

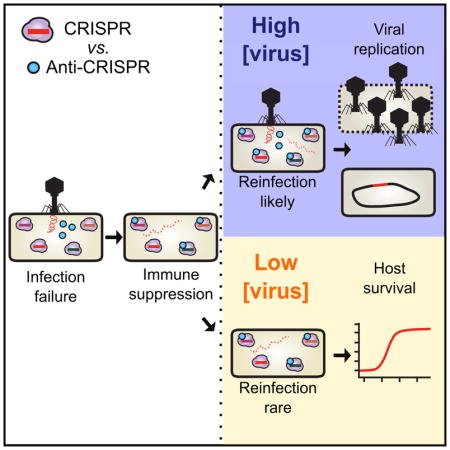
SUMMARY
Bacteria utilize CRISPR-Cas adaptive immune systems for protection from bacteriophages (phages), and some phages produce anti-CRISPR (Acr) proteins that inhibit immune function. Despite thorough mechanistic and structural information for some Acr proteins, how they are deployed and utilized by a phage during infection is unknown. Here, we show that Acr production does not guarantee phage replication when faced with CRISPR-Cas immunity, but instead, infections fail when phage population numbers fall below a critical threshold. Infections succeed only if a sufficient Acr dose is contributed to a single cell by multiple phage genomes. The production of Acr proteins by phage genomes that fail to replicate leave the cell immunosuppressed, which predisposes the cell for successful infection by other phages in the population. This altruistic mechanism for CRISPR-Cas inhibition demonstrates inter-virus cooperation that may also manifest in other host-parasite interactions.
In Brief
A critical threshold level of phage anti-CRISPR proteins is required for CRISPR resistance and infection of immune hosts, suggesting that anti-CRISPR systems may have evolved under conditions where the likelihood of multiple or sequential infection is high.
INTRODUCTION
Bacteria and the viruses that infect them (phages) are engaged in an ancient evolutionary arms race, which has resulted in the emergence of a diversity of CRISPR-Cas (clustered regularly interspaced short palindromic repeats and CRISPR-associated genes) adaptive immune systems (Koonin et al., 2017). CRISPR-Cas immunity is powered by the acquisition of small fragments of phage genomes into the bacterial CRISPR array, the subsequent transcription and processing of these arrays to generate small CRISPR RNAs, and the RNA-guided destruction of the phage genome (Barrangou et al., 2007; Brouns et al., 2008; Garneau et al., 2010; Levy et al., 2015). The destruction of foreign DNA by CRISPR-Cas has been shown to prevent the acquisition of plasmids, DNA from the environment, phage lytic replication, and prophage integration (Barrangou et al., 2007; Bikard et al., 2012; Cady et al., 2012; Edgar and Qimron, 2010; Garneau et al., 2010; Marraffini and Sontheimer, 2008). In bacterial populations, these systems provide a fitness advantage to their host microbe when phage are present in the environment (van Houte et al., 2016; Westra et al., 2015).
To combat the potent action of RNA-guided CRISPR-Cas nucleases, phages have developed inhibitor proteins called anti-CRISPRs (Acrs). Acr proteins have been discovered in phages, prophages, mobile islands, and core genomes across many distinct bacteria and archaea (Borges et al., 2017; He et al., 2018; Pawluk et al., 2017). Specific Acr proteins that inhibit Type I-F, I-E, and I-D CRISPR-Cas3 systems have been identified (Bondy-Denomy et al., 2013; He et al., 2018; Pawluk et al., 2014; 2016b), as well as proteins that inhibit Type II-A and II-C CRISPR-Cas9 systems (Hynes et al., 2017; Pawluk et al., 2016a; Rauch et al., 2017). Phylogenetic studies indicate that these proteins are likely ubiquitous in coevolving populations of bacteria and phages (Pawluk et al., 2017) and provide a significant replicative advantage to phages in the presence of CRISPR-Cas immunity (van Houte et al., 2016).
Anti-CRISPRs were first identified in phages that neutralize the Pseudomonas aeruginosa Type I-F system (anti-CRISPR type I-F, AcrIF1-5)(Bondy-Denomy et al., 2013), and five more I-F anti-CRISPRs (AcrIF6-10) were subsequently identified in various mobile genetic elements (Pawluk et al., 2016b). The I-F Csy surveillance complex (also called I-F Cascade) is comprised of an unequal stoichiometry of four proteins (Csy1-4) that assemble with a 60 nt CRISPR RNA (crRNA) guide (Chowdhury et al., 2017; Guo et al., 2017; Haurwitz et al., 2010; Peng et al., 2017; Przybilski et al., 2011; Richter et al., 2012; Wiedenheft et al., 2011). The Csy complex locates and binds foreign dsDNA targets complementary to the crRNA, then recruits a trans-acting nuclease/helicase protein called Cas2/3 to degrade the target (Rollins et al., 2017; Wang et al., 2016a; 2016b). Anti-CRISPR proteins function by interacting directly with the Csy complex and inhibiting DNA binding, or bind to Cas2/3 and prevent nuclease-mediated degradation (Bondy-Denomy et al., 2015). The structures of Type I-F Acr proteins AcrIF1, AcrIF2, AcrIF3, and AcrIF10 have been solved in complex with their target proteins, revealing mechanistically distinct inhibitors that bind tightly to their targets (Chowdhury et al., 2017; Guo et al., 2017; Maxwell et al., 2016; Peng et al., 2017; Wang et al., 2016a; 2016b). Together with the recent identification and characterization of proteins that inhibit Cas9, all characterized Acr proteins block phage DNA binding or cleavage (Dong et al., 2017; Harrington et al., 2017; Pawluk et al., 2016a; Rauch et al., 2017; Shin et al., 2017; Yang and Patel, 2017).
All AcrIF proteins are robust inhibitors of CRISPR-Cas activity when expressed from high copy plasmids prior to phage challenge, however this method of CRISPR-inactivation is not reflective of anti-CRISPR deployment by phages in nature. When phage DNA cleavage has been assessed in vivo, it occurs in as little as 2 minutes (Garneau et al., 2010), suggesting that phage genome degradation may outpace de novo Acr synthesis and function. We therefore hypothesized that successful inhibition of CRISPR-Cas immunity by Acr proteins during phage infection would be challenging, as all components of the P. aeruginosa immune system are expressed prior to phage infection (Bondy-Denomy et al., 2013; Cady et al., 2012).
Here, we demonstrate that complete CRISPR-Cas inactivation by a phage-produced Acr protein is challenging, and that the concentration of Acr proteins required to inactivate CRISPR-Cas is contributed by multiple phage genomes. While initial phage infections fail due to rapid genome degradation by the CRISPR-Cas system, Acr deposition prior to phage destruction causes cellular immunosuppression. If the cell is re-infected, Acr proteins from the initial phage infection enhance the likelihood of subsequent phage replication. We propose that pathogens can contribute to the “remodeling” of their host cell via rapid protein production, even if the initial infecting genomes are cleared, opening the door for their clones.
RESULTS
Anti-CRISPR proteins are imperfect CRISPR-Cas inhibitors
We utilized the diversity of acr genes encoded by phages infecting P. aeruginosa to determine the mechanism of CRISPR-Cas neutralization during infection. Five natural phages, each encoding a single acrIF gene, were selected to represent acrIF1-IF4 and acrIF7 (acrIF5 does not exist as the sole acrIF gene on any phage, acrIF6, F8–F10 are not encoded by this phage family). Three of the five phages exhibited reduced efficiency of plaquing (EOP) on P. aeruginosa strain PA14, which possesses a naturally active Type I-F CRISPR-Cas system with 1 or 2 spacers targeting these phages (Figure 1A, WT:pEmpty normalized to plaquing on ΔCRISPR). Overexpression of a targeting crRNA (WT:pSp1) exacerbated anti-CRISPR inefficiency, limiting the replication of all phages by at least one order of magnitude. This suggests that Acr proteins are unable to fully protect their associated phage genome.
Figure 1. Anti-CRISPRs are imperfect CRISPR-Cas inhibitors.

(A) Efficiency of plaquing (EOP) of 5 related phages bearing distinct acrIF genes (JBD30acrIF1, MP29acrIF2, JBD88aacrIF3, JBD24acrIF4, LPB1acrIF7) on Pseudomonas aeruginosa strain PA14. Plaque forming units (PFUs) were quantified on wild-type PA14 with 1–2 natural targeting spacers (WT + pEmpty) or on PA14 overexpressing 1 targeting spacer (WT + pSp1), then normalized to the number of PFUs measured on a non-targeting PA14 derivative (0sp). Data are represented as the mean of 3 biological replicates +/− SD.
(B) EOP of isogenic DMS3macr phages with acrIF1-7 or acrIE3 in the DMS3m acr locus. EOP was calculated as PFU counts measured on WT PA14 with 1 targeting spacer (1sp) or a laboratory evolved PA14 derivative with 5 targeting spacers (5sp) normalized to PFU counts measured on non-targeting PA14 (0sp). Data are represented as the mean of 3 biological replicates +/− SD. ND, not detectable.
(C) Plot of association (ka) and dissociation (kd) rates for AcrIF1 (data adapted from Chowdhury et al. 2017) and AcrIF4 binding the PA14 Csy complex. AcrIF1 rate constants: ka = 5 × 104 (1/Ms), kd = 2 × 10−7 (1/s), KD = 3 × 10−11 M. AcrIF4 rate constants: ka = 1 × 103 (1/Ms), kd = 5 × 10−4 (1/s), KD = 4 × 10−7 (M). See Figure S2 for AcrIF4 SPR sensogram.
To assess anti-CRISPR strength directly, an isogenic phage panel was generated by replacing the acrIE3 gene in the anti-CRISPR locus of phage DMS3m with single acrIF genes F1–F7 (DMS3macrIF1-DMS3macrIF7). acrIF1-F5, and acrIF7 are all encoded by DMS3m-like phages in syntenic anti-CRISPR loci, while acrIF6 was discovered in a distinct type of P. aeruginosa phage. WT PA14 (1 spacer targeting DMS3m, “1sp”) and a PA14 derivative which acquired 4 more spacers against DMS3m through laboratory evolution (“5sp”) were challenged with this panel of recombinant phages. For phages encoding acrIF1, F2, F3, F6 or F7, >90% of phage in the population failed to replicate (EOP=10−1) when faced with 5 targeting spacers (Figure 1B). acrIF4 and acrIF5 were very weak, with 99.0–99.99% of phages failing to replicate, depending on the CRISPR spacer content. Phages must rely on acrIF genes when infecting the 5sp strain, as the acrIE3-encoding phage is unable to escape CRISPR targeting via protospacer mutation alone. We conclude that phages encoding anti-CRISPRs remain sensitive to CRISPR-Cas immunity, suggesting that anti-CRISPR deployment and action is an imperfect process.
The observations above identified groups of “strong” and “weak” Acr proteins. We selected one representative from each group for downstream experiments, and a third Acr that does not target the I-F CRISPR system (i.e. AcrIE3), as a negative control. AcrIF1 was selected as a model strong inhibitor, as its mechanism and binding affinity are known (Csy complex binding, KD = 3 × 10−11 M (Bondy-Denomy et al., 2015; Chowdhury et al., 2017)). In contrast, AcrIF4 is a weak inhibitor that also binds the Csy complex (Bondy-Denomy et al., 2015), but with a significantly slower on-rate and faster off-rate compared to AcrIF1 (Figure 1C, Figure S1).
Lytic replication requires a critical Acr protein concentration
We next assessed the survival of bacterial populations when infected with phages that rely on apparently imperfect Acr proteins for survival. To assay the lytic cycle only, phages were prevented from entering lysogeny by knocking out the C repressor gene (gp1) in DMS3macrIF1, DMS3macrIF4, and DMS3macrIE3. The virulent (vir) phages were used to infect the 5sp strain in liquid culture, and bacterial growth measured. Given that AcrIF4 has a KD for its binding partner that is >4 orders of magnitude weaker than AcrIF1 for its binding partner, we reasoned that a higher concentration of phages encoding AcrIF4 may be required to inactivate CRISPR-Cas function. In the presence of CRISPR-Cas immunity, bacterial death only occurred at multiplicities of infection (MOI, input plaque forming units per colony forming unit) greater than 0.02 (≥105 PFU) for acrIF1 (Figure 2A) and greater than an MOI of 2.0 (≥107 PFU) for acrIF4 (Figure 2B). Phage replication observed here was due to Acr function, and not a result of phage escape mutations, as output phages remained as sensitive to CRISPR-Cas immunity as the input phage population (Figure S2A–C). Furthermore, the phage encoding acrIE3 had no impact on bacterial survival when faced with CRISPR immunity (Figure 2C), while in the absence of CRISPR, phages at all concentrations cleared bacterial cultures (Figure 2D–2F). These data demonstrate that Acr-mediated CRISPR-Cas inactivation requires a critical phage concentration that is inversely proportional to Acr strength.
Figure 2. Anti-CRISPR success requires cooperative infections during lytic growth.

(A–F) 12 hour growth curves of P. aeruginosa strain PA14 with 5 targeting spacers (+CRISPR, panels A–C) or no CRISPR-Cas function (ΔCRISPR, D–F) infected with virulent variants of DMS3macrIF1, DMS3macrIF4, or DMS3macrIE3 at multiplicities of infection (MOI) increasing in 10-fold steps from 2×10−5 to 2×101 (rainbow colors) or uninfected (black). Colors correspond to the MOI legend and growth curves. OD600nm is represented as the mean of 3 biological replicates +/− SD (vertical lines). ND, not detectable.
(G–I) Replication of virulent DMS3macr phages (acceptor phage) in the presence of 106 PFU (MOI 0.2) hybrid phage (donor) in PA14 with 5 targeting spacers (5sp) expressing the JBD30 C repressor. Phages were harvested after 24 hours of co-culture and DMS3macr phage PFUs were quantified on PA14 0sp expressing the JBD30 C repressor. Phage output is represented as the mean of 3 biological replicates +/− SD. ND, not detectable.
(J) Schematic of the experimental design in G–I, where a high MOI of non-replicative “donor” phages is used to rescue a low MOI infection of wild-type “acceptor” phages.
We hypothesized that the phage concentration dependence that dictates Acr success is achieved by the contribution of Acr proteins from multiple phage genomes in a single cell, which is not achieved at low MOIs. To this end, we rendered a subset of phages in the population non-replicative Acr donors to test if Acr donation alone is sufficient to rescue a failing (i.e. low MOI) infection. The C repressor gene (gp1) and surrounding immunity region from a DMS3m-like phage (JBD30) was introduced into DMS3m phages, generating a hybrid phage. The replication of the hybrid phage could be specifically prevented by overexpression of the JBD30 C repressor (gp1, Figure S3A), a protein that does not interfere with DMS3m phage with wild-type immunity regions (Figure S3B). This enabled the mixing of two independent phage populations: a sacrificial Acr “donor” that cannot replicate and a wild-type (replication competent) Acr “acceptor”.
In the presence of donor phages encoding AcrIF1 (106 PFU, MOI = 0.2), we observed a striking contribution to CRISPR-Cas neutralization, despite the inability of this phage to replicate (Figure S3C). The acceptor phages DMS3macrIF1 (Figure 2G) and DMS3macrIF4 (Figure 2H) replicated robustly from input MOIs that are unsuccessful in the absence of an AcrIF1 donor phage (Figure 2G, 2H, see “IE3” and “buffer”). The presence of AcrIF1 donor phages had a mildly protective effect on the DMS3macrIE3 acceptor phage (Figure 2I), though it was not able to reach high titers. Notably, the acceptor phage output from these experiments remain as sensitive to CRISPR-Cas targeting by the 5sp host as the original input phages, demonstrating escape mutations do not arise under these conditions (Figure S2D–G). Additionally, any potential lysogens formed by the donor phage in this experiment would not have amplified the replicating phage, as these lysogens are resistant to superinfection (Figure S3D). These data demonstrate that the determinant of phage replicative success is the concentration of Acr proteins reached in single cells, which is achievable by Acr production from independent phage genomes (Figure 2J).
Lysogeny requires Acr proteins contributed by transient intracellular genomes
All phages encoding Acr proteins that infect P. aeruginosa are naturally temperate, and can form lysogens by integrating into the bacterial genome. We therefore measured the impact of CRISPR and Acr proteins on lysogeny establishment during a single round of infection. While previous experiments examined cumulative phage replication in the lytic cycle over many hours, assaying lysogen formation over a short time frame is ideal for understanding the initial events that determine phage genome survival or cleavage. Additionally, lysogeny provides a direct readout for phage genome survival (i.e. a cell with an integrated prophage), while in lytic replication, phage survival leads to a dead cell that cannot be recovered. For these experiments, we selected the weak AcrIF4 protein as it provided the largest dynamic range of inefficiency in a single round of infection.
We generated derivatives of DMS3macrIF4 and DMS3macrIE3 marked with a gentamicin resistance cassette at the end of the genome, replacing a nonessential gene, gp52. This allowed the independent titration of two distinct replication-competent phage populations and the selection and analysis of stable lysogens after the experiment. These phages were used to infect ΔCRISPR cells (0sp) for a time span less than a single round of infection (50 minutes, data not shown), and the number of gentamicin resistant lysogens was assessed. In the absence of CRISPR selection, a linear increase in the number of lysogens with increasing MOI was observed, over ~4 orders of magnitude (Figure 3A–3B, circles). In the presence of spacers targeting DMS3m (5sp), CRISPR immunity reduced the number of lysogen forming units (LFUs) for the weak acr phage DMS3macrIF4 (Figure 3A, triangles). DMS3macrIF4 demonstrated concentration dependence for successful lysogeny, with efficiency of lysogeny (EOL) values below or at the limit of detection for lower MOIs, increasing to EOL = 0.01 at higher MOIs (Figure 3C). Phage DMS3macrIE3 formed no lysogens at all input concentrations tested, demonstrating that Acr-mediated immune suppression is required to establish lysogeny (Figure 3B, 3D).
Figure 3. Immunosuppression facilitates acquisition of a marked prophage.

(A,B) Acquisition of a marked DMS3macrIF4 gp52::gent or DMS3macrIE3 gp52::gent prophage by PA14 with 0 spacers (0sp, circles) or 5 targeting spacers (5sp, triangles). This experiment was performed in biological triplicate, and individual replicate values are displayed. LoD, limit of detection.
(C,D) Efficiency of lysogeny (EOL) of DMS3macrIF4 gp52::gent and DMS3macrIE3 gp52::gent in the presence of CRISPR targeting. EOL was calculated by dividing the output lysogens forming units (LFUs) from the strain with 5 targeting spacers (5sp) to the number of LFUs in PA14 with 0 targeting spacers (0sp). Data are represented as the mean of 3 biological replicates +/− SD. ND, not detectable.
(E,F) EOL of 103 LFUs of DMS3macrIF4 gp52::gent and DMS3macrIE3 gp52::gent in the presence of 107 PFU of the indicated DMS3macr phage. Data are represented as the mean of 3 biological replicates +/− SD. ND, not detectable. See Figure S4 for analysis of lysogen prophage content.
(G) Schematic of the experimental design in E–F, where a high MOI of wild-type “donor” phages is used to rescue a low MOI infection of marked “acceptor” phages.
We hypothesized that phage concentration dependence for CRISPR neutralization during lysogeny could also be explained by phage cooperation, and that below-threshold concentrations of DMS3macrIF4 gp52::gent could be rescued by the addition of wild-type (replication competent) Acr donor phages in trans. To test this hypothesis, we infected the 5sp strain with a mixture of 103 LFU marked acceptor phage and 107 PFU of unmarked Acr donor phages, and measured the EOL of the acceptor phage. The EOL of the acceptor phage DMS3macrIF4 gp52::gent increased by 2 orders of magnitude with Acr donor phage DMS3macrIF1, and by 1 order of magnitude with the DMS3macrIF4 donor phage (Figure 3E). The addition of Acr donor phages DMS3macrIE3, or an escaper phage DMS3macrIE3* had no effect on the EOL of the acceptor phage, demonstrating that the donor phage must be an Acr-producer. A marked acceptor phage lacking an acrIF gene (DMS3macrIE3 gp52::gent) only established rare lysogens in the presence of the AcrIF1 donor phage (Figure 3F).
To determine the specific mechanism of anti-CRISPR donation leading to survival of the acceptor phages, we used the resulting lysogens as a genetic record of infection success for both the marked acceptor phage and the unmarked donor phage (Figure 3G). This family of Mu-like phages integrates randomly into the host genome, allowing for the formation of strains with multiple prophages (Bondy-Denomy et al., 2016). We assayed the lysogens resulting from the experiment described above (Figures 3E, 3F) for the presence of the donor prophage genome in addition to the acceptor prophage. All resulting DMS3macrIF4 gp52::gent lysogens (n=48) possessed only the marked acceptor prophage, with none possessing the Acr donor prophage (Figure S4A,B). Furthermore, the DMS3macrIF4 gp52::gent acceptor phages induced from the lysogens that formed only in the presence of Acr donor phages remained sensitive to CRISPR-Cas targeting, indicating these lysogens did not arise due to phage protospacer mutation (Figure S4B). Double lysogens only emerged when the marked acrIE3 phage was used as an acceptor phage, which would be incapable of maintaining lysogeny alone due to CRISPR-Cas self-targeting (Figure 3F). These results demonstrate that the transient presence (i.e. no lysogeny) of an Acr donor phage genome in a cell was sufficient to generate enough Acr protein to protect the marked acceptor phage, leading to the establishment of lysogens that would not exist if not for the Acr donor (Figure 3E, compare “Buffer” to “IF1”). Collectively, these data demonstrate that the production of Acr proteins from a phage genome prior to its cleavage generates an immunosuppressed cell that can be successfully parasitized by another phage upon re- or co-infection(s).
Cas9 inhibitors require bacteriophage cooperation
The intrinsic inefficiency of stoichiometric inhibitors is likely due to the requirement for the rapid synthesis of a high concentration of inhibitors before phage genome cleavage. To determine whether this model generally applies to other stoichiometric inhibitors of bacterial immunity, we engineered a P. aeruginosa strain to express the Cas9 protein from Streptococcus pyogenes (SpyCas9) and a DMS3m phage to express a previously identified Cas9 inhibitor, AcrIIA4 (Dong et al., 2017; Rauch et al., 2017). With this entirely heterologous system, we again observed inefficiency for a phage relying on an Acr protein. Spot-titration of phage lysates on a strain expressing a single guide RNA (sgRNA) targeting DMS3m decreased the titer of DMS3macrIE3 by >3 orders of magnitude, while DMS3macrIIA4 was protected (Figure 4A). However, EOP quantification again revealed that relying on an Acr protein for replication is imperfect, with an EOP = 0.4 (Figure 4B). In lytic replication infection experiments, DMS3macrIIA4 displayed concentration-dependent bacterial lysis in the presence of CRISPR targeting (Figure 4C), while DMS3macrIE3 did not affect bacterial growth (Figure 4D). The replication of DMS3macrIIA4 was not due to protospacer mutation leading to phage escape because the output phage population displayed the same EOP as the input (Figure S2H–I). In the absence of CRISPR-Cas targeting, however, both phages killed their hosts at all phage concentrations tested (Figure 4E, 4F).
Figure 4. Cas9 anti-CRISPR AcrIIA4 requires cooperative infection to neutralize Type II-A CRISPR immunity.

(A) 10-fold serial dilutions of DMS3macrIE3 or DMS3macrIIA4 plated on a lawn of Pseudomonas aeruginosa strain PAO1 expressing Streptococcus pyogenes Type II-A Cas9 (PAO1::SpyCas9) and single guide RNA (+ sgRNA) or non-targeting control (+ vector).
(B) Efficiency of plaquing of DMS3macrIIA4 and DMS3macrIE3 was calculated by normalizing PFU counts on a targeting strain of PAO1::SpyCas9 (+sgRNA) to PFU counts on a non-targeting strain of PAO1::SpyCas9 (+vector). Data are represented as the mean of 3 biological replicates +/− SD. ND, not detectable.
(C–F) 12 hour growth curves of PAO1::SpyCas9 expressing a targeting sgRNA (+ sgRNA, panels C–D) or a non-targeting vector control (+vector, E–F) that were infected with virulent DMS3macrIIA4 or DMS3macrIE3 at multiplicities of infection (MOI, rainbow colors) from 2×10−5 to 2×10−2. Growth curves of uninfected cells are shown in black. OD600nm values are represented as the mean of 3 biological replicates +/− SD (vertical lines).
(G–H) Replication of virulent DMS3macr phages (acceptor phage) in the presence of 107 PFU (MOI 2) hybrid phage (donor) in PAO1::SpyCas9 + sgRNA expressing the JBD30 C repressor. Phages were harvested after 24 hours and DMS3macr phage PFUs quantified on PAO1::SpyCas9 + vector expressing the JBD30 C repressor. Phage output is represented as the mean of 3 biological replicates +/− SD. ND, not detectable.
(I) Schematic of the experimental design in G–H, where a high MOI of non-replicative “donor” phages is used to rescue a low MOI infection of wild-type “acceptor” phages.
To determine whether this concentration dependence for Cas9 inhibition was also a result of insufficient intracellular Acr dose, a non-replicative hybrid DMS3macrIIA4 phage was generated and used as an Acr donor during infection. Indeed, increased delivery of AcrIIA4 to cells enhanced replication of the wild-type DMS3macrIIA4 acceptor phage by 4 orders of magnitude (Figure 4G), demonstrating phage cooperation neutralizes CRISPR-Cas9. AcrIIA4 donation was able to slightly rescue an acceptor phage without a II-A Acr, DMS3macrIE3 (Figure 4H), however this phage was unable to replicate to high titers. Furthermore, the effect of an AcrIIA4 donor rescuing either DMS3macrIIA4 or DMS3macrIE3 was not due to mutational escape of the acceptor phage (Figure S2J–L), demonstrating these phages had survived solely due to the immunosuppressive effect of AcrIIA4 donation (Figure 4I). Collectively, these data demonstrate that phage-phage cooperation via cellular immunosuppression is a broadly useful strategy to overcome bacterial immunity.
DISCUSSION
Here we demonstrate that the necessary intracellular concentration of an anti-CRISPR protein to achieve inactivation of CRISPR-Cas immunity depends on the relative strengths of both the inhibitor and CRISPR immunity, which dictates the number of infecting viruses required in the population. We conclude that a single cell can become immunosuppressed by Acr protein contributions from independent infection events. In the absence of viral replication, these infection events serve to contribute to the inactivation of cellular immunity, thus enhancing the probability of successful infection events in the future. We expect that cooperation of this sort is necessary when the immune process acts rapidly and irreversibly on the infecting viral genome, as CRISPR-Cas immunity does.
Anti-CRISPR deployment and successful CRISPR-Cas inactivation requires a critical concentration of phage in the population to allow replication in the lytic or lysogenic cycle. We used three distinct genetic strategies to monitor phage-phage cooperation within an otherwise clonal population, allowing the independent titration and tracking of isogenic phages: i) non-replicative Acr donor phages, ii) marked and unmarked phages to follow the fate of only one phage, and iii) the prophage status of lysogens, as a genetic record of phage success. In the presence of non-replicative Acr donor phages, we observed the successful lytic amplification of a low-dose of wild-type phages, otherwise destined for replication failure (Figure 2G, 2H). This provided an explanation for the observed phage inefficiencies during plaque assays (Figure 1A, 1B) and population concentration thresholds in liquid infections (Figure 2A, 2B). Next, the acquisition of a marked prophage was monitored in the presence of wild-type Acr-donor phages. AcrIF proteins provided in trans caused cellular immunosuppression, enabling the formation of lysogens that were not established in their absence (Figure 3E, 3F). The presence of only a single, marked prophage in the bacterial genome demonstrates that the donor phage neither entered the lytic cycle (this would kill the cell), nor lysogenized (prophage would be integrated), but had been present in the cell transiently.
The key result here is the observation that phages can remodel their host cell, even in the absence of a replicating or integrated genome. It has long been known that integrated prophages modulate host phenotypes via gene expression, including superinfection exclusion, toxin production, and the production of Acr proteins (Bondy-Denomy et al., 2013; Bondy-Denomy and Davidson, 2014; Bondy-Denomy et al., 2016; Waldor and Mekalanos, 1996; Weigle and Delbruck, 1951). Furthermore, the Imm protein produced by the lytic phage T4 prevents other phages in the environment from infecting the cell that one phage is currently replicating within (Lu and Henning, 1989). This has been attributed to preventing sequential infections and the disruption of the carefully timed phage replication cycle. In contrast to these examples, we propose a new model of phage-induced host remodeling, whereby a transient, unsuccessful infection produces proteins that inactivate defense, enabling future infections.
Consistent with our observations of viral cooperation, beneficial virus-virus interactions in both eukaryotic and prokaryotic systems have been previously observed. Broadly, these phenomena can be separated into 2 categories: i) cooperative interactions between distinct viral genotypes and ii) group behaviors manifested in clonal viral populations: i) Similar to Acr proteins functioning as a public good, genetically distinct viruses can share protein products during coinfection (Xue et al., 2016), even bypassing deleterious mutations in cis via functional complementation in trans (Aguilera et al., 2017; Cicin-Sain et al., 2005; Vignuzzi et al., 2005). Additionally, the direct exchange of viral genetic material can also increase viral fitness. The mosaic nature of phage genomes (Botstein, 1980; Hatfull and Hendrix, 2011; Hendrix et al., 1999) and the high abundance of chimeric viruses in nature highlights the importance of coinfection and genetic exchange in viral evolution (Diemer and Stedman, 2012; Krupovic et al., 2015; Roux et al., 2013). In fact, CRISPR-targeted phages can evade CRISPR-Cas immunity via homologous recombination with genetically distinct phages, disrupting protospacers (Andersson and Banfield, 2008; Paez-Espino et al., 2015). ii) Group behaviors manifesting in clonal populations of virus is less frequently reported, likely because they leave no genetic signature. However, the lambda lytic/lysogeny switch is a famous example of phage group behavior: during lambda phage co-infection, high concentrations of the CII protein product derived from multiple infecting clones drives the cooperative decision to enter lysogeny (Kourilsky and Knapp, 1974; Trinh et al., 2017; Zeng et al., 2010). In more recent literature, the discovery of the widespread arbitrium system as the first phage-phage communication mechanism demonstrates the potential of phages to act as a group and manifest cooperative behaviors (Erez et al., 2017). The immunosuppressive mechanism of anti-CRISPR function further exemplifies cooperation within clonal populations of phages, which may occur more often than was previously appreciated.
The distinct aspect here is the altruistic nature of immunosuppression: to neutralize CRISPR-Cas immunity, many infections must fail such that a few can succeed. To our knowledge this is the first documented example of true viral altruism, which is evolutionary beneficial only through kin selection.
A distinct, but notable observation from this work is that not all Acr proteins operate at equivalent strengths. However, encoding even a weak inhibitor (e.g. AcrIF4) still provides a significant advantage to the phage, compared to lacking them entirely (Figure 1B). We show that AcrIF4 binds the Csy complex with affinities that are orders of magnitude weaker (Figure 1C) than Acr proteins like AcrIF1 and AcrIF2 (Chowdhury et al., 2017). We selected AcrIF1 as a model strong Acr protein because of its comparable mechanism of action to AcrIF4 (i.e. Csy complex binding), and consider it representative of other strong Acr proteins (AcrIF2, F3, F6, F7), based on EOP data. Going forward, we speculate that the strongest Acr proteins would be enzymatic in nature, allowing rapid and efficient inactivation of CRISPR complexes in a sub-stoichiometric manner, although no such Acr mechanism has been discovered. While not an enzyme, the recent demonstration of the AcrIIC3 protein inactivating two Cas9 proteins at the same time would likely be a more efficient path towards CRISPR neutralization (Harrington et al., 2017). It is also interesting to consider individual bacterial strains that encode multiple CRIPSR-Cas system subtypes (Carte et al., 2014; Patterson et al., 2016; van Belkum et al., 2015), all which must be neutralized in order for a targeted phage to replicate. A dual-activity inhibitor is likely at a specific disadvantage in this scenario, as one protein would be tasked with inhibiting Cas proteins produced by two different systems. This may in part explain why DMS3m-like Pseudomonas phages often encode dedicated Type I-E and Type I-F Acrs in the same Acr locus (Bondy-Denomy et al., 2013; Pawluk et al., 2014), instead of employing dual I-F and I-E inhibitors such as AcrIF6 (Pawluk et al., 2016b). Although encoding multiple Acrs comes with the burden of more genetic cargo in a phage’s genome, this strategy could be advantageous on a biochemical level when infecting a bacterial strain with multiple CRISPR-Cas subtypes.
The challenge of neutralizing a pre-expressed CRISPR-Cas system likely explains why stoichiometric inhibitors like Acr proteins are imperfect, and phages relying on them are partially targeted by CRISPR. The sacrificial, population-level aspect of CRISPR inhibition is reminiscent of the manifestations of CRISPR adaptation in populations of bacterial cells. The majority of infected naïve host cells die, before a clone with a new spacer emerges (Barrangou et al., 2007; Hynes et al., 2014). In the case of anti-immunity, many phages die in order to inhibit CRISPR on a single cell level, and this must happen at a sufficient frequency within a community for phage to prevail. We suspect that this mechanism of cellular immunosuppression and inter-parasite cooperation may have parallels in other host-pathogen interactions, where concentration dependence manifests at predictable levels due the strengths of immune and anti-immune processes.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Please direct any requests for further information or reagents to the Lead Contact, Joseph Bondy-Denomy (joseph.bondy-denomy@ucsf.edu), Department of Microbiology and Immunology, University of California, San Francisco.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Microbes
Pseudomonas aeruginosa strains (UCBPP-PA14 and PAO1) and Escherichia coli strains (DH5α, for plasmid maintenance) were cultured on lysogeny broth (LB) agar or liquid media at 37 °C. LB was supplemented with gentamicin (50 μg/mL for P. aeruginosa, 30 μg/mL for E. coli) to maintain the pHERD30T plasmid or carbenicillin (250 μg/mL for P. aeruginosa, 100 μg/mL for E. coli) to maintain pHERD20T or pMMB67HE. To maintain pHERD30T and pMMB67HE in the same strain of P. aeruginosa, double selection of 30 μg/mL gentamicin and 100 μg/mL carbenicillin was employed. In all P. aeruginosa experiments, expression from pHERD20/30T was induced with 0.1% arabinose and expression from pMMB67HE was induced with 1mM Isopropyl β-D-1-thiogalactopyranoside (IPTG). Escherichia coli strains BL21 (DE3) were grown in LB broth supplemented with ampicillin (100 μg/mL) to maintain pAcrIF4, or with ampicillin (100 μg/mL) and kanamycin (50 μg/mL) to maintain pCsy and pCRISPR together.
Phages
Pseudomonas aeruginosa DMS3m-like phages (JBD30, MP29, JBD88a, JBD24, LPB1, DMS3m and DMS3m derivatives) were amplified on PA14 ΔCRISPR or PAO1 and stored in SM buffer at 4 °C.
METHOD DETAILS
Construction of PA14 crRNA overexpression strains
PA14 CRISPR2 spacer-17 or CRISPR2 spacer-20 sequences flanked by PA14 Type I-F direct repeats were ordered as complementary ssDNA oligos (IDT), annealed, and ligated into the NcoI/HindIII site in pHERD30T to make pAB02 and pAB03, respectively. These constructs were transformed into PA14 WT, and expression induced with 0.1% arabinose.
Construction of PAO1::SpyCas9 expression strain
SpyCas9 expressed from the PLAC promoter of pUC18T-mini-Tn7T-Gm (pBAO95) was integrated into the P. aeruginosa strain PAO1 chromosome by electroporation and Flp-mediated marker excision as previously described (Choi and Schweizer, 2006). To generate the heterologous Type II-A PAO1 strain the PAO1-attTn7::pUC18T-miniTn7T-PLAC-SpyCas9 strain was transformed with pMMB67HE-PLAC-sgRNA (pBAO72) by electroporation. In all experiments with this strain, SpyCas9 and the sgRNA were induced with 1mM IPTG.
Construction of recombinant DMS3macr phages
DMS3macrIF1 was generated previously (Bondy-Denomy et al., 2013) by infecting cells containing a recombination plasmid bearing JBD30 genes 34–38 (the anti-CRISPR locus with large flanking regions). JBD30 naturally carries acrIF1 and has high genetic similarity to DMS3macrIE3, permitting for the selection of recombinant DMS3m phages that acquired acrIF1. To generate the extended panel of DMS3macr phages in this work, recombination cassettes were generated with regions from up and downstream the anti-CRISPR gene from JBD30 and these fragments were assembled to flank the acr gene of interest on pHERD20T or pHERD30T (see Table S1 for acr gene sources, Table S2 for recombination plasmids) using Gibson assembly methods. In the case of AcrIF5, AcrIF6, and AcrIIA4 recombination cassettes, a ribosomal binding site was introduced between the acr and the downstream gene aca1 to ensure proper expression of the aca1 gene. Recombinant phages were generated by infecting cells bearing these recombination substrates. DMS3macr phages were screened for their ability to resist CRISPR targeting, and the insertion of the anti-CRISPR gene was confirmed by PCR. Virulent derivatives of DMS3macr phages were constructed by deleting gp1 (C repressor) using materials and methods previously generated (Cady et al., 2012).
Construction of DMS3macr gp52::gent phages
A recombination substrate (pAB45) with a gentamicin resistance cassette flanked by homology arms matching the DMS3m genome up and downstream of gp52 (450 bp and 260 bp, respectively) was assembled into pHERD20T using Gibson assembly. This recombination cassette was transformed into PA14 ΔCRISPR lysogenized with either DMS3macrIE3 or DMS3macrIF4. These transformed lysogens were grown under gentamicin selection for 16 hours, then sub-cultured 1:100 into LB with gentamicin and 0.2 μg/mL mitomycin C to induce the DMS3macr prophage. Supernatants were harvested after 24 hours of induction, and used to infect PA14 ΔCRISPR in liquid culture for 24 hours. These cells were then plated on gentamicin plates to select for cells that had acquired a prophage bearing the gentamicin resistance cassette, and gentamicin resistant lysogens were then re-induced with 0.2 μg/mL mitomycin C to recover the recombinant phage.
Construction of DMS3macr gp1-JBD30 Hybridacr phages
DMS3macrIE3 and JBD30acrIE3 were used to co-infect PA14 ΔCRISPR and the infected cells were mixed with molten top agar and poured onto solid plates. After 24 hours of growth at 30 °C, the phages were harvested by flooding the plate with SM buffer and collecting and clarifying the supernatant. Phages were then used to infect PA14 ΔCRISPR expressing the DMS3m C repressor from pHERD30T (pAB80), and the infections were mixed with molten top agar and poured onto solid plates. After 24 hours of growth at 30 °C, individual plaques with DMS3m morphology were picked, purified 3× by passage in PA14 ΔCRISPR and screened as shown in Figure S3B. The acrIF1 gene was then knocked in to this hybrid phage using methods described above to generate DMS3macrIF1 gp1-JBD30.
Plaque forming unit quantification
Phage plaque forming units (PFU) were quantified by mixing 10 μl of phage with 150 μl of an overnight culture of host bacteria. The infection mixture was incubated at 37 °C for 10 minutes to promote phage adsorption, then mixed with 3 mL molten top agar and spread on an LB agar plate supplemented with 10 mM MgSO4. After 16 hours of growth 30 °C, PFUs were quantified.
Phage titering
A bacterial lawn was generated by spreading 3 mL of top agar seeded with 150 μl of host bacteria on a LB agar plate supplemented with 10 mM MgSO4. 3 μl of phage serially diluted in SM buffer was then spotted onto the lawn, and incubated at 30 °C for 16 hours.
Liquid culture phage infections
A P. aeruginosa overnight culture was diluted 1:100 in LB supplemented with 10 mM MgSO4, required antibiotics and inducer. 140 μl of diluted bacteria were then infected with 10 μl of phage diluted in SM buffer in a 96 well Costar plate. These infections proceeded for 24 hours in a Synergy H1 microplate reader (BioTek, using Gen5 software) at 37 °C with continuous shaking. After 24 hours, phage was extracted by treating each sample with chloroform followed by centrifugation at 21,000 × g for 2 minutes.
Prophage acquisition and lysogen analysis
Overnight cultures of PA14 were subcultured at 1:100 for ~3 hours (OD600nm = 0.3) in LB supplemented with 10 mM MgSO4. 1 mL of cells was infected with 10 μl DMS3macr gp52::gent and incubated for 50 minutes at 37 °C, shaking at 100 rpm. The sample was then treated with a 10% volume of 10X gentamicin, spun down at 8,000×g, and resuspended in 200 μl of LB with 50 μg/mL gentamicin. 100 μl of sample was then plated (after further dilution, if required) on gentamicin selection plates and incubated at 37 °C. To analyze the lysogens, the resulting colonies were grown for 16 hours in LB + 10 mM MgSO4 (no selection), the supernatants harvested, and serial dilutions spotted onto lawns of non-targeting PA14 (0sp) or PA14 with 5 targeting spacers (5sp). Crude genomic DNA for PCR analysis was harvested from the lysogens by boiling 10 μl of culture in 0.02% SDS for 10 minutes.
Lysogen PCR
PCR amplification of 2 μl of crude genomic DNA harvested from lysogens was used to screen for the presence of DMS3m-gp52 and the gent cassette using MyTaq (Bioline) polymerase with MyTaq GC buffer under standard conditions.
Csy complex purification
Csy genes and a synthetic CRISPR array were co-expressed on separate vectors (pCsy, pCRISPR) in E. coli BL21 (DE3) cells as previously described (Rollins et al., 2017). Expression was induced with 0.5 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) at OD600 ~0.5. Cells were incubated overnight at 16°C, then pelleted by centrifugation (5,000 × g for 15 min at 4 °C), and resuspended in lysis buffer [50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) pH 7.5, 300 mM potassium chloride, 5% glycerol, 1 mM Tris(2-carboxyethyl) phosphine hydrochloride (TCEP), 1× protease inhibitor mixture (Thermo Scientific)]. Pellets were sonicated on ice for 3 × 2.5 min (1 s on, 3 s off), and then the lysate was clarified by centrifugation at 22,000 × g for 30 min at 4 °C. The Csy complex self-assembles in vivo, and the intact complex was affinity-purified over NiNTA Superflow resin (Qiagen) using 6× his tags on Cas7f. Protein was eluted with lysis buffer supplemented with 300 mM imidazole and then concentrated (Corning Spin-X concentrators) at 4 °C before further purification over a Superdex 200 size-exclusion column (GE Healthcare) in 20 mM Hepes pH 7.5, 100 mM KCl, 5% glycerol, and 1 mM TCEP.
AcrIF4 purification
Gene 37 from phage JBD26 (AcrIF4) was cloned into a p15TV-L vector with N-terminal His6 tags (pAcrIF4) and expressed in E. coli BL21 (DE3) cells. Expression was induced with 0.5 mM IPTG at OD600 ~ 0.5. Cells were incubated overnight at 16°C, then pelleted by centrifugation (5,000 × g for 15 min at 4 °C), and resuspended in lysis buffer containing 50 mM Tris, pH 7.5, 300 mM NaCl, 5% glycerol, 0.5× protease inhibitor cocktail (Thermo Scientific) and 1 mM TCEP. Cells were lysed by sonication and lysate was clarified by centrifugation as described above. AcrIF4 protein was affinity-purified over NiNTA Superflow resin (Qiagen) and eluted in lysis buffer supplemented with 300 mM imidazole, then concentrated (Corning Spin-X concentrators) at 4 °C before further purification over a Superdex 75 size-exclusion column (GE Healthcare) in 20 mM Tris, pH 7.5, 250 mM NaCl, 5% glycerol, 1 mM TCEP.
Surface plasmon resonance
Purified Csy complex was covalently immobilized by amine coupling to the surface of a carboxymethyldextran-modified (CM5) sensor chip (GE Healthcare). Purified 6his-tagged AcrIF4 was injected into the buffer flow in increasing concentrations (1.85 nM, 55.6 nM, 167 nM, 500 nM, 1.5 uM), and Csy complex-AcrIF4 binding events were recorded in real time. Experiments were conducted at 37°C, in 20 mM HEPES pH 7.5, 100 mM KCl, 1mM TCEP, 0.005% Tween.
QUANTIFICATION AND STATISTICAL ANALYSIS
All numerical data, with the exception of the surface plasmon resonance (SPR) data, were analyzed and plotted using GraphPad Prism 6.0 software. The SPR data were analyzed and plotted using Biocore evaluation software (GE). Below, we provide the details of the number of biological replicates as well as data quantification and presentation for the experimental methods utilized in this manuscript.
Efficiency of plaquing (Figure 1A–B, Figure 4B, Figure S2A–L)
Efficiency of plaquing (EOP) was calculated as the ratio of the number of plaque forming units (PFUs) that formed on a targeting (+CRISPR, +sgRNA) strain of bacteria divided by the number of PFUs that formed on a non-targeting (ΔCRISPR, vector) strain. Each PFU measurement was performed in biological triplicate. The EOP data in Figure 1A–B and Figure 4B are displayed as the mean EOP +/− standard deviation (error bars) whereas the EOP data in Figure S2A–L are displayed as individual replicate values overlayed with the mean EOP value +/− standard deviation.
Bacterial growth curves (Figure 2A–F, Figure 4C–F)
OD600nm values were measured in biological triplicate for each experimental condition over a period of 12 hours, and the data displayed as the mean OD600nm as a function of time (hours) +/− standard deviation (error bars).
Quantification of phage lytic replication (Figure 2G–I, Figure 4G–H, Figure S3C)
Phage infections were performed in biological triplicate, and the phages harvested from each infection were quantified as plaque forming units (PFUs) on a non-targeting (ΔCRISPR, vector) strain. Values are displayed as the mean number of PFUs from the 3 experimental replicates, +/− standard deviation (error bars).
Quantification of phage lysogeny (Figure 3A–B)
Phage lysogeny was measured as the number of lysogen forming units (LFUs) that formed under a given experimental condition. In our experimental setup, each sample was diluted at least 2-fold before quantification, meaning that the limit of detection (LoD) of this assay is 2 LFUs. Phage lysogeny experiments were performed in biological triplicate, and each replicate value is displayed.
Efficiency of lysogeny (Figure 3C–F)
Efficiency of lysogeny (EOL) was calculated as ratio of the number of lysogen forming units (LFUs) that form under the targeting condition (5sp) divided by the number of LFUs that form under the non-targeting condition (0sp). Phage lysogeny experiments were performed in biological triplicate, and EOL is displayed as mean EOL +/− standard deviation (error bars).
Analysis of AcrIF4 binding kinetics (Figure 1C, Figure S2)
Data were fit with a model describing Langmuir binding (i.e. 1:1 binding between free analyte and immobilized ligand). Plotted residual data points scattered around zero and were <10% of Rmax, indicating good model fit. Kinetic rate constants were extracted from this curve fit using Biacore evaluation software (GE). Parameter significance was evaluated by assessing standard error (SE)/T-value (T-value = parameter value/standard error). This value provides a measure of how sensitive the model fit is to changes in the parameter value; high SE/low T-value indicates poor significance. SEs for ka and kd were both >21-fold lower than T-values, indicating good significance.
Supplementary Material
Table S1. DMS3macr phage genotypes, Related to STAR Methods
Table S2. Plasmids used in this study, Related to STAR Methods
Sensogram showing real-time binding of increasing concentrations of free AcrIF4 (1.85 nM, 55.6 nM, 167 nM, 500 nM, 1.5 μM) to immobilized Csy complex. A model describing Langmuir binding (black line) was fit to the data to calculate binding constants (ka, kd, and KD; boxed inset).
(A–C) Efficiency of plaquing (EOP) of the original stocks of virulent DMS3macr (input) on PA14 5sp compared to EOP of DMS3macr harvested from high MOI infections in Figure 2A–C (MOI 2×10−2, MOI 2×10−1 output for DMS3macrIF1 and MOI 2 and MOI 20 output for DMS3macrIF4 and DMS3macrIE3).
(D–F) EOP of acceptor output phages that amplified in the presence of AcrIF1 donor phages from Figure 2D–F on PA14 5sp compared to the original stock of virulent DMS3macr (input).
(G) EOP of a laboratory evolved DMS3macrIE3 5 spacer escaper (DMS3macrIE3*) on PA14 5sp relative to the parental DMS3macrIE3 stock.
(H–I) Efficiency of plaquing (EOP) of the original stocks of virulent DMS3macr (input) on PAO1::SpyCas9 + sgRNA compared to EOP of DMS3macr harvested from high MOI infections in Figure 4C–D (MOI 2×10−3, MOI 2×10−2).
(J–K) EOP of acceptor output phages that amplified in the presence of AcrIIA4 donor phages from Figure 4G–H on PAO1::SpyCas9 + sgRNA compared to the original stock of virulent DMS3macr (input).
(L) EOP of a laboratory evolved DMS3macrIE3 sgRNA escaper (DMS3macrIE3**) on PAO1::SpyCas9 + sgRNA relative to the parental DMS3macrIE3 stock.
All data are represented as the mean of 3 biological replicates +/− SD.
(A) 10-fold serial dilutions of hybrid DMS3macrIE3 gp1-JBD30 plated on lawns of non-targeting (0sp) Pseudomonas aeruginosa PA14 expressing the DMS3m C repressor (gp1-DMS3m), the JBD30 C repressor (gp1-JBD30), or a crRNA which uniquely targets JBD30 (2sp17) outside of the immunity region.
(B) 10-fold serial dilutions of DMS3macrIE3 and JBD30acrIF1 spotted on lawns of non-targeting (0sp) Pseudomonas aeruginosa PA14 expressing the DMS3m C repressor (gp1-DMS3m), the JBD30 C repressor (gp1-JBD30), or a vector control.
(C) Hybrid phage (HybridacrIF1 or HybridacrIE3) harvested from infections of PA14 5sp expressing the JBD30 C repressor from experiments shown in Figure 2G–I. Hybrid PFUs were quantified on the 0sp PA14 strain. Data are represented as the mean of 3 biological replicates +/− SD.
(D) 10-fold serial dilutions of JBD30acrIF1 or virulent DMS3macrIE3 spotted on lawns of PA14 Δcsy3 or PA14 Δcsy3 lysogenized with HybridacrIE3 or HybridacrIF1. Despite being heteroimmune with respect to JBD30, the DMS3m phage is unable to replicate well on this lysogens due to other superinfection exclusion properties of DMS3m.
(A) PCR of genomic DNA harvested from overnight cultures of lysogens from Figure 3E (1–48) and 3F (49–51) amplified with primers targeting gp52-DMS3m (top) or the gentamicin resistance cassette used to replace gp52 in DMS3macr gp52::gent derivatives (bottom).
(B) 10-fold serial dilutions of supernatant harvested from overnight cultures of lysogens from Figure 3E (1–48) and 3F (49–51), spotted on a non-targeting (0sp) strain of PA14 and the 5 spacer (5sp) targeting strain of PA14. A faint clearing corresponds to induction of the gentamicin marked phage, while strong plaquing (i.e. 50, 51) reflects the presence of the gentamicin marked phage and the donor phage. Loss of plaquing efficiency in the presence of 5 targeting spacers corresponds to CRISPR sensitivity, and the plaquing efficiency of the input DMS3macrIF4 gp52::gent on the 5sp host is shown on the right for comparison.
Highlights.
Phages deploy anti-CRISPR proteins in host bacteria to inhibit CRISPR-Cas immunity
Anti-CRISPR proteins do not fully protect their associated phage genome
Anti-CRISPR proteins produced during failed infections immunosuppress the host
Cellular immunosuppression facilitates successful phage replication upon reinfection
Acknowledgments
The Bondy-Denomy lab is supported by the University of California San Francisco Program for Breakthrough in Biomedical Research, funded in part by the Sandler Foundation, and an NIH Office of the Director Early Independence Award (DP5-OD021344). Research in the Wiedenheft lab is supported by the National Institutes of Health (P20GM103500, P30GM110732, R01GM110270, R01GM108888 and R21 AI130670), the National Science Foundation EPSCoR (EPS-110134), the M. J. Murdock Charitable Trust, a young investigator award from Amgen, and the Montana State University Agricultural Experimental Station (USDA NIFA). The laboratory evolved PA14 strain with 5 spacers targeting DMS3m was generously provided by Stineke van Houte and Edze Westra (University of Exeter).
Footnotes
AUTHOR CONTRIBUTIONS
A.L.B. and J.B.D. designed experiments and wrote the manuscript. A.L.B. engineered phage variants and conducted all experiments with the exception of binding affinity measurements. J.Y.Z. assisted with experimental design and execution, phage engineering, and data collection. B.A.O. built the Cas9 strain in Pseudomonas aeruginosa. J.B.D. supervised experiments. M.R. conducted Acr binding affinity measurements under the supervision of B.W. All authors contributed to data analysis and edited the manuscript.
DECLARATION OF INTERESTS
The authors declare no competing interests.
Figures S1–S4 and Tables S1–2 are provided separately.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguilera ER, Erickson AK, Jesudhasan PR, Robinson CM, Pfeiffer JK. Plaques Formed by Mutagenized Viral Populations Have Elevated Coinfection Frequencies. mBio. 2017;8:e02020–16. 12. doi: 10.1128/mBio.02020-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson AF, Banfield JF. Virus population dynamics and acquired virus resistance in natural microbial communities. Science. 2008;320:1047–1050. doi: 10.1126/science.1157358. [DOI] [PubMed] [Google Scholar]
- Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- Bikard D, Hatoum-Aslan A, Mucida D, Marraffini LA. CRISPR interference can prevent natural transformation and virulence acquisition during in vivo bacterial infection. Cell Host Microbe. 2012;12:177–186. doi: 10.1016/j.chom.2012.06.003. [DOI] [PubMed] [Google Scholar]
- Bondy-Denomy J, Pawluk A, Maxwell KL, Davidson AR. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature. 2013;493:429–432. doi: 10.1038/nature11723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondy-Denomy J, Davidson AR. When a virus is not a parasite: the beneficial effects of prophages on bacterial fitness. J Microbiol. 2014;52:235–242. doi: 10.1007/s12275-014-4083-3. [DOI] [PubMed] [Google Scholar]
- Bondy-Denomy J, Garcia B, Strum S, Du M, Rollins MF, Hidalgo-Reyes Y, Wiedenheft B, Maxwell KL, Davidson AR. Multiple mechanisms for CRISPR-Cas inhibition by anti-CRISPR proteins. Nature. 2015;526:136–139. doi: 10.1038/nature15254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondy-Denomy J, Qian J, Westra ER, Buckling A, Guttman DS, Davidson AR, Maxwell KL. Prophages mediate defense against phage infection through diverse mechanisms. The ISME Journal. 2016;10:2854–2866. doi: 10.1038/ismej.2016.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges AL, Davidson AR, Bondy-Denomy J. The Discovery, Mechanisms, and Evolutionary Impact of Anti-CRISPRs. Annual Review of Virology. 2017;4 doi: 10.1146/annurev-virology-101416-041616. annurev–virology–101416–041616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botstein D. A theory of modular evolution for bacteriophages. Ann N Y Acad Sci. 1980;354:484–490. doi: 10.1111/j.1749-6632.1980.tb27987.x. [DOI] [PubMed] [Google Scholar]
- Brouns SJJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJH, Snijders APL, Dickman MJ, Makarova KS, Koonin EV, van der Oost J. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321:960–964. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cady KC, Bondy-Denomy J, Heussler GE, Davidson AR, O’Toole GA. The CRISPR/Cas adaptive immune system of Pseudomonas aeruginosa mediates resistance to naturally occurring and engineered phages. 2012;194:5728–5738. doi: 10.1128/JB.01184-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carte J, Christopher RT, Smith JT, Olson S, Barrangou R, Moineau S, Glover CVC, Graveley BR, Terns RM, Terns MP. The three major types of CRISPR-Cas systems function independently in CRISPR RNA biogenesis in Streptococcus thermophilus. Molecular Microbiology. 2014;93:98–112. doi: 10.1111/mmi.12644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury S, Carter J, Rollins MF, Golden SM, Jackson RN, Hoffmann C, Nosaka L, Bondy-Denomy J, Maxwell KL, Davidson AR, et al. Structure Reveals Mechanisms of Viral Suppressors that Intercept a CRISPR RNA-Guided Surveillance Complex. Cell. 2017;169:47–57. e11. doi: 10.1016/j.cell.2017.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicin-Sain L, Podlech J, Messerle M, Reddehase MJ, Koszinowski UH. Frequent Coinfection of Cells Explains Functional In Vivo Complementation between Cytomegalovirus Variants in the Multiply Infected Host. Journal of Virology. 2005;79:9492–9502. doi: 10.1128/JVI.79.15.9492-9502.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diemer GS, Stedman KM. A novel virus genome discovered in an extreme environment suggests recombination between unrelated groups of RNA and DNA viruses. Biol Direct. 2012;7:13. doi: 10.1186/1745-6150-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong D, Guo M, Wang S, Zhu Y, Wang S, Xiong Z, Yang J, Xu Z, Huang Z. Structural basis of CRISPR-SpyCas9 inhibition by an anti-CRISPR protein. Nature. 2017 doi: 10.1038/nature22377. [DOI] [PubMed] [Google Scholar]
- Edgar R, Qimron U. The Escherichia coli CRISPR system protects from λ lysogenization, lysogens, and prophage induction. J Bacteriol. 2010;192:6291–6294. doi: 10.1128/JB.00644-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erez Z, Steinberger-Levy I, Shamir M, Doron S, Stokar-Avihail A, Peleg Y, Melamed S, Leavitt A, Savidor A, Albeck S, et al. Communication between viruses guides lysis–lysogeny decisions. Nature. 2017;541:488–493. doi: 10.1038/nature21049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garneau JE, Dupuis M-È, Villion M, Romero DA, Barrangou R, Boyaval P, Fremaux C, Horvath P, Magadán AH, Moineau S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 2010;468:67–71. doi: 10.1038/nature09523. [DOI] [PubMed] [Google Scholar]
- Guo TW, Bartesaghi A, Yang H, Falconieri V, Rao P, Merk A, Eng ET, Raczkowski AM, Fox T, Earl LA, et al. Cryo-EM Structures Reveal Mechanism and Inhibition of DNA Targeting by a CRISPR-Cas Surveillance Complex. Cell. 2017;171:414–426. e12. doi: 10.1016/j.cell.2017.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington LB, Doxzen KW, Ma E, Liu J-J, Knott GJ, Edraki A, Garcia B, Amrani N, Chen JS, Cofsky JC, et al. A Broad-Spectrum Inhibitor of CRISPR-Cas9. Cell. 2017 doi: 10.1016/j.cell.2017.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatfull GF, Hendrix RW. Bacteriophages and their genomes. Curr Opin Virol. 2011;1:298–303. doi: 10.1016/j.coviro.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haurwitz RE, Jinek M, Wiedenheft B, Zhou K, Doudna JA. Sequence- and Structure-Specific RNA Processing by a CRISPR Endonuclease. Science. 2010;329:1355–1358. doi: 10.1126/science.1192272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Bhoobalan-Chitty Y, Van LB, Kjeldsen AL, Dedola M, Makarova KS, Koonin EV, Brodersen DE, Peng X. Anti-CRISPR proteins encoded by archaeal lytic viruses inhibit subtype I-D immunity. Nature Microbiology. 2018;71:1. doi: 10.1038/s41564-018-0120-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrix RW, Smith MC, Burns RN, Ford ME, Hatfull GF. Evolutionary relationships among diverse bacteriophages and prophages: all the world’s a phage. Proc Natl Acad Sci USA. 1999;96:2192–2197. doi: 10.1073/pnas.96.5.2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes AP, Rousseau GM, Lemay ML, Horvath P, Romero DA, Fremaux C, Moineau S. An anti-CRISPR from a virulent streptococcal phage inhibits Streptococcus pyogenes Cas9. Nature Microbiology. 2017;315:1. doi: 10.1038/s41564-017-0004-7. [DOI] [PubMed] [Google Scholar]
- Hynes AP, Villion M, Moineau S. Adaptation in bacterial CRISPR-Cas immunity can be driven by defective phages. Nature Communications. 2014;5:4399. doi: 10.1038/ncomms5399. [DOI] [PubMed] [Google Scholar]
- Koonin EV, Makarova KS, Wolf YI. Evolutionary Genomics of Defense Systems in Archaea and Bacteria. Annu Rev Microbiol. 2017;71 doi: 10.1146/annurev-micro-090816-093830. annurev–micro–090816–093830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourilsky P, Knapp A. Lysogenization by bacteriophage lambda. III. Multiplicity dependent phenomena occuring upon infection by lambda. Biochimie. 1974;56:1517–1523. [PubMed] [Google Scholar]
- Krupovic M, Zhi N, Li J, Hu G, Koonin EV, Wong S, Shevchenko S, Zhao K, Young NS. Multiple Layers of Chimerism in a Single-Stranded DNA Virus Discovered by Deep Sequencing. Genome Biol Evol. 2015;7:993–1001. doi: 10.1093/gbe/evv034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy A, Goren MG, Yosef I, Auster O, Manor M, Amitai G, Edgar R, Qimron U, Sorek R. CRISPR adaptation biases explain preference for acquisition of foreign DNA. 2015;520:505–510. doi: 10.1038/nature14302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu MJ, Henning U. The immunity (imm) gene of Escherichia coli bacteriophage T4. Journal of Virology. 1989;63:3472–3478. doi: 10.1128/jvi.63.8.3472-3478.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini LA, Sontheimer EJ. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science. 2008;322:1843–1845. doi: 10.1126/science.1165771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell KL, Garcia B, Bondy-Denomy J, Bona D, Hidalgo-Reyes Y, Davidson AR. The solution structure of an anti-CRISPR protein. Nature Communications. 2016;7:13134. doi: 10.1038/ncomms13134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez-Espino D, Sharon I, Morovic W, Stahl B, Thomas BC, Barrangou R, Banfield JF. CRISPR Immunity Drives Rapid Phage Genome Evolution in Streptococcus thermophilus. mBio. 2015;6:e00262–15. 9. doi: 10.1128/mBio.00262-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson AG, Jackson SA, Taylor C, Evans GB, Salmond GPC, Przybilski R, Staals RHJ, Fineran PC. Quorum Sensing Controls Adaptive Immunity through the Regulation of Multiple CRISPR-Cas Systems. Mol Cell. 2016;64:1102–1108. doi: 10.1016/j.molcel.2016.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawluk A, Amrani N, Zhang Y, Garcia B, Hidalgo-Reyes Y, Lee J, Edraki A, Shah M, Sontheimer EJ, Maxwell KL, et al. Naturally Occurring Off-Switches for CRISPR-Cas9. Cell. 2016a;167:1829–1838. e1829. doi: 10.1016/j.cell.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawluk A, Bondy-Denomy J, Cheung VHW, Maxwell KL, Davidson AR. A new group of phage anti-CRISPR genes inhibits the type I-E CRISPR-Cas system of Pseudomonas aeruginosa. mBio. 2014;5:e00896-e00896-14. doi: 10.1128/mBio.00896-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawluk A, Davidson AR, Maxwell KL. Anti-CRISPR: discovery, mechanism and function. Nat Rev Micro. 2017;1 doi: 10.1038/nrmicro.2017.120. nrmicro.2017.120. [DOI] [PubMed] [Google Scholar]
- Pawluk A, Staals RHJ, Taylor C, Watson BNJ, Saha S, Fineran PC, Maxwell KL, Davidson AR. Inactivation of CRISPR-Cas systems by anti-CRISPR proteins in diverse bacterial species. Nature Microbiology. 2016b;1:1–6. doi: 10.1038/nmicrobiol.2016.85. [DOI] [PubMed] [Google Scholar]
- Peng R, Xu Y, Zhu T, Li N, Qi J, Chai Y, Wu M, Zhang X, Shi Y, Wang P, et al. Alternate binding modes of anti-CRISPR viral suppressors AcrF1/2 to Csy surveillance complex revealed by cryo-EM structures. Cell Res. 2017;132:712. doi: 10.1038/cr.2017.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przybilski R, Richter C, Gristwood T, Clulow JS, Vercoe RB, Fineran PC. Csy4 is responsible for CRISPR RNA processing in Pectobacterium atrosepticum. RNA Biol. 2011;8:517–528. doi: 10.4161/rna.8.3.15190. [DOI] [PubMed] [Google Scholar]
- Rauch BJ, Silvis MR, Hultquist JF, Waters CS, McGregor MJ, Krogan NJ, Bondy-Denomy J. Inhibition of CRISPR-Cas9 with Bacteriophage Proteins. Cell. 2017;168:150–158. e10. doi: 10.1016/j.cell.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter H, Zoephel J, Schermuly J, Maticzka D, Backofen R, Randau L. Characterization of CRISPR RNA processing in Clostridium thermocellum and Methanococcus maripaludis. Nucleic Acids Research. 2012;40:9887–9896. doi: 10.1093/nar/gks737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollins MF, Chowdhury S, Carter J, Golden SM, Wilkinson RA, Bondy-Denomy J, Lander GC, Wiedenheft B. Cas1 and the Csy complex are opposing regulators of Cas2/3 nuclease activity. Proceedings of the National Academy of Sciences. 2017;23:201616395–E5121. doi: 10.1073/pnas.1616395114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux S, Enault FCO, le Bronner GE, Vaulot D, Forterre P, Krupovič M. Chimeric viruses blur the borders between the major groups of eukaryotic single-stranded DNA viruses. Nature Communications. 2013;4:1–10. doi: 10.1038/ncomms3700. [DOI] [PubMed] [Google Scholar]
- Shin J, Jiang F, Liu JJ, Bray NL, Rauch BJ, Baik SH, Nogales E, Bondy-Denomy J, Corn JE, Doudna JA. Disabling Cas9 by an anti-CRISPR DNA mimic. Sci Adv. 2017;3:e1701620. doi: 10.1126/sciadv.1701620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh JT, Székely T, Shao Q, Balázsi G, Zeng L. Cell fate decisions emerge as phages cooperate or compete inside their host. Nature Communications. 2017;8:14341–13. doi: 10.1038/ncomms14341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Belkum A, Soriaga LB, LaFave MC, Akella S, Veyrieras JB, Barbu EM, Shortridge D, Blanc B, Hannum G, Zambardi G, et al. Phylogenetic Distribution of CRISPR-Cas Systems in Antibiotic-Resistant Pseudomonas aeruginosa. mBio. 2015;6:e01796–15. doi: 10.1128/mBio.01796-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Houte S, Ekroth AKE, Broniewski JM, Chabas H, Ben Ashby Bondy-Denomy J, Gandon S, Boots M, Paterson S, Buckling A, et al. The diversity-generating benefits of a prokaryotic adaptive immune system. Nature. 2016;532:385–388. doi: 10.1038/nature17436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignuzzi M, Stone JK, Arnold JJ, Cameron CE, Andino R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature. 2005;439:344–348. doi: 10.1038/nature04388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldor MK, Mekalanos JJ. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science. 1996;272:1910–1914. doi: 10.1126/science.272.5270.1910. [DOI] [PubMed] [Google Scholar]
- Wang J, Ma J, Cheng Z, Meng X, You L, Wang M, Zhang X, Wang Y. A CRISPR evolutionary arms race: structural insights into viral anti-CRISPR/Cas responses. Cell Res. 2016a;26:1165–1168. doi: 10.1038/cr.2016.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Yao D, Xu J-G, Li A-R, Xu J, Fu P, Zhou Y, Zhu Y. Structural basis of Cas3 inhibition by the bacteriophage protein AcrF3. Nat Struct Mol Biol. 2016b;23:868–870. doi: 10.1038/nsmb.3269. [DOI] [PubMed] [Google Scholar]
- Weigle JJ, Delbruck M. Mutual exclusion between an infecting phage and a carried phage. J Bacteriol. 1951;62:301–318. doi: 10.1128/jb.62.3.301-318.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westra ER, van Houte S, Oyesiku-Blakemore S, Makin B, Broniewski JM, Best A, Bondy-Denomy J, Davidson A, Boots M, Buckling A. Parasite Exposure Drives Selective Evolution of Constitutive versus Inducible Defense. Curr Biol. 2015;25:1043–1049. doi: 10.1016/j.cub.2015.01.065. [DOI] [PubMed] [Google Scholar]
- Wiedenheft B, van Duijn E, Bultema JB, Bultema J, Waghmare SP, Waghmare S, Zhou K, Barendregt A, Westphal W, Heck AJR, et al. RNA-guided complex from a bacterial immune system enhances target recognition through seed sequence interactions. Proceedings of the National Academy of Sciences. 2011;108:10092–10097. doi: 10.1073/pnas.1102716108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue KS, Hooper KA, Ollodart AR, Dingens AS, Bloom JD. Cooperation between distinct viral variants promotes growth of H3N2 influenza in cell culture. Elife. 2016;5:e13974. doi: 10.7554/eLife.13974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Patel DJ. Inhibition Mechanism of an Anti-CRISPR Suppressor AcrIIA4 Targeting SpyCas9. Mol Cell. 2017:1–17. doi: 10.1016/j.molcel.2017.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L, Skinner SO, Zong C, Sippy J, Feiss M, Golding I. Decision Making at a Subcellular Level Determines the Outcome of Bacteriophage Infection. Cell. 2010;141:682–691. doi: 10.1016/j.cell.2010.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. DMS3macr phage genotypes, Related to STAR Methods
Table S2. Plasmids used in this study, Related to STAR Methods
Sensogram showing real-time binding of increasing concentrations of free AcrIF4 (1.85 nM, 55.6 nM, 167 nM, 500 nM, 1.5 μM) to immobilized Csy complex. A model describing Langmuir binding (black line) was fit to the data to calculate binding constants (ka, kd, and KD; boxed inset).
(A–C) Efficiency of plaquing (EOP) of the original stocks of virulent DMS3macr (input) on PA14 5sp compared to EOP of DMS3macr harvested from high MOI infections in Figure 2A–C (MOI 2×10−2, MOI 2×10−1 output for DMS3macrIF1 and MOI 2 and MOI 20 output for DMS3macrIF4 and DMS3macrIE3).
(D–F) EOP of acceptor output phages that amplified in the presence of AcrIF1 donor phages from Figure 2D–F on PA14 5sp compared to the original stock of virulent DMS3macr (input).
(G) EOP of a laboratory evolved DMS3macrIE3 5 spacer escaper (DMS3macrIE3*) on PA14 5sp relative to the parental DMS3macrIE3 stock.
(H–I) Efficiency of plaquing (EOP) of the original stocks of virulent DMS3macr (input) on PAO1::SpyCas9 + sgRNA compared to EOP of DMS3macr harvested from high MOI infections in Figure 4C–D (MOI 2×10−3, MOI 2×10−2).
(J–K) EOP of acceptor output phages that amplified in the presence of AcrIIA4 donor phages from Figure 4G–H on PAO1::SpyCas9 + sgRNA compared to the original stock of virulent DMS3macr (input).
(L) EOP of a laboratory evolved DMS3macrIE3 sgRNA escaper (DMS3macrIE3**) on PAO1::SpyCas9 + sgRNA relative to the parental DMS3macrIE3 stock.
All data are represented as the mean of 3 biological replicates +/− SD.
(A) 10-fold serial dilutions of hybrid DMS3macrIE3 gp1-JBD30 plated on lawns of non-targeting (0sp) Pseudomonas aeruginosa PA14 expressing the DMS3m C repressor (gp1-DMS3m), the JBD30 C repressor (gp1-JBD30), or a crRNA which uniquely targets JBD30 (2sp17) outside of the immunity region.
(B) 10-fold serial dilutions of DMS3macrIE3 and JBD30acrIF1 spotted on lawns of non-targeting (0sp) Pseudomonas aeruginosa PA14 expressing the DMS3m C repressor (gp1-DMS3m), the JBD30 C repressor (gp1-JBD30), or a vector control.
(C) Hybrid phage (HybridacrIF1 or HybridacrIE3) harvested from infections of PA14 5sp expressing the JBD30 C repressor from experiments shown in Figure 2G–I. Hybrid PFUs were quantified on the 0sp PA14 strain. Data are represented as the mean of 3 biological replicates +/− SD.
(D) 10-fold serial dilutions of JBD30acrIF1 or virulent DMS3macrIE3 spotted on lawns of PA14 Δcsy3 or PA14 Δcsy3 lysogenized with HybridacrIE3 or HybridacrIF1. Despite being heteroimmune with respect to JBD30, the DMS3m phage is unable to replicate well on this lysogens due to other superinfection exclusion properties of DMS3m.
(A) PCR of genomic DNA harvested from overnight cultures of lysogens from Figure 3E (1–48) and 3F (49–51) amplified with primers targeting gp52-DMS3m (top) or the gentamicin resistance cassette used to replace gp52 in DMS3macr gp52::gent derivatives (bottom).
(B) 10-fold serial dilutions of supernatant harvested from overnight cultures of lysogens from Figure 3E (1–48) and 3F (49–51), spotted on a non-targeting (0sp) strain of PA14 and the 5 spacer (5sp) targeting strain of PA14. A faint clearing corresponds to induction of the gentamicin marked phage, while strong plaquing (i.e. 50, 51) reflects the presence of the gentamicin marked phage and the donor phage. Loss of plaquing efficiency in the presence of 5 targeting spacers corresponds to CRISPR sensitivity, and the plaquing efficiency of the input DMS3macrIF4 gp52::gent on the 5sp host is shown on the right for comparison.