

Abstract
Target identification and mechanistic studies of cytotoxic agents are challenging processes that are both time-consuming and costly. Here we describe an approach to mechanism of action studies for potential anticancer compounds by utilizing the simple prokaryotic system, E. coli, and we demonstrate its utility with the characterization of a ruthenium polypyridyl complex [Ru(bpy)2dmbpy2+]. Expression of the photoconvertible fluorescent protein Dendra2 facilitated both high throughput studies and single-cell imaging. This allowed for simultaneous ratiometric analysis of inhibition of protein production and phenotypic investigations. The profile of protein production, filament size and population, and nucleoid morphology revealed important differences between inorganic agents that damage DNA vs more selective inhibitors of transcription and translation. Trace metal analysis demonstrated that DNA is the preferred nucleic acid target of the ruthenium complex, but further studies in human cancer cells revealed altered cell signaling pathways compared to the commonly administrated anticancer agent cisplatin. This study demonstrates E. coli can be used to rapidly distinguish between compounds with disparate mechanisms of action and also for more subtle distinctions within in studies in mammalian cells.
Keywords: cancer, drug discovery, cisplatin, ruthenium, bacterial cytological profiling
Introduction
The fortuitous observation of filamentous growth of E. coli by Barnett Rosenberg led to the discovery of cisplatin, one of the most important and widely used chemotherapeutic agents.1−3 Cisplatin, and its later generation analogues, are essential components in clinical treatments of ovarian, testicular, small-cell lung, and head and neck cancers.4−6 The administration of platinum drugs, however, is limited by adverse side effects, including nephrotoxicity, neurotoxicity, ototoxicity, and other complications.7,8 Drug resistance (either intrinsic or acquired) compromises the efficacy of platinum drugs as well.9−11 These deficiencies have necessitated the development of new chemotherapeutic agents to overcome such obstacles.
Significant efforts have been applied in the field of medicinal inorganic chemistry to identify cytotoxic agents that replicate the efficacy of cisplatin, with the hope of adding to our current arsenal of chemotherapeutic drugs.12−14 While many of the new chemical entities show promising efficacy, the understanding of their biological activities is often incomplete. The very nature of inorganic agents (with variable charge states, geometries, and coordination numbers, all of which can be altered by speciation) adds to the challenge and can result in polypharmacology.13,15 As a result, elucidation of the biological effects of potential medicinal inorganic agents has lagged far behind chemical innovation. For example, oxaliplatin, which has been in clinical use for over 20 years, was recently reported to induce ribosome biogenesis stress,16 rather than the previously accepted mechanism similar to cisplatin involving DNA damage. Organic or inorganic agents developed through target-based drug discovery avoid some of these pitfalls, but undesired off-target effects are prevalent for these systems as well. Thus, mechanistic studies are necessary even for compounds designed to inhibit single, well-validated targets.17−19
Despite multiple technological advances, the identification of the mechanism of action for cytotoxic compounds remains a time-consuming and challenging process. While simple in vitro systems such as purified enzymes and nucleic acids can provide key insights, there are undeniable advantages to working in living cells. Bacteria are intrinsically simpler systems than eukaryotic cells, with E. coli containing only 4288 genes,20,21 as opposed to the approximately 30 000 genes found in the human genome.22,23 Essential processes are homologues between bacteria and eukaryote, including DNA replication, transcription, and translation. It is well-known that many agents that are toxic to eukaryotic systems also have antibacterial activities, such as classical antitumor antibiotics, though many orthogonal variations do exist between the two.24,25
Rosenberg’s classical experiment illustrated that a simple prokaryotic system could be employed to discover anticancer agents. Recently, other groups, including those of Lippard and Brabec, have utilized E. coli phenotypic assays as qualitative means to characterize potential anticancer agents, and as with cisplatin, a good correlation was shown between activity in the prokaryotic system and cancer cells.26−28 We also have an interest in simple biological systems, but our motivation is instead to utilize them as a tool to investigate mechanistic details of anticancer agents. Our premise is that compounds that are found to be active in mammalian cells but not in E. coli can be expected to affect processes or targets absent in the simpler biological system. Alternatively, compounds that show similar activities in the two cell types can be deduced to inhibit processes common to both. Thus, it should be possible to use E. coli as a first-pass screen to radically reduce the number of likely biological entities or processes targeted by cytotoxic agents. Furthermore, E. coli is readily amenable to the incorporation of genetically encoded reporter systems, allowing for additional phenotypic analysis to be used to rapidly parse mechanistic features of active compounds.18,29 This approach could greatly expedite mechanism of action studies.
Here we describe studies that demonstrate that E. coli is an excellent model for mammalian systems for investigating the effect of metal complex inhibition of cell growth and phenotypic changes consistent with DNA damage.30 A promising light-activated ruthenium complex developed in our laboratory31 (compound 1, Scheme 1) was compared to cisplatin, along with three organic antibiotics. Noteworthy differences were observed between the inorganic compounds and organic compounds in the bacterial system; these differences directly correlate with their different mechanisms of action. Moreover, differences between compound 1 and cisplatin in mammalian cells suggest more subtle disparities in their mechanistic features, which offers the possibility to maintain anticancer efficacy without experiencing the same resistance profile by altering the metal center from platinum to ruthenium.
Scheme 1. Thermal Hydrolysis of Cisplatin and the Photochemical Hydrolysis of Compound 1.

Methods
E. coli Culture Maintenance
The Dendra2 gene was cloned into a pCW-ori plasmid modified to contain an N-terminal 6× histidine tag with multiple restriction enzyme cloning sites. Escherichia coli BL21(DE3) competent cells transformed with pCWori plasmid containing Dendra2 gene (pCWori-Dendra2) were cultured in Luria Broth (LB) at 37 °C with 180 rpm shaking.
Mammalian Cell Maintenance
Human promyelocytic leukemia HL60 cells were purchased from ATCC. Dulbecco’s modified eagle medium (DMEM), Iscove’s modified Dulbecco’s medium (IMDM), Opti-MEM I reduced serum medium, heat inactivated fetal bovine serum (FBS), penicillin/streptomycin (5000 U/mL), trypsin–EDTA (0.5%), Dulbecco’s phosphate buffered saline (DPBS), and Trypan Blue Solution (0.4%) were purchased from Life Technologies.
HL60 cells were maintained in IMDM supplemented with 10% FBS and 50 U/mL of penicillin/streptomycin. A549 cells were maintained in DMEM with the same supplements. Cells were maintained at 37 °C with 5% CO2.
Cytotoxicity Determination
E. coli BL21(DE3) cells transformed with pCWori-Dendra2 plasmid were plated in M63 minimal medium at 4 × 106 cells per well in 96 well flat bottom transparent tissue culture treated plates (Greiner Bio One). Compounds were dosed from 0–300 μM, followed by 3 min of light irradiation (7 J/cm2 blue light (>400 nm)). The cells were then incubated for 16 h with the compounds, and cell growth was determined by measurement of the optical density at 600 nm using a SpectraMax Multiwell Plate Reader (Molecular Devices).
HL60 cells were plated in Opti-MEM supplemented with 1% FBS and 50 U/mL of penicillin/streptomycin at 30 000 cells per well in 96 well flat bottom transparent tissue culture treated plates (Greiner Bio One). Compounds were dosed from 0–300 μM and incubated for 16 h, followed by light irradiation with 7 J/cm2 blue light (>400 nm) in 30 s pulses for a total light exposure of 3 min. The cells were then incubated for 72 h, and cell viability was determined by conversion of resazurin to resorufin. Dark controls were run in parallel. The emission of resorufin was measured on a SpectraFluor Plus Plate Reader (Tecan).
The data were normalized to the untreated control and fitted to a sigmoidal dose response model using Prism 6.02 to determine IC50 values. Minimal inhibitory concentration (MIC) was fitted to the model published by Lambert et al. using Prism 6.02.32
Protein Synthesis Inhibition
E. coli BL21DE3 cells transformed with pCWori-Dendra2 were cultured in LB medium to an OD600 of 0.8. Cells were then resuspended in M63 minimal media and induced with 0.5 mM IPTG for 3 h at 37 °C with 180 rpm shaking. Photoconversion of Dendra2 was carried out with a 405 nm LED flood array (Loctite) with a total light exposure time of 2 min. Cells were then plated in 96 well plates at 6 × 107 cells per well. Green and red emission was measured directly after photoconversion using a SpectraMax Multiwell Plate Reader (Molecular Devices) for a baseline evaluation of Dendra2 protein (t = 0 h). For green emission, an excitation wavelength of 491 nm and emission wavelength of 538 nm was used; for red emission, the excitation wavelength was 544 nm and emission wavelength was 590 nm. Compounds were then dosed from 0 to 300 μM, and compound 1 was activated with light as described above. The cells were incubated for 16 h before the green and red emission was measured again for an evaluation of protein synthesis with compound treatment (t = 16 h).
The average fluorescence ratio of green/red at t = 0 and 16 h was calculated, the values were normalized, and the data fitted to a sigmoidal dose response.
E. coli Filamentous Growth
E. coli were cultured as above and plated at 3 × 108 cells per well in 24 well flat bottom transparent tissue culture treated plates (Greiner Bio One). IPTG was added at a concentration of 0.5 mM for induction of Dendra2 production. Compound treatment was then carried out, with cells dosed at the MIC or 10× MIC for each compound and cultured at 37 °C with 180 rpm of shaking for 6 and 16 h before imaging.
E. coli Cell Imaging
After compound treatment, E. coli cells were centrifuged at 8000 rpm for 2 min, washed twice with PBS, and 3 × 107 cells were resuspended in 1 mL of PBS. The fluorescent dyes FM4–64 and Hoechst 33342 were added to a final concentration of 5 and 10 μg/mL respectively. The cells were protected from light for 20 min, and then, 2 μL of cell suspension was placed on a slide, and a cover glass was applied before imaging. Imaging was carried out on an Olympus IX2-RFAEVA-2 microscope with the following filter settings:
Dendra2 (green), excitation filter: 473/10 nm BrightLine single-band bandpass filter, FF01–473/10–25 (Semrock, Rochester, NY, USA); emission filter: 525/50 nm BrightLine single-band bandpass filter, FF03–525/50–25 (Semrock, Rochester, NY, USA). Dendra2 (red) and FM4–64, excitation filter: HQ 550/30 (Chroma, Bellows Falls, VT, USA); emission filter: 664 nm EdgeBasic long-pass edge filter, BLP01–664R-25 (Semrock, Rochester, NY, USA). Hoechst 33342, excitation filter: BP 360–390 (Chroma, Bellows Falls, VT, USA); emission filter, HQ470/30 M (Chroma, Bellows Falls, VT, USA).
Imaging data was processed and analyzed with ImageJ.
Metal Uptake in Bacterial Cells
E. coli were cultured in M63 minimal medium as discussed above and dosed with 20 μM compound 1 or cisplatin. Cells treated with compound 1 were irradiated with 7 J/cm2 blue filtered light (>400 nm) for a total of 3 min or were protected from light. Cells were collected 24 h after compound addition by centrifugation at 8000 rpm for 5 min. The culture medium was separated for analysis, and cells were washed twice with PBS and pelleted. Both cell content and medium were heated at 110 °C for 3 h with 20% (v/v) HNO3.
Total RNA and genomic DNA were isolated using Qiagen kits. RNA and DNA samples were digested in HNO3 as described above. Following sample digestion, the metal content was analyzed using a Varian AAS with a replicate reading and a spiked reading.
Cellular uptake was calculated as follows
Genomic DNA and total RNA were quantified by measuring their absorbance at 260 nm. Mass to DNA nucleotide pair conversion was calculated using the average molecular weight of DNA nucleotide pairs. The number of DNA nucleotide bases per metal center was calculated as follows
Mass to RNA nucleotide base conversion was calculated using the average molecular weight of RNA nucleotide bases. Number of RNA nucleotide bases per metal center was calculated as follows
Metal Uptake in HL60 Cells
HL60 cells were plated in Opti-MEM supplemented with 1% FBS and 50 U/mL penicillin/streptomycin at a density of 1 × 106 cells/mL in 25 cm2 cell culture flasks and dosed with 20 μM compound 1 or cisplatin. Cells treated with compound 1 were incubated 12 h, protected from light, before irradiating with 7 J/cm2 blue filtered light (>400 nm) in 30 s pulses for a total of 3 min or protected from light. Cells were collected 24 h after compound addition by centrifugation at 124 × g for 5 min. The culture media was separated for analysis, and cells were washed twice with PBS. Total RNA and genomic DNA were also isolated, and the nucleic acids, cell content, and media were prepared for analysis as described above.
Immunoblotting
HL60 cells were harvested 0, 1, 3, 6, 12, 24, 30, and 48 h after treatment, pelleted by centrifugation at 124 × g for 5 min, and washed twice with DPBS. A549 cells were plated at 2 × 105 cells per well in 6 well flat bottom transparent tissue culture treated multiwell plates and in the same treatment conditions detailed for HL60 cells. Cells were harvested at 0, 6, 12, 24, and 48 h after treatment.
All cells were lysed in RIPA buffer supplemented with 5 mM sodium pyrophosphate (2 × 106 cells/100 μL) for 15 min on ice. The insoluble fraction was removed by centrifugation at 2017 × g for 10 min at 4 °C. The supernatant was collected, and the protein concentration was determined by BCA assay. Protein (20 μg) was loaded onto 4–12% bis–tris gels and followed by transfer to nitrocellulose membranes. After blocking with 2.5% BSA in DPBS with 0.1% Tween20 (PBST) for 1 h at room temperature, the membrane was immunoblotted with the following primary antibodies and corresponding dilutions.
Cleaved caspase 3, cleaved PARP, p-p53, p21, p-Chk1, p-JNK and γ-H2AX at 1:1000 dilutions; p53 and p-ERK at 1:500 dilutions; and GAPDH at a 1:2000 dilution in 2.5% BSA overnight at 4 °C. Immunoblots were washed with PBST for 10 min four times and incubated for 1 h with secondary antibodies at a 1:10 000 dilution for GAPDH and 1:5000 dilutions for all other antibodies. Detection was carried out with Clarity Western ECL Substrate and imaged with a ChemiDoc MP System (Bio-Rad).
DNA Fragmentation
HL60 cells were cultured and treated as described above. Cells were harvested at 0, 3, 8, 12, 24, and 30 h after treatment, pelleted by centrifugation at 124 × g for 5 min, washed twice with DPBS, and prepared with an apoptotic DNA-ladder kit as per manufacturer instructions (Rosch). Gel electrophoresis was carried out using a 1% agarose gel containing 0.5 μg/mL ethidium bromide for 90 min at 75 V. Gel imaging was performed with the ChemiDoc MP.
Flow Cytometry
HL60 cells were cultured and treated as detailed previously. Cells were harvested at 24 h after treatment, pelleted by centrifugation at 124 × g for 5 min, and washed twice with DPBS. For cell death mechanism analysis, cells were stained 15 min with FITC–Annexin V and PI; for cell cycle analysis, cells were stained 15 min with PI only. Cells were analyzed with a FACSCalibur (Becton-Dickenson). A minimum of 20 000 events were measured for each sample.
Results
Comparison of Compound Efficacies in E. coli and Mammalian Cancer Cells
The capacity of E. coli to serve as a model system for cancer cells was first evaluated by comparing the relative cytotoxicities of the metal complexes in the two cell types. The ruthenium complex prodrug, compound 1, and cisplatin were tested in dose response, along with the antibiotics rifampicin, tetracycline, and nalidixic acid. Optical density was used to quantify the response in E. coli. The activity of 1 was evaluated both in the absence of light and after light activation (described as “dark” and “light”; irradiation results in the formation of compound 2; Scheme 1). The half maximal inhibitory concentration (IC50) value was compared with the minimum inhibitory concentration (MIC),33 an important clinical standard parameter32−35 that effectively defines the lowest concentration to achieve a complete inhibition effect. As shown in Table 1, IC50 values of 2.6 and 2.0 μM for light-activated 1 and cisplatin were obtained in E. coli, with MIC values that were 2–3-fold higher.
Table 1. Cytotoxicity Values and Inhibition of Protein Production for Various Compounds in E. coli and HL60 Cells.
|
E. coli |
HL60 | |||
|---|---|---|---|---|
| MIC (μM) | growth inhibition IC50 (μM) | Dendra2 production inhibition IC50 (μM) | cytotoxicity IC50 (μM) | |
| 1 light | 6.1 ± 0.8 | 2.6 ± 0.4 | 77 ± 3 | 3.4 ± 0.3 |
| 1 dark | >300 | >300 | >300 | >300 |
| cisplatin | 4.4 ± 0.5 | 2.0 ± 0.1 | 85 ± 11 | 2.6 ± 0.4 |
| rifampicin | 0.6 ± 0.3 | 0.3 ± 0.1 | 2.8 ± 0.3 | n.d. |
| tetracycline | 10 ± 1.1 | 4.8 ± 0.7 | 1.3 ± 0.2 | n.d. |
| nalidixic acid | 5.2 ± 2.0 | 2.6 ± 0.4 | 4.6 ± 0.1 | n.d. |
The biological activity of cisplatin and compound 1 was also studied in human promyelocytic leukemia HL60 cells. This relatively fast growing suspension cell line was chosen over adherent cell lines to more closely resemble bacterial growth conditions. Upon light irradiation, 1 exhibited an IC50 of 3.4 μM, similar to the IC50 of 2.6 μM for cisplatin. No cytotoxic effect was seen for compound 1 at 300 μM in the dark, resulting in a phototoxicity index (PI) of >88. As expected, the cytotoxicity of cisplatin was not affected by treatment with light. These experiments demonstrated that light irradiated 1, like cisplatin, is cytotoxic in both prokaryotic and eukaryotic cells, and with very similar potencies, suggesting the mechanism of action is through general cellular targets or biological processes present in both cell types.
Cellular Uptake and Nucleic Acid Metalation
Cellular uptake of the metals in E. coli was measured by atomic absorption spectroscopy (AAS; Table 2). Light irradiation of compound 1 resulted in a 5-fold increase in cellular uptake, with a total of 10% of the dosed compound localized in E. coli cells. Only 6% of the dosed cisplatin was found in the cells.
Table 2. Cellular Metal Uptake and Metal Content with Different Nucleic Acids Measured by AAS.
|
E.
coli |
HL60 |
|||||
|---|---|---|---|---|---|---|
| cellular uptakea | DNA nt/mcb | RNA nt/mcc | cellular uptakea | DNA nt/mcb | RNA nt/mcc | |
| 1 light | 10% | 2000 ± 200 | 3800 ± 600 | 0.64% | 4800 ± 400 | 5000 ± 700 |
| 1 dark | 2% | -d | -d | 0.11% | -d | -d |
| cisplatin | 6% | 3000 ± 200 | 4700 ± 900 | 0.72% | 7000 ± 200 | 7800 ± 700 |
Cellular uptake was calculated as metal content measured in cells divided by total metal content in both cell samples and cell culture media samples.
DNA nt/mc was calculated as DNA nucleotide bases (μmol) divided by metal content measured in DNA sample (μmol).
RNA nt/mc was calculated as RNA nucleotide bases (μmol) divided by metal content measured in DNA sample (μmol).
Ruthenium levels in DNA and RNA samples were under the detection limit (<2 ppb).
Genomic DNA and total RNA isolation was performed after 24 h of treatment, followed by AAS analysis for ruthenium or platinum. While no ruthenium was found with either of the nucleic acids for compound 1 in the dark, 1.3% of the ruthenium was found with the DNA when the compound had been exposed to light. This corresponds to a ratio of 2000 nucleotide bases per metal center (nt/mc). Only 0.5% of ruthenium was found with the RNA, providing a ratio of 3800 nt/mc. As a result, the active compound 2 appears to be slightly more reactive with DNA than RNA, with about a 1.5–2-fold difference between the metal levels in the two nucleic acids. A similar trend of increased reactivity with DNA over RNA was observed for cisplatin, with 3000 nc/mc in DNA and 4700 nt/mc in RNA, to give a 1.6-fold difference in apparent reactivity.
The uptake of the compounds was also assessed in mammalian cells. After 24 h of treatment with 20 μM of compound 1, 0.64% of dosed ruthenium was found in HL60 cells with light irradiation, in contrast to only 0.11% present when the cells were kept in the dark. These results indicate that the prodrug form is taken up much less effectively than the active species. The metal content of the active compound in cells is comparable to the 0.72% of cisplatin that accumulated under the same conditions. Isolation of DNA and RNA and metal content analysis revealed that no nucleic-acid-bound ruthenium was observed for 1 in the dark, but treatment of 1 and irradiation resulted in 4800 nt/mc in DNA and 5000 nt/mc in RNA. This corresponds to 1.3% of the cellular ruthenium found with the DNA and 2.0% in the RNA. Quantification of the metal binding of cisplatin gave 7000 nt/mc in DNA and 7800 nt/mc in RNA (1.1 and 1.5%). The nucleotide base to metal center ratios were close, but consistently a slightly higher reactivity was observed with DNA for both irradiated compound 1 and cisplatin.
It has been reported by DeRose et al. that platinum accumulates more in the cellular RNA than DNA.36 This is partly due to the higher abundance of RNA in the cell (10–50-fold). Despite this difference in abundance for the different nucleic acids, DeRose demonstrated that there is a 3.8-fold preference for cisplatin to react with DNA vs RNA in S. cerevisiae, with 1661 nt/mc in DNA and 6369 nt/mc in RNA after 12 h of treatment at 100 μM.36 While our study used 20 μM of cisplatin treatment for 24 h, we observed the same preferential metal binding with DNA over RNA in both E. coli and mammalian cancer cells, though we observed a closer nt/mc ratio between DNA and RNA. This similar binding trend across different cell types reveals once more that cisplatin exhibits a general DNA damaging ability in both eukaryotic and prokaryotic systems. The similar biological accumulation characteristics of compound 1 and cisplatin in bacterial, yeast, and mammalian cells suggest a mechanism of action through common biological targets or processes present in both prokaryotic and eukaryotic cell types. In combination with extensive in vitro DNA damaging assays,31,37 this supports a DNA-based mechanism of action, though multiple subsequent events may be involved that induce the cytotoxic effects.
Phenotypic Analysis of E. coli following Compound Treatment
1. Filament Size
For imaging studies, the MIC was used in order to more closely mimic physiological treatment conditions; data was also taken at 10× MIC. The cytological characteristics of the E. coli were assessed, and elongated cells were observed after treatment with cisplatin and compound 1 with irradiation (Figure 1). Treatment with 1 in the absence of light did not induce E. coli filamentous growth, and the cells were characterized by the same short rod shaped morphology as the untreated control.
Figure 1.

Complex 1 induces filamentous growth and decreased protein production in E. coli. Bright field and fluorescent imaging of E. coli cells. (A) N. C. control, (B) cisplatin, (C) compound 1 with light, (D) compound 1 in the dark. Size distribution histograms of E. coli cells associated with the conditions for (A–D): (E) N. C. control, (F) cisplatin, (G) compound 1 with light, (H) compound 1 in the dark. Histograms of average fluorescence intensity correlated to cell size with the different treatments: (I) N. C. control, (J) cisplatin, (K) compound 1 with light, (L) compound 1 in the dark. Cells were treated with 100 μM of each compound for 6 h before imaging.
To gain a more quantitative understanding of filament formation in populations, cells in multiple views (∼200 per condition) were chosen for size analysis. Treatment at 10× MIC with cisplatin and compound 1 with irradiation caused a shift in population distribution, where 68 and 73% of cells were filamentous for cisplatin and 1, respectively. The major population group at 100 μM 1 were cells over 40 μm long (29%), and only 5% of the cells were in the ≤5 μm size range. The same trend was seen after cisplatin treatment, and the histograms for the population of filaments are remarkably similar (Figure 1F,G). In contrast, at the MIC, both the filament length and % filamentous population were lower, with only 30 and 41% of cells forming filaments for cisplatin and 1 (Figures 2E and S4). As shown in the histograms in Figure 1E,H, both the no treatment control and dark control for compound 1 exhibited a dominant population (over 97%) of cells in the ≤5 μm size range, which represents the normal E. coli cell size. Thus, filamentation is only associated with the light-activated form of 1.
Figure 2.

Phenotypic profiles of compounds with different mechanisms of action in E. coli cells. Fluorescent imaging: (A) top: N. C. control; middle: cisplatin; bottom: compound 1 with light; (B) top: rifampicin; middle: tetracycline; bottom: nalidixic acid. The merge is the combination of the Hoechst and FM4–64 membrane stain. Colony forming experiment with various compounds: (C) left: N. C. control; middle: cisplatin; right: compound 1 with light; (D) left: nalidixic acid; middle: rifampicin; right: tetracycline. Cells were treated with each compound at MIC for 6 h before imaging or colony forming. (E) Quantitative and qualitative analysis of E. coli filamentous growth and nucleoid morphology phenotypes in response to compound treatment.
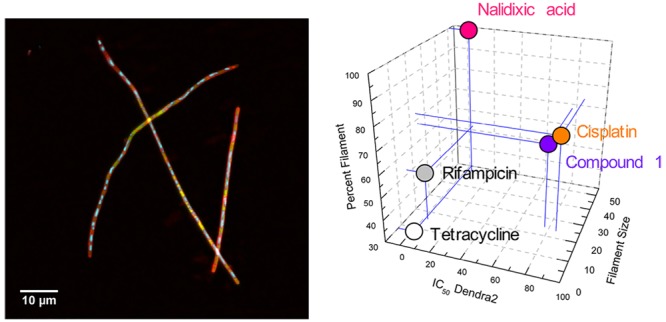
It has been observed that many compounds induce filamentous growth of E. coli. In order to determine if this morphological feature corresponds to the compounds’ mechanisms of action, we compared the metal-based compounds, preliminarily classified as DNA cross-linkers, to two commonly used antibiotics that inhibit transcription or translation, and one gyrase inhibitor. Rifampacin prevents transcription by binding and inhibiting the bacterial DNA-dependent RNA polymerase (RNAP),38−40 and tetracycline inhibits translation via binding to the 30S subunit of the ribosome, preventing entrance of aminoacyl-tRNAs to the A-site. These compounds were selected as agents that do not induce DNA damage.41−43 Nalidixic acid, which inhibits gyrase and induces DNA double-strand breaks, was investigated as a DNA damaging agent with a distinct mechanism of action from cisplatin.44,45
All antibiotics were able to induce E. coli filaments, but the populational size analysis revealed that the major populations of E. coli varied significantly in length. Tetracycline treatment at the MIC resulted in a large fraction (88%) of the cell population of normal length, with only 22% forming short (5–10 μm) filaments. In marked contrast, nalidixic acid induced very long filamentation, and the filaments were the only population (100%; average length of 51 μm). For rifampicin, a concentration of 10× MIC was required to induce any filaments. Treatment at this concentration resulted in 30% of the population forming short filaments of 5–10 μm; 70% of cells were normal length (≤5 μm; Figure S2). This initial analysis made clear that compound 1, cisplatin, and nalidixic acid all induced longer filaments that were a larger portion of the population under all treatment conditions than antibiotics that inhibited transcription or translation.
2. Membrane Integrity
The membrane stain FM4–64 was used to confirm that the observed filaments were single cells and to visualize membrane integrity. As shown in Figure 2A,B, filaments were formed by single cells upon compound treatment. No disruption of the cell membrane was observed, indicating that the phenotypic changes were not associated with cell lysis. This is consistent with results that were obtained utilizing Trypan blue staining of HL60 cells, which indicated that neither of the two metal compounds act as membrane damaging agents. Thus, the abnormal features observed occur in live cells and are not an artifact resulting from physical disruptions of cellular integrity. In addition, the mechanism of action does not entail membrane damage.
3. Nucleoid Morphology and Number
As E. coli contain a single chromosome, DNA staining and analysis allows for detection of DNA fragmentation or other morphological changes due to compound treatment. Over 30 cells per treatment condition were analyzed, and distinct effects were observed for the impact of the different compounds on E. coli nucleoids (Figure 2A,B).
Both rifampicin and tetracycline treatment produced filaments with a regular distribution of DNA. Rifampicin treatment (at 10× MIC) produced the fewest nucleoids, with the majority of filaments containing a single nucleoid that spread along the length of the cell. Tetracycline, in contrast, produced a number of nucleoids in each of the filaments, and the nucleoids were compact and regularly distributed throughout the cell.
In marked contrast, nalidixic acid, cisplatin, and light irradiated 1 caused expansion, fragmentation, and irregular distribution of nucleoids. Both the size and distribution of the nucleoids within the cells were quite varied. In order to quantify this observation, the % STD (the ratio of the standard deviation to the average nucleoid size, used as a measurement of variability; Figure S12) was calculated. The % STD was 38% for the no treatment control and was 36–49% for the transcription and translation inhibitors. In the metal complex and nalidixic acid treated systems, however, the % STD was 121–160%. Values greater than 100% indicate that the standard deviation of nucleoid size exceeded the average size of the nucleoids. This large range of nucleoid size implicates issues of DNA fragmentation and failure of DNA segregation after DNA replication.46 The morphological changes in the bacterial nucleoids treated with the metal compounds and nalidixic acid demonstrate a multifaceted process as a consequence of DNA damage, in contrast with compounds that act to inhibit transcription or translation, which did not result in DNA fragmentation.
It is well-established that the processes of transcription and translation are closely coordinated in E. coli, and the “transertion model” posits, in part, that coupled transcription–translation and membrane association of the growing protein impacts nucleoid morphology. Thus, any process that interferes with mRNA production and protein synthesis could be reflected in the nucleoids. A recent report demonstrated that transcription and translation inhibitors affected E. coli nucleoid shape and spatial distribution, with expansion observed with treatment of rifampicin and compaction with tetracycline.46 This is qualitatively similar to our results. In addition, treatment with nalidixic acid resulted in the observation of fragmented nucleoids,46 similar to our imaging results with this compound and the metal complexes. This supports our hypothesis that nucleoid morphology can be used as a phenotypic indicator of DNA damage.
A colony forming assay was performed to provide further support for the assignment of a DNA damaging mechanism of action (Figure 2C,D). Cells were treated at the MIC for each compound, and then, the media was removed, and the cells were spread on an agarose plate. Only cells treated with the transcription and translation inhibitors were able to form colonies; the metal complexes and nalidixic acid were clearly cytotoxic at their MIC. This supports a conclusion that these three compounds induce irreversible damage to the E. coli, likely through DNA.
4. Protein Production
Cisplatin and other platinum-based agents are known to interfere with protein production. Some question remains, however, if this is an important feature that induces cell death, or simply a side effect of the DNA damage. Several experiments have quantified the impact on protein production after transfection of already metalated plasmids into living systems.47,48 To study the process and impact of DNA metalation, we treated E. coli with the metal complexes and subsequently monitored protein production. This experiment couples the quantitation of protein levels in the detection of the fluorescent protein to the preceding natural sequence of events that impact transcription/translation and allows for observation of important features that may play a role, such as compound uptake, localization within the cell, or sequence-dependent interactions with the nucleic acid.
Cells undergoing death will slow or cease protein production, which produces a similar phenotype to cells that are under the influence of a transcription or translation inhibitor. To discriminate between inhibition of protein production and induction of cell death, we used a photoconvertible protein, Dendra2, as a reporter, since it is able to provide information on both aspects of cell viability and new protein production simultaneously.49 Dendra2 undergoes a photochemical reaction, transforming from a green fluorescent protein to a red fluorescent protein when exposed to 405 nm light. The photoconverted “Red” Dendra2 emission provided a stable internal reference for cell health and cell number for all samples, while new protein production (after light exposure) is reflected in the “Green” Dendra2 emission. Both forms are stable and persist in living cells with half-lives (t1/2) on the order of 50 to 70 h.50−52 The two forms of the protein thus provide spatial and temporal tracking of Dendra2 formed before and after light exposure.
Dendra2 production was induced in E. coli with IPTG and allowed to proceed for 3 h before photoconversion, followed by compound treatment. A clear negative correlation was seen between protein production and cell size, where filamentous cells with longer filament lengths exhibited a lower fluorescence intensity, reflecting a reduction in the amount of new Dendra2 protein being produced. As shown in Figure 1, after 24 h of treatment with compound 1, the average fluorescence intensity of the cell population with the largest length (>40 μm) dropped by over 70% compared to the control population. Other populations with increased cell lengths exhibited a 30–70% decrease in fluorescence intensity. The same trend was seen in cisplatin treated cells, where the fluorescence intensity decreased by 15–80%, depending on the length of the filament. Both compound dose and the time of treatment was found to have an effect on filament formation and protein production (see Figure S3–S5).
The production of Dendra2 was quantified by dose response, providing IC50 values for inhibition of protein production. Protein production was quantified using the ratio of the average fluorescent intensity of the two forms of Dendra2, as shown in Figure 3. The transcription inhibitor rifampicin and translation inhibitor tetracycline exhibited IC50 values for inhibition of Dendra2 production that matched well with growth inhibition (within 3–10-fold; see Table 1 and Figure 3). In contrast, both compound 1 and cisplatin displayed a greater disparity between inhibition of protein production and cell growth inhibition. The 30–40-fold decrease in potency reflects that the mechanism of action of cisplatin and compound 1 is not solely (or primarily) through transcription or translation inhibition. In contrast, nalidixic acid, which induces DNA double-strand breaks, was far more effective at inhibiting protein production.
Figure 3.

Dendra2 expression in E. coli cells. (A) Dendra2 distribution in cells and filaments; Red Dendra2 (left), Green Dendra2 (middle), merge (right). From top to bottom: N. C. control; cisplatin (100 μM); compound 1, light (100 μM); rifampicin (3 μM); tetracycline (48 μM). Cells were treated for 6 h before imaging. The scale bar is 20 μM. (B) Dendra2 production inhibition after 16 h of treatment. Compound treatment only affects the production of new, green Dendra2. (C) Dose response of Dendra2 production inhibition measured at 0 and 16 h after treatment with cisplatin (blue), compound 1 with light (red), rifampicin (green), tetracycline (black).
While cisplatin has been described as a transcription inhibitor, it was the least effective of the five compounds tested for inhibition of protein production. The impact of the DNA damage induced by platinum compounds on protein production has been comprehensively and conclusively proven, along with the restoration of protein production when the appropriate DNA repair mechanisms are activated to remove the lesions. However, our studies suggest that the inhibition of protein production by cisplatin is of secondary importance for the health of E. coli, as the concentrations required to observe a significant impact on this process far exceeded the toxic dose for the compound.
In an analogous study, Lippard and co-workers tested cisplatin in mammalian cells containing a genetically encoded fluorescent reporter system.53 Very good agreement was observed between the concentrations required to inhibit protein synthesis and to induce cytotoxicity evaluated via a colony counting assay in that report. The reason for the disparity in the ability of cisplatin to inhibit protein production in E. coli compared to the HeLa cells used by Lippard is unclear. It is particularly surprising, given the very similar values we found for DNA and RNA metalation in E. coli and HL60 cells, as described above. However, due to the sensitivity of DNA damage detection mechanisms in mammalian cells, it seems unlikely that translation would be affected before initiation of the DNA damage response.
5. Protein Distribution
The use of a photoconvertible protein allows for a spatiotemporal analysis of protein content. This provided the opportunity to address intriguing questions such as the impact of interruption of cell division and filamentous growth on the activity of ribosomes for new protein production, and the redistribution of existing protein within a filamentous cell. Fluorescent imaging was performed to probe the effects of the different compounds on protein distribution in single cells. Compounds were dosed after Dendra2 photoconversion, and imaging was performed 6 h later. As shown in Figure 3A, both the “Red” Dendra2 (the internal control of pretreatment protein level) and the “Green” Dendra2 (reflecting protein synthesis after treatment) were distributed throughout the cell, as the healthy cells underwent multiple cell divisions. Both Red and Green forms of Dendra2 were also found within the filamentous cells where cell division was blocked by either DNA damaging agents or transcription/translation inhibitors. It has been reported that disruption of DNA replication and double-strand breaks resulting from nalidixic acid treatment could lead to uneven distribution of ribosomes in filamentous cells.46 However, we did not observe any particular spatial sequestration of active ribosomes; alternatively, protein diffusion is sufficiently rapid to prevent observation of any localization during the time scale of the experiment.
Comparison of in Vitro and in Cell Protein Production
Previously, we reported an in vitro transcription and translation assay (IVTT) with compound 1 and cisplatin using Green Fluorescent Protein (GFP) as a fluorescent reporter.37 In this assay, either a plasmid containing the GFP gene or the mRNA transcript for GFP were allowed to react with varying concentrations of cisplatin or compound 1, with irradiation, before addition of the nucleic acids to a cancer cell lysate containing transcription and translation machinery. Both metal compounds inhibited GFP production with a clear dose response. Interestingly, the IC50 value for inhibition of protein production was ∼3 μM for both compounds. The ratio of DNA nucleotides or RNA nucleotides to each metal center was calculated at the IC50 for protein synthesis inhibition. The values for compound 1 were 1140:1 and 820:1, while the values for cisplatin were 600:1 for DNA and 610:1 for RNA, respectively.
In the current uptake studies, E. coli cells were dosed at 20 μM, which is approximately 10× higher than the IC50 value for growth inhibition, but well-below the IC50 value for inhibition of protein synthesis as determined by Dendra2 production (see Table 1). In order to compare the in vitro experiment to the cell data, extrapolation of the ratio for DNA and RNA nucleotides per metal center at the IC50 value for in vivo protein synthesis inhibition was performed as detailed in the Supporting Information. The calculated values were remarkably close to the values from the IVTT assay, with DNA nucleotides to metal center ratios of 520:1 for compound 1 and 700:1 for cisplatin. The RNA nucleotide to metal center ratio was 1000:1 for compound 1 and 1090:1 for cisplatin.
This analysis of the ratio of DNA or RNA bases to metal centers suggests the functional inhibition of protein synthesis by covalent adducts to DNA and mRNA by compound 1 and cisplatin is similar in E. coli and the in vitro assay. It is notable that in a living cell, where the reaction conditions are much more complex than those of the buffered system of IVTT assay, the IC50 values to inhibit protein synthesis were diminished by over 60-fold relative to the IVTT assay. However, the ratio between DNA or RNA bases and the metal center for inhibition of protein production remain quite consistent. The increase in the IC50 values in cells suggests two conclusions: (1) inhibition of protein synthesis is not the factor that induces cell death and (2) both compounds suffer from off-target binding to biological molecules. The later is known to be a major issue for many currently administered drugs, especially cisplatin.
The role of off-target binding was also supported by the AAS analysis of metal content with the different nucleic acids in E. coli and mammalian cells, as only a minor component of the metal compounds entered the cells, and of this, only 1.30 and 1.26% of cellular ruthenium from compound 1 and 0.98 and 1.12% of the cellular platinum from cisplatin were found with genomic DNA in the two systems. If one includes the <2% of metal present in the RNA as on-target damage, this means that over 96% of the cellular metal is reacting with potentially nonrelevant targets. Extending this argument, if off-target binding could be eliminated, cytotoxicity IC50 values would be reduced to nanomolar concentrations if the same levels of cellular uptake could be maintained. This may lead to another method to improve the potency of currently used drugs: instead of focusing on the generation of analogues that are more potent against nucleic acids, analogues with reduced off-target binding could be more effective.
It has been shown that the vast majority of cisplatin and other Pt species bind to plasma proteins and do not reach their target in vivo.54−56 It was anticipated that the ruthenium compound would fare better than cisplatin in avoiding off-target binding, due to its lower affinity for hydrophobic proteins such as human serum albumin (HSA)37 and thiols such as glutathione (GSH), but this has not been found to be the case in cells.31,57 As the preferred binding partners are not the same for the platinum and ruthenium complexes, it will be important to identify the primary off-target biomolecules responsible for sequestering the ruthenium in order to rationally design derivatives that avoid these species to increase the potency of these inorganic compounds.
Ru and Pt Compounds Induce Distinct Cellular Responses in Mammalian Cells
The cellular effect of compound 1 was also studied in mammalian cells, with a focus on proteins involved in cell signaling and cell death. As shown in Figure 4, effects on cell cycle and apoptosis were studied using flow cytometry, immunoblotting for apoptotic markers, and DNA fragmentation. No cell-cycle-specific arrest point was observed with compound 1 treatment, while a sub G1 population of 20% of cells was observed after 24 h. Flow cytometry analysis of apoptosis vs necrosis using FITC–Annexin V and propidium iodide indicated that compound 1 induced cell death through apoptosis as the dominant mechanism. While cisplatin induced necrosis in a small fraction (5%), less than 2% of cells treated with compound 1 were characterized as necrotic. Immunoblotting of caspase 3 and PARP showed a time-dependent induction of apoptosis; in addition, isolation of genomic DNA showed fragmentation, which is consistent with apoptotic cell death. All apoptotic reporters were clearly observed at 24 h.
Figure 4.

Compound 1 induces apoptosis in HL60 cells without cell cycle arrest. (A) Flow cytometry by PI/Annexin V in HL60 cells; red = apoptotic cells, blue = necrotic cells, black = dead cells. (B) Flow cytometry by PI in HL60 cells; black = G1; red = G2; white = S phase. (C) Immunoblotting of cleaved PARP and cleaved caspase 3 in HL60 cells. GAPDH was blotted as loading control. (D) Agarose gel electrophoresis of DNA laddering. HL60 cells were treated for 24 h for flow cytometry. All panels: compound 1, 20 μM; cisplatin, 20 μM; doxorubicin, 1 μM.
The tumor suppressor protein p53 regulates cell growth and cell cycle checkpoints to eliminate proliferation. It is one of the most commonly mutated genes in cancer, resulting in loss of its regulatory function.58,59 Both p53/p21 and chk1 are involved in G1/S and G2/M cell cycle checkpoints in response to DNA damage.60,61 In order to probe the role of p53 in response to compound 1, immunoblotting was performed in A549 cells. This nonsmall-cell lung cancer cell contains functional p53, in contrast with the p53 deficient HL60 cell line. While both cisplatin and doxorubicin were able to induce apoptosis in the absence of functional p53 in HL60 cells (Figure 4), the A549 cell line demonstrated clear induction of p53 for these two compounds. In contrast, compound 1 did not induce elevated expression of p53, and did not significantly alter its phosphorylation or expression of p21 (Figure 5A).
Figure 5.

Immunoblotting of apoptotic markers and cell signaling proteins in (A,B) A549 cells; C) HL60 cells. Cells were treated with 20 μM of compound 1 for specified time periods; cisplatin (20 μM) and doxorubicin (2 μM) at 24 h of treatment were used as controls. GAPDH was used as the loading control.
Another surprising difference observed between the platinum and ruthenium compounds is that p-chk1, which is involved in G2/M cell cycle arrest in response to DNA damage, was not induced by treatment of compound 1 in either cell line, while both cisplatin and doxorubicin induced phosphorylation of chk1. This finding is consistent with the fact that no cell cycle arrest point was seen with compound 1 in HL60 cells, in contrast to cisplatin and doxorubicin. However, phosphorylation of γ-H2AX, an early sensor of DNA damage, was observed after 6 h of treatment with 1, indicating DNA damage even in the absence of chk1 activation.
Prosurvival and proapoptotic pathways, including MAPK pathways, were examined, and consistent signaling behaviors after compound 1 treatment were observed in both cell lines. The ERK pathway has been reported to facilitate cell survival and prevent apoptosis.62,63 As shown in Figure 5, this pathway was inactivated by compound 1 in both A549 and HL60 cell lines; in contrast, both cisplatin and doxorubicin did not downregulate this prosurvival pathway. The JNK pathway has been reported to act as a proapoptotic pathway in response to cellular stress induced by DNA damage and is mainly activated by mismatch repair signals.62,64 Both cisplatin and doxorubicin were able to induce phosphorylation of JNK in A549 and HL60 cells at 24 h, though different phosphorylation levels in HL60 cells were observed, which might indicate possible phosphorylation time course differences. Phospho-JNK was seen as early as 6 h after doxorubicin treatment in HL60 cells, while cisplatin induced phospho-JNK was seen to increase to its maximum level at 24 h.65,66 Surprisingly, compound 1 did not activate the JNK pathway to the same extent as cisplatin or doxorubicin. The phosphorylation level was slightly increased within 6 to 12 h of treatment with compound 1 in both cell lines but then decreased over time. This, along with the previously discussed markers, indicate a different DNA damage response for compound 1 either from altered cell signaling pathways or by a different class of DNA damage.
Recent reports have raised questions as to the source of the biological effects of compound 1 and analogous Ru(II) systems, with data suggesting that it is the liberated bipyridyl ligand, rather than the Ru(II) metal center 2, that is responsible for activity.67,68 This study demonstrates such striking similarities between the behavior of the light-activated Ru(II) complex and cisplatin, both in E. coli and mammalian cancer cells, that we find it improbable that the ligand, rather than the metal center, is responsible for the phenotypic effects. The cytological profile, nucleic acid metalation, and DNA damage response is consistent with metal-mediated DNA damage. However, it is possible that the ligand is inducing other effects that are not observed with these assays. A more detailed investigation is underway.
Discussion
This work demonstrates that a combination of phenotypic screening based on E. coli imaging and protein production using Dendra2 as a fluorescent reporter allows for rapid investigations of mechanisms of action for cytotoxic agents that may have similar activities in mammalian cells. We found that a combination of these two experimental parameters facilitates discrimination of DNA damaging agents from agents that work solely as transcription or translation inhibitors. While filaments are formed by all classes of compound, filament size and population distribution was radically different depending on the mechanism of action. Furthermore, the observation of irregular bacterial nucleoids, easily visualized using Hoechst staining, was associated with DNA damage, while regular nucleoid size, shape, and distribution was found with compounds that do not directly affect DNA. Such experiments could not be performed in eukaryotic cells, given the number of chromosomes.
E. coli have been used in the past as model systems to probe binding characteristics of drugs, such as cisplatin, with proteins using NMR.69,70 Phenotypic analysis in bacteria by microscopy is now gaining more attention, primarily to identify the cellular pathways impacted by antibiotics,71,72 but a recent report identified anticancer activity for a molecule characterized by cytological profiling in bacteria.73 In that case, identification of the mechanism of action of the molecule under investigation motivated later studies in cancer cells, and the authors characterized bacterial cytological profiling (BCP) as a tool for drug repurposing.
We are taking the reverse approach by using bacteria to elucidate mechanistic features of compounds already identified as promising anticancer agents. We believe that these studies in bacterial and mammalian cells highlight the capacity of performing rapid studies of anticancer agents in a simple biological model system. Phenotypic studies and quantitative analysis reveal similarities in biological activities between complex 1 and cisplatin. Some limitations are apparent, though, as the cellular response to the compounds in cancer cells suggests diverse pathway regulation following the DNA damage. Altering the metal center thus offers a possibility to maintain efficacy without experiencing the same resistance.
While these studies bring us closer to understanding the mechanism of action of a particular cytotoxic ruthenium complex, it also raised several provocative questions. These include the following: (1) why is the cytotoxic potency of cisplatin and other inorganic putative DNA damaging agents the same in E. coli and mammalian cells when DNA damage recognition and repair pathways that are unique to eukaryotic systems are implicated as playing a key role in their mechanism of action? (2) While more of the metal complexes are taken up in E. coli than the HL60 cells, the nt/mc ratio remains quite consistent for both DNA and RNA. What biological entities are responsible for the enhanced sequestration of the metals in the E. coli, and is it possible that similar molecules play a role in cisplatin-resistant cancer cells? (3) Why does DNA packing not play a greater role in determining the degree of metalation? DNA is packaged in different ways and to different degrees of compaction in the two cell types, and if the more highly exposed, transcriptionally active sequences were the primary target, we would anticipate greater potency for inhibition of Dendra2 production. We believe that addressing these basic questions may be very important to the rational development of improved DNA targeting agents, and we see E. coli as a excellent system to seek the answers.
Acknowledgments
This work is supported by the National Institutes of Health (R01GM107586). YS was the recipient of a University of Kentucky Opportunity Fellowship. All metal uptake studies are done with technical support of Environmental Research Training Laboratories (ERTL) at the University of Kentucky. Experiments were performed at the UK Flow Cytometry & Cell Sorting core facility, which is supported in part by the Office of the Vice President for Research, the Markey Cancer Center, and an NCI Center Core Support Grant (P30 CA177558) to the University of Kentucky Markey Cancer Center.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.molpharmaceut.8b00407.
Supporting figures and tables (PDF)
Author Contributions
‡ YS and DKH contributed equally. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Rosenberg B.; Vancamp L.; Krigas T. Inhibition of cell division in Eschrichia coli by electrolysis products from a platinum electrode. Nature 1965, 205, 698–699. 10.1038/205698a0. [DOI] [PubMed] [Google Scholar]
- Rosenberg B.; Van Camp L.; Grimley E. B.; Thomson A. J. The inhibition of growth or cell division in Escherichia coli by different ionic species of platinum(IV) complexes. J. Biol. Chem. 1967, 242, 1347–1352. [PubMed] [Google Scholar]
- Rosenberg B.; VanCamp L.; Trosko J. E.; Mansour V. H. Platinum compounds: a new class of potent antitumour agents. Nature 1969, 222 (5191), 385–386. 10.1038/222385a0. [DOI] [PubMed] [Google Scholar]
- Cepeda V.; Fuertes M. A.; Castilla J.; Alonso C.; Quevedo C.; Perez J. M. Biochemical mechanisms of cisplatin cytotoxicity. Anti-Cancer Agents Med. Chem. 2007, 7 (1), 3–18. 10.2174/187152007779314044. [DOI] [PubMed] [Google Scholar]
- Boulikas T.; Vougiouka M. Cisplatin and platinum drugs at the molecular level. (Review). Oncol. Rep. 2003, 10 (6), 1663–1682. 10.3892/or.10.6.1663. [DOI] [PubMed] [Google Scholar]
- Wang D.; Lippard S. J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discovery 2005, 4 (4), 307–320. 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7 (8), 573–584. 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- Ciarimboli G. Membrane transporters as mediators of cisplatin side-effects. Anticancer Res. 2014, 34 (1), 547–550. [PubMed] [Google Scholar]
- Kartalou M.; Essigmann J. M. Mechanisms of resistance to cisplatin. Mutat. Res., Fundam. Mol. Mech. Mutagen. 2001, 478 (1–2), 23–43. 10.1016/S0027-5107(01)00141-5. [DOI] [PubMed] [Google Scholar]
- Stewart D. J. Mechanisms of resistance to cisplatin and carboplatin. Critical Reviews in Oncology/Hematology 2007, 63 (1), 12–31. 10.1016/j.critrevonc.2007.02.001. [DOI] [PubMed] [Google Scholar]
- Galluzzi L.; Senovilla L.; Vitale I.; Michels J.; Martins I.; Kepp O.; Castedo M.; Kroemer G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31 (15), 1869–1883. 10.1038/onc.2011.384. [DOI] [PubMed] [Google Scholar]
- Chen D.; Milacic V.; Frezza M.; Dou Q. P. Metal complexes, their cellular targets and potential for cancer therapy. Curr. Pharm. Des. 2009, 15 (7), 777–791. 10.2174/138161209787582183. [DOI] [PubMed] [Google Scholar]
- Frezza M.; Hindo S.; Chen D.; Davenport A.; Schmitt S.; Tomco D.; Ping Dou Q. Novel metals and metal complexes as platforms for cancer therapy. Curr. Pharm. Des. 2010, 16 (16), 1813–1825. 10.2174/138161210791209009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone T. C.; Suntharalingam K.; Lippard S. J. Third row transition metals for the treatment of cancer. Philos. Trans. R. Soc., A 2015, 373 (2037), 20140185. 10.1098/rsta.2014.0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Canelon I.; Sadler P. J. Next-generation metal anticancer complexes: multitargeting via redox modulation. Inorg. Chem. 2013, 52 (21), 12276–12291. 10.1021/ic400835n. [DOI] [PubMed] [Google Scholar]
- Bruno P. M.; Liu Y.; Park G. Y.; Murai J.; Koch C. E.; Eisen T. J.; Pritchard J. R.; Pommier Y.; Lippard S. J.; Hemann M. T. A subset of platinum-containing chemotherapeutic agents kills cells by inducing ribosome biogenesis stress. Nat. Med. 2017, 23 (4), 461–471. 10.1038/nm.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anighoro A.; Bajorath J.; Rastelli G. Polypharmacology: challenges and opportunities in drug discovery. J. Med. Chem. 2014, 57 (19), 7874–7887. 10.1021/jm5006463. [DOI] [PubMed] [Google Scholar]
- Moffat J. G.; Rudolph J.; Bailey D. Phenotypic screening in cancer drug discovery - past, present and future. Nat. Rev. Drug Discovery 2014, 13 (8), 588–602. 10.1038/nrd4366. [DOI] [PubMed] [Google Scholar]
- Schenone M.; Dancik V.; Wagner B. K.; Clemons P. A. Target identification and mechanism of action in chemical biology and drug discovery. Nat. Chem. Biol. 2013, 9 (4), 232–240. 10.1038/nchembio.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T.; Ara T.; Hasegawa M.; Takai Y.; Okumura Y.; Baba M.; Datsenko K. A.; Tomita M.; Wanner B. L.; Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2006, 2 (1), 0008. 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattner F. R.; Plunkett G. 3rd; Bloch C. A.; Perna N. T.; Burland V.; Riley M.; Collado-Vides J.; Glasner J. D.; Rode C. K.; Mayhew G. F. The complete genome sequence of Escherichia coli K-12. Science 1997, 277 (5331), 1453–1462. 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- Frazer K. A. Decoding the human genome. Genome Res. 2012, 22 (9), 1599–1601. 10.1101/gr.146175.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann B.; Walter T.; Heriche J. K.; Bulkescher J.; Erfle H.; Conrad C.; Rogers P.; Poser I.; Held M.; Liebel U.; Cetin C.; Sieckmann F.; Pau G.; Kabbe R.; Wunsche A.; Satagopam V.; Schmitz M. H.; Chapuis C.; Gerlich D. W.; Schneider R.; Eils R.; Huber W.; Peters J. M.; Hyman A. A.; Durbin R.; Pepperkok R.; Ellenberg J. Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature 2010, 464 (7289), 721–727. 10.1038/nature08869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catrina S. B.; Refai E.; Andersson M. The cytotoxic effects of the anti-bacterial peptides on leukocytes. J. Pept. Sci. 2009, 15 (12), 842–848. 10.1002/psc.1185. [DOI] [PubMed] [Google Scholar]
- Vaucher R. A.; Teixeira M. L.; Brandelli A. Investigation of the cytotoxicity of antimicrobial peptide P40 on eukaryotic cells. Curr. Microbiol. 2010, 60 (1), 1–5. 10.1007/s00284-009-9490-z. [DOI] [PubMed] [Google Scholar]
- Johnstone T. C.; Alexander S. M.; Lin W.; Lippard S. J. Effects of Monofunctional Platinum Agents on Bacterial Growth: A Retrospective Study. J. Am. Chem. Soc. 2014, 136 (1), 116–118. 10.1021/ja411742c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janovska E.; Novakova O.; Natile G.; Brabec V. Differential genotoxic effects of antitumor trans-[PtCl(2)(E-iminoether)(2)] and cisplatin in Escherichia coli. J. Inorg. Biochem. 2002, 90 (3–4), 155–158. 10.1016/S0162-0134(02)00403-8. [DOI] [PubMed] [Google Scholar]
- Brabec V.; Pracharova J.; Novakova O.; Gibson D.; Kasparkova J. The induction of lysis in lysogenic strains of Escherichia coli by a new antitumor transplatin derivative and its DNA interactions. Dalton Trans 2015, 44 (8), 3573–3582. 10.1039/C4DT02603A. [DOI] [PubMed] [Google Scholar]
- Carragher N. O.; Brunton V. G.; Frame M. C. Combining imaging and pathway profiling: an alternative approach to cancer drug discovery. Drug Discovery Today 2012, 17 (5–6), 203–214. 10.1016/j.drudis.2012.02.002. [DOI] [PubMed] [Google Scholar]
- A comprehensive study correlating the mechanism of action of multiple chemotherapeutics on the cytological profile of bacterial reporter systems will be published separately.
- Howerton B. S.; Heidary D. K.; Glazer E. C. Strained ruthenium complexes are potent light-activated anticancer agents. J. Am. Chem. Soc. 2012, 134 (20), 8324–8327. 10.1021/ja3009677. [DOI] [PubMed] [Google Scholar]
- Lambert R. J.; Pearson J. Susceptibility testing: accurate and reproducible minimum inhibitory concentration (MIC) and non-inhibitory concentration (NIC) values. J. Appl. Microbiol. 2000, 88 (5), 784–790. 10.1046/j.1365-2672.2000.01017.x. [DOI] [PubMed] [Google Scholar]
- Peitz I.; van Leeuwen R. Single-cell bacteria growth monitoring by automated DEP-facilitated image analysis. Lab Chip 2010, 10 (21), 2944–2951. 10.1039/c004691d. [DOI] [PubMed] [Google Scholar]
- Carson C. F.; Hammer K. A.; Riley T. V. Broth micro-dilution method for determining the susceptibility of Escherichia coli and Staphylococcus aureus to the essential oil of Melaleuca alternifolia (tea tree oil). Microbios 1995, 82 (332), 181–185. [PubMed] [Google Scholar]
- Davison H. C.; Low J. C.; Woolhouse M. E. What is antibiotic resistance and how can we measure it?. Trends Microbiol. 2000, 8 (12), 554–559. 10.1016/S0966-842X(00)01873-4. [DOI] [PubMed] [Google Scholar]
- Hostetter A. A.; Osborn M. F.; DeRose V. J. RNA-Pt adducts following cisplatin treatment of Saccharomyces cerevisiae. ACS Chem. Biol. 2012, 7 (1), 218–225. 10.1021/cb200279p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidary D. K.; Glazer E. C. A light-activated metal complex targets both DNA and RNA in a fluorescent in vitro transcription and translation assay. ChemBioChem 2014, 15 (4), 507–511. 10.1002/cbic.201300681. [DOI] [PubMed] [Google Scholar]
- Calvori C.; Frontali L.; Leoni L.; Tecce G. Effect of rifamycin on protein synthesis. Nature 1965, 207 (995), 417–418. 10.1038/207417a0. [DOI] [PubMed] [Google Scholar]
- Wehrli W. Rifampin: Mechanisms of Action and Resistance. Clin. Infect. Dis. 1983, 5, S407–S411. 10.1093/clinids/5.Supplement_3.S407. [DOI] [PubMed] [Google Scholar]
- Campbell E. A.; Korzheva N.; Mustaev A.; Murakami K.; Nair S.; Goldfarb A.; Darst S. A. Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell 2001, 104 (6), 901–912. 10.1016/S0092-8674(01)00286-0. [DOI] [PubMed] [Google Scholar]
- Speer B. S.; Shoemaker N. B.; Salyers A. A. Bacterial resistance to tetracycline: mechanisms, transfer, and clinical significance. Clin. Microbiol. Rev. 1992, 5 (4), 387–399. 10.1128/CMR.5.4.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salyers A. A.; Speer B. S.; Shoemaker N. B. New perspectives in tetracycline resistance. Mol. Microbiol. 1990, 4 (1), 151–156. 10.1111/j.1365-2958.1990.tb02025.x. [DOI] [PubMed] [Google Scholar]
- Nguyen F.; Starosta A. L.; Arenz S.; Sohmen D.; Donhofer A.; Wilson D. N. Tetracycline antibiotics and resistance mechanisms. Biol. Chem. 2014, 395 (5), 559–575. 10.1515/hsz-2013-0292. [DOI] [PubMed] [Google Scholar]
- Drlica K.; Malik M.; Kerns R. J.; Zhao X. Quinolone-mediated bacterial death. Antimicrob. Agents Chemother. 2008, 52 (2), 385–392. 10.1128/AAC.01617-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldred K. J.; Kerns R. J.; Osheroff N. Mechanism of quinolone action and resistance. Biochemistry 2014, 53 (10), 1565–1574. 10.1021/bi5000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Q.; Singh B.; Peisker K.; Metzendorf N.; Ge X.; Dasgupta S.; Sanyal S. Organization of ribosomes and nucleoids in Escherichia coli cells during growth and in quiescence. J. Biol. Chem. 2014, 289 (16), 11342–11352. 10.1074/jbc.M114.557348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang W. H.; Brown W. W.; Lippard S. J. Preparation of Mammalian Expression Vectors Incorporating Site-Specifically Platinated-DNA Lesions. Bioconjugate Chem. 2009, 20 (5), 1058–1063. 10.1021/bc900031a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang W. H.; Myint M.; Lippard S. J. Transcription Inhibition by Platinum DNA Cross-links in Live Mammalian Cells. J. Am. Chem. Soc. 2010, 132 (21), 7429–7435. 10.1021/ja101495v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidary D. K.; Fox A.; Richards C. I.; Glazer E. C. A High-Throughput Screening Assay Using a Photoconvertable Protein for Identifying Inhibitors of Transcription, Translation, or Proteasomal Degradation. SLAS Discovery 2017, 22 (4), 399–407. 10.1177/2472555216684333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fron E.; Van der Auweraer M.; Moeyaert B.; Michiels J.; Mizuno H.; Hofkens J.; Adam V. Revealing the excited-state dynamics of the fluorescent protein Dendra2. J. Phys. Chem. B 2013, 117 (8), 2300–2313. 10.1021/jp309219m. [DOI] [PubMed] [Google Scholar]
- Adam V.; Nienhaus K.; Bourgeois D.; Nienhaus G. U. Structural basis of enhanced photoconversion yield in green fluorescent protein-like protein Dendra2. Biochemistry 2009, 48 (22), 4905–4915. 10.1021/bi900383a. [DOI] [PubMed] [Google Scholar]
- Woods E.; Courtney J.; Scholz D.; Hall W. W.; Gautier V. W. Tracking protein dynamics with photoconvertible Dendra2 on spinning disk confocal systems. J. Microsc. 2014, 256 (3), 197–207. 10.1111/jmi.12172. [DOI] [PubMed] [Google Scholar]
- Sandman K. E.; Marla S. S.; Zlokarnik G.; Lippard S. J. Rapid fluorescence-based reporter-gene assays to evaluate the cytotoxicity and antitumor drug potential of platinum complexes. Chem. Biol. 1999, 6 (8), 541–551. 10.1016/S1074-5521(99)80086-6. [DOI] [PubMed] [Google Scholar]
- Timerbaev A. R.; Hartinger C. G.; Aleksenko S. S.; Keppler B. K. Interactions of antitumor metallodrugs with serum proteins: advances in characterization using modern analytical methodology. Chem. Rev. 2006, 106 (6), 2224–2248. 10.1021/cr040704h. [DOI] [PubMed] [Google Scholar]
- Ossipov K.; Scaffidi-Domianello Y. Y.; Seregina I. F.; Galanski M.; Keppler B. K.; Timerbaev A. R.; Bolshov M. A. Inductively coupled plasma mass spectrometry for metallodrug development: albumin binding and serum distribution of cytotoxic cis- and trans-isomeric platinum(II) complexes. J. Inorg. Biochem. 2014, 137, 40–45. 10.1016/j.jinorgbio.2014.04.008. [DOI] [PubMed] [Google Scholar]
- Harper B. W.; Friedman-Ezra A.; Sirota R.; Petruzzella E.; Aldrich-Wright J. R.; Gibson D. Probing the Interactions of Cytotoxic [Pt(1S,2S-DACH)(5,6-dimethyl-1,10-phenanthroline)] and Its Pt(IV) Derivatives with Human Serum. ChemMedChem 2017, 12 (7), 510–519. 10.1002/cmdc.201700092. [DOI] [PubMed] [Google Scholar]
- Domotor O.; Hartinger C. G.; Bytzek A. K.; Kiss T.; Keppler B. K.; Enyedy E. A. Characterization of the binding sites of the anticancer ruthenium(III) complexes KP1019 and KP1339 on human serum albumin via competition studies. J. Biol. Inorg. Chem. 2013, 18 (1), 9–17. 10.1007/s00775-012-0944-6. [DOI] [PubMed] [Google Scholar]
- Muller P. A.; Vousden K. H. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell 2014, 25 (3), 304–317. 10.1016/j.ccr.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcel V.; Catez F.; Diaz J. J. p53, a translational regulator: contribution to its tumour-suppressor activity. Oncogene 2015, 34, 5513–5523. 10.1038/onc.2015.25. [DOI] [PubMed] [Google Scholar]
- Elias J.; Dimitrio L.; Clairambault J.; Natalini R. The p53 protein and its molecular network: modelling a missing link between DNA damage and cell fate. Biochim. Biophys. Acta, Proteins Proteomics 2014, 1844 (1 Pt B), 232–247. 10.1016/j.bbapap.2013.09.019. [DOI] [PubMed] [Google Scholar]
- Woods D.; Turchi J. J. Chemotherapy induced DNA damage response: convergence of drugs and pathways. Cancer Biol. Ther. 2013, 14 (5), 379–389. 10.4161/cbt.23761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brozovic A.; Osmak M. Activation of mitogen-activated protein kinases by cisplatin and their role in cisplatin-resistance. Cancer Lett. 2007, 251 (1), 1–16. 10.1016/j.canlet.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Woessmann W.; Chen X.; Borkhardt A. Ras-mediated activation of ERK by cisplatin induces cell death independently of p53 in osteosarcoma and neuroblastoma cell lines. Cancer Chemother. Pharmacol. 2002, 50 (5), 397–404. 10.1007/s00280-002-0502-y. [DOI] [PubMed] [Google Scholar]
- Mansouri A.; Ridgway L. D.; Korapati A. L.; Zhang Q.; Tian L.; Wang Y.; Siddik Z. H.; Mills G. B.; Claret F. X. Sustained activation of JNK/p38 MAPK pathways in response to cisplatin leads to Fas ligand induction and cell death in ovarian carcinoma cells. J. Biol. Chem. 2003, 278 (21), 19245–19256. 10.1074/jbc.M208134200. [DOI] [PubMed] [Google Scholar]
- Peidis P.; Papadakis A. I.; Muaddi H.; Richard S.; Koromilas A. E. Doxorubicin bypasses the cytoprotective effects of eIF2α phosphorylation and promotes PKR-mediated cell death. Cell Death Differ. 2011, 18 (1), 145–154. 10.1038/cdd.2010.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persons D. L.; Yazlovitskaya E. M.; Cui W.; Pelling J. C. Cisplatin-induced Activation of Mitogen-activated Protein Kinases in Ovarian Carcinoma Cells: Inhibition of Extracellular Signal-regulated Kinase Activity Increases Sensitivity to Cisplatin. Clin. Cancer Res. 1999, 5 (5), 1007–1014. [PubMed] [Google Scholar]
- Cuello-Garibo J. A.; Meijer M. S.; Bonnet S. To cage or to be caged? The cytotoxic species in ruthenium-based photoactivated chemotherapy is not always the metal. Chem. Commun. (Cambridge, U. K.) 2017, 53 (50), 6768–6771. 10.1039/C7CC03469E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azar D. F.; Audi H.; Farhat S.; El-Sibai M.; Abi-Habib R. J.; Khnayzer R. S. Phototoxicity of strained Ru(II) complexes: is it the metal complex or the dissociating ligand?. Dalton Trans 2017, 46 (35), 11529–11532. 10.1039/C7DT02255G. [DOI] [PubMed] [Google Scholar]
- Arnesano F.; Banci L.; Bertini I.; Felli I. C.; Losacco M.; Natile G. Probing the interaction of cisplatin with the human copper chaperone Atox1 by solution and in-cell NMR spectroscopy. J. Am. Chem. Soc. 2011, 133 (45), 18361–18369. 10.1021/ja207346p. [DOI] [PubMed] [Google Scholar]
- Luchinat E.; Banci L. A Unique Tool for Cellular Structural Biology: In-cell NMR. J. Biol. Chem. 2016, 291 (8), 3776–3784. 10.1074/jbc.R115.643247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonejuie P.; Burkart M.; Pogliano K.; Pogliano J. Bacterial cytological profiling rapidly identifies the cellular pathways targeted by antibacterial molecules. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (40), 16169–16174. 10.1073/pnas.1311066110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quach D. T.; Sakoulas G.; Nizet V.; Pogliano J.; Pogliano K. Bacterial Cytological Profiling (BCP) as a Rapid and Accurate Antimicrobial Susceptibility Testing Method for Staphylococcus aureus. EBioMedicine 2016, 4, 95–103. 10.1016/j.ebiom.2016.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson M. Z.; Wang R.; Gitai Z.; Seyedsayamdost M. R. Mode of action and resistance studies unveil new roles for tropodithietic acid as an anticancer agent and the gamma-glutamyl cycle as a proton sink. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (6), 1630–1635. 10.1073/pnas.1518034113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.