.5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vazquez D. FEBS Lett. 1974;40,Suppl.:S63–S84. doi: 10.1016/0014-5793(74)80689-7. [DOI] [PubMed] [Google Scholar]
- 26.Furukawa K, Smith-Swintosky V L, Mattson M P. Exp Neurol. 1995;133:153–163. doi: 10.1006/exnr.1995.1018. [DOI] [PubMed] [Google Scholar]
- 27.Piston D W, Patterson G H, Knobel S M. Methods Cell Biol. 1999;58:31–48. doi: 10.1016/s0091-679x(08)61947-0. [DOI] [PubMed] [Google Scholar]
- 28.Heim R, Prasher D C, Tsien R Y. Proc Natl Acad Sci USA. 1994;91:12501–12504. doi: 10.1073/pnas.91.26.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Makino Y, Amada K, Taguchi H, Yoshida M. J Biol Chem. 1997;272:12468–12474. doi: 10.1074/jbc.272.19.12468. [DOI] [PubMed] [Google Scholar]
- 30.Lujan R, Roberts J D, Shigemoto R, Ohishi H, Somogyi P. J Chem Neuroanat. 1997;13:219–241. doi: 10.1016/s0891-0618(97)00051-3. [DOI] [PubMed] [Google Scholar]
- 31.Ong W Y, Yeo T T, Balcar V J, Garey L J. J Neurocytol. 1998;27:719–730. doi: 10.1023/a:1006946717065. [DOI] [PubMed] [Google Scholar]
- 32.Ong W Y, He Y, Tan K K, Garey L J. Exp Brain Res. 1998;119:367–374. doi: 10.1007/s002210050352. [DOI] [PubMed] [Google Scholar]
- 33.Kang H, Schuman E M. Science. 1996;273:1402–1406. doi: 10.1126/science.273.5280.1402. [DOI] [PubMed] [Google Scholar]
- 34.Huber K M, Kayser M S, Bear M F. Science. 2000;288:1254–1256. doi: 10.1126/science.288.5469.1254. [DOI] [PubMed] [Google Scholar]
- 35.Mayford M, Bach M E, Kandel E. Cold Spring Harbor Symp Quant Biol. 1996;61:219–224. [PubMed] [Google Scholar]
- 36.Ouyang Y, Rosenstein A, Kreiman G, Schuman E M, Kennedy M B. J Neurosci. 1999;19:7823–7833. doi: 10.1523/JNEUROSCI.19-18-07823.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sheetz A J, Nairn A C, Constantine-Paton M. Nat Neurosci. 2000;3:211–216. doi: 10.1038/72915. [DOI] [PubMed] [Google Scholar]
- 38.Fischer M, Kaech S, Knutti D, Matus A. Neuron. 1998;20:847–854. doi: 10.1016/s0896-6273(00)80467-5. [DOI] [PubMed] [Google Scholar]
- 39.Alberts B, Bray D, Lewis J, Raff M, Roberts K, Watson J D. Molecular Biology of the Cell. 3rd Ed. Hamden, CT: Garland; 1994. [Google Scholar]
- 40.Manolova A, Manolov S. Adv Exp Med Biol. 1991;296:21–28. doi: 10.1007/978-1-4684-8047-4_3. [DOI] [PubMed] [Google Scholar]
- 41.Martone M E, Pollock J A, Ellisman M H. Mol Neurobiol. 1998;18:227–246. doi: 10.1007/BF02741301. [DOI] [PubMed] [Google Scholar]
- 42.Phillips L L, Pollack A E, Steward O. J Neurosci Res. 1990;26:474–482. doi: 10.1002/jnr.490260410. [DOI] [PubMed] [Google Scholar]
- 43.Steward O, Davis L, Dotti C, Phillips L L, Rao A, Banker G. Mol Neurobiol. 1988;2:227–261. doi: 10.1007/BF02935634. [DOI] [PubMed] [Google Scholar]
- 44.Bartlett W P, Banker G A. J Neurosci. 1984;4:1954–1965. doi: 10.1523/JNEUROSCI.04-08-01954.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Steward O, Falk P M. J Neurosci Res. 1985;13:75–88. doi: 10.1002/jnr.490130106. [DOI] [PubMed] [Google Scholar]