

Abstract
An 18-month-old boy was diagnosed with late-onset ornithine transcarbamylase deficiency. Genetic analysis revealed a mosaic frameshift mutation (p.Q279fs) in the OTC gene. Despite the presence of a null mutation, he exhibited a milder phenotype, suggesting that the wild-type allele could rescue the function of OTC. The presence of mosaicism has great effects on the clinical phenotype and recurrence-risk assessment, which should be taken into consideration for genetic counseling.
Ornithine transcarbamylase (OTC) deficiency (OTCD) (MIM#311250) is the most common urea cycle disorder caused by mutations in the OTC gene located on chromosome Xp211,2. OTCD causes hyperammonemia, presenting with clinical manifestations such as lethargy, vomiting, coma, and in severe cases, death3. Most of the OTCD patients are hemizygous males, and approximately 20% of the female carriers of OTC mutations are also symptomatic4–6.
OTCD is classified into two groups: severe neonatal-onset and late-onset phenotypes. Neonatal-onset OTCD presents with acute hyperammonemia within the first few days of life, whereas late-onset OTCD can present later from infancy to adulthood7. Genotype−phenotype correlations have been well defined. Null mutations, such as large deletions, frameshift, and nonsense mutations, and some missense mutations resulting in a complete loss of OTC function cause severe neonatal-onset diseases in hemizygous males and in most symptomatic heterozygous females3,8–10. Missense mutations that retain some OTC activity cause late-onset disease in hemizygous males3,8–10.
Mosaicism is a well-established biological phenomenon. The frequency of mosaicism appears to vary among different disease genes. To date, somatic mosaicism in the OTC gene has been reported in only a few cases11–14.
Here, we investigate late-onset OTCD in a boy caused by a somatic mosaic frameshift mutation.
An 18-month-old Japanese boy born to nonconsanguineous parents as the third child with a healthy elder sister and a brother was referred to the hospital due to vomiting and unconsciousness. His mental and physical development was normal to date. Blood examination revealed elevated ammonia levels (435 µg/dl, normal range: 30–70) and glutamine (1011.5 nmol/ml, normal range: 422.1–703.8), whereas citrulline (7.4 nmol/ml, normal range: 17.1–42.6) and arginine (19.8 nmol/ml, normal range: 53.6–133.6) levels were decreased. Urinary orotic acid was extremely increased to 1735.7 mmol/molCr. These results confirmed the diagnosis of OTCD biochemically. Glucose was administered intravenously, and protein intake was restricted. Sodium benzoate, arginine, and citrulline were also administered. These treatments dramatically improved his clinical and biochemical abnormalities. After recovering from the first episode of hyperammonemia, he was treated with a protein-restricted diet (1.8 g/kg/day), sodium phenylbutyrate, and citrulline. Although he had four additional episodes of hyperammonemia due to infections or excessive protein intake, he developed and grew normally at the age of 3 years. His clinical course was consistent with a late-onset OTCD phenotype.
This study was approved by the Ethics Committee at Hyogo College of Medicine (Approval No. rinhi 113), and informed consent was obtained from the patient’s parents.
Genomic DNA from the peripheral blood was extracted using standard phenol-chloroform extraction methods, and genomic DNA from oral mucosa was extracted using the QlAamp DNA Mini Kit (Qiagen, Venlo, The Netherlands). A mutation analysis of 57 genes involved in urea cycle diseases was performed by multiplex PCR-based library preparation (1824 primers) and next-generation sequencing (NGS) (Miseq Sequencing System, Illumina, San Diego, CA, USA) at the Kazusa DNA Research Institute. Polymerase chain reaction (PCR) and direct sequencing analysis were also performed as previously described15.
To quantify the ratio between the normal and mutant alleles, a semiquantitative analysis was performed. gDNA was amplified by 27 cycles of PCR with a fluorescein-labeled reverse primer. Quantification was performed by measuring its area using GeneMapper software (Applied Biosystems by Thermo Fisher Scientific, Waltham, MA, USA) after PCR amplification.
NGS analysis and direct sequence analysis of genomic DNA from lymphocytes identified a mosaic frameshift mutation (c.834_840delCCAGGCT, p.Q279fs) in the OTC gene (Fig. 1a), revealing two different types of alleles, including a wild type and mutant with a frameshift mutation (c.834_840delCCAGGCT), in the patient. Only the wild-type allele was identified in the patient’s mother (Fig. 1a). His karyotype was 46, XY. These results suggested that a de novo somatic mosaicism caused OTCD. Because this mutation was a frameshift mutation, his clinical phenotype was expected to be a severe neonatal-onset phenotype. However, he exhibited a milder late-onset clinical course. This finding suggested that the wild-type allele could maintain OTC function despite the presence of a null mutation.
Fig. 1. Nucleotide sequence and semiquantitative analysis of the OTC gene.
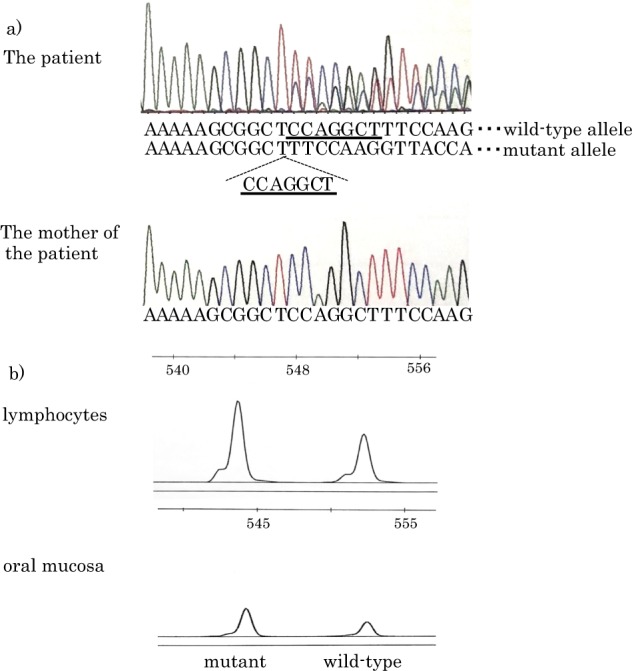
a The results of a direct sequencing analysis of the OTC gene using genomic DNA from lymphocytes of the patient and his mother. Upper figure: Direct sequencing analysis revealed two different types of alleles: the wild-type and the mutant alleles with a frameshift mutation (c.834_840delCCAGGCT). Lower figure: Only the wild-type allele was identified in his mother. b Semiquantitative analysis using the GeneMapper software for lymphocytes and oral mucosa. The mutant allele was in 60.38 and 65.19% of the lymphocytes and oral mucosa cells, respectively
Next, we examined the mosaic ratio of the wild-type and mutant alleles. The mosaic ratio identified by NGS (62.73%, AmpliVar analysis) in the current case was doubtful due to a T homopolymer close to the mutation, which possibly caused misreading. Therefore, we performed semiquantitative analysis using GeneMapper software to determine the mosaic ratio in the lymphocytes and oral mucosa. The mosaic ratio was 60.38% in the lymphocytes and 65.19% in the oral mucosa (Fig. 1b). Unfortunately, other tissues were not investigated.
The mutation identified in the current patient was a novel mutation16. Despite the presence of a null mutation, this patient exhibited a milder late-onset phenotype due to mosaicism. The mutant allele was present in 60–65% of the lymphocytes and oral mucosa cells, which may vary among different tissues. Given that OTC is mainly expressed in the liver, the degree of mosaicism in the liver is interesting; however, this information was beyond the scope of the present study.
Postzygotic mutations with germline and somatic mosaics have been recognized as the cause of various genetic diseases17. In the patient in the present study, only somatic cells were investigated; therefore, somatic and gonosomal mosaicism can be considered.
To date, greater than 400 disease-causing mutations in the OTC gene have been reported16; however, somatic mosaic mutations have only been reported in only a few patients (Table 1). To the best of our knowledge, these six mutations have not been previously reported in male patients without somatic mosaicism. Only the mutation of patient #5 (L349P) was reported as female OTCD without detailed information16. Patients #1 and #2 exhibited milder late-onset phenotypes despite a large deletion mutation. Patient #4 and the current case also exhibited late-onset phenotypes despite a splice site and frameshift mutation, respectively. Despite the presence of a null mutation, these cases exhibited milder phenotypes, suggesting that the wild-type allele could rescue the function of OTC. For X-linked disorders in particular, such somatic mosaicism can explain mild forms of the disease in male patients11. Surprisingly, patients #3 and #5 with mosaic missense mutations exhibited no symptoms; however, their daughters exhibited severe phenotypes.
Table 1.
Patients with OTCD caused by somatic mosaicism
| Patient | Sex | Mutation in the OTC gene | Phenotype | Reference |
|---|---|---|---|---|
| #1 | Male | exon 5–7 (8) deletion | Late | Maddalena et al.11 |
| #2 | Male | exon7–9 deletion | Late | Legius et al.12 |
| #3 | Male | c.444 G > C (p.L148F) | Asymptomatic | Komaki et al.13 |
| #4 | Male | c.386+1 G > T | Late | Qin et al.14 |
| #5 | Male | c.1046 T > C (p.L349P) | Asymptomatic | Qin et al.14 |
| #6 | Male | c.834_840delCCAGGCT (p.Q279fs) | Late | Current patient |
Three daughters of patient #3 died during childhood due to hyperammonemia, and a heterozygous mutation (p.L148F) in the OTC gene was identified in the third daughter. Although it is unclear why all three daughters had severe symptomatic OTCD, biases for the inactivation of the maternal normal X chromosome apparently occurred13. The daughter of patient #5 experienced hyperammonemia, and a heterozygous mutation (p.L349P) was identified in the OTC gene. Unexpectedly, NGS analysis confirmed 21% mosaic for the same mutation in her father (patient #5) but not in her mother.
Interestingly, the daughters in these two families inherited mutations from the asymptomatic father with a mosaic mutation. Heterozygous female patients present a wide range of clinical manifestations due to random X-chromosome inactivation or the severity of the mutation18. In theory, heterozygous female patients exhibit milder phenotypes compared with male patients who are hemizygous for the same mutation. However, if the male patients have a mosaic mutation, this theory is not applicable. Therefore, female patients can have more severe phenotypes compared with male patients depending on skewed X-chromosome inactivation.
Therefore, for accurate genetic counseling, whether the mosaicism is also present in the gonads of this patient should be taken into consideration as a future daughter of this patient might become a carrier of severe OTCD. In particular, a future daughter of this patient can have a more severe phenotype than the daughters of patients #3 and #5.
These results also suggested that if de novo mutations in the OTC gene occur in females, efforts should be made to determine whether there is mosaicism in the father even if he appears to be asymptomatic. There can be cases with undetected mosaicism in the OTC gene. Determination of the presence of mosaicism is important for accurate recurrence-risk assessment and genetic counseling.
In summary, a somatic mosaic frameshift mutation was identified in a male patient with milder late-onset OTCD. Despite the presence of a null mutation, the current patient exhibited a milder phenotype. The presence of mosaicism can significantly impact the clinical phenotype and accurate recurrence-risk assessment, which should be taken into consideration for genetic counseling.
Acknowledgements
We greatly appreciate the cooperation of the patient and his family for this study. We thank Mrs. Yukako Yamashita for technical assistance. This research was partially supported by the Practical Research Project for Rare/Intractable Diseases from the Japan Agency for Medical Research and Development, AMED under Grant Number JP17ek0109276.
HGV Database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at 10.6084/m9.figshare.hgv.2363
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Lindgren V, de Martinville B, Horwich AL, Rosenberg LE, Francke U. Human ornithine transcarbamylase locus mapped to band Xp21.1 near the Duchenne muscular dystrophy locus. Science. 1984;226:698–700. doi: 10.1126/science.6494904. [DOI] [PubMed] [Google Scholar]
- 2.Gordon N. Ornithine transcarbamylase deficiency: a urea cycle defect. Eur. J. Pediatr. Neurol. 2003;7:115–121. doi: 10.1016/S1090-3798(03)00040-0. [DOI] [PubMed] [Google Scholar]
- 3.Yamaguchi S, Brailey LL, Morizono H, Bale AE, Tuchman M. Mutations and polymorphisms in the human ornithine transcarbamylase (OTC) gene. Hum. Mutat. 2006;27:626–632. doi: 10.1002/humu.20339. [DOI] [PubMed] [Google Scholar]
- 4.Maestri NE, Brusilow SW, Clissold DB, Bassett SS. Long-term treatment of girls with orinithine transcarbamylase deficiency. N. Engl. J. Med. 1996;335:855–859. doi: 10.1056/NEJM199609193351204. [DOI] [PubMed] [Google Scholar]
- 5.Maestri NE, Lord C, Glynn M, Bale A, Brusilow SW. The phenotype of ostensibly healthy women who are carriers for ornithine transcarbamylase deficiency. Medicine (Baltimore) 1998;77:389–397. doi: 10.1097/00005792-199811000-00004. [DOI] [PubMed] [Google Scholar]
- 6.Hudak ML, Jones MD, Brusilow SW. Differentiation of transient hyperammmonemia of the newborn and urea cycle enzyme defects by clinical presentation. J. Pediatr. 1985;107:712–719. doi: 10.1016/S0022-3476(85)80398-X. [DOI] [PubMed] [Google Scholar]
- 7.McCullough BA, et al. Genotype spectrum of ornithine transcarbamylase deficiency: correlation with the clinical and biochemical phenotype. Am. J. Med. Genet. 2000;93:313–319. doi: 10.1002/1096-8628(20000814)93:4<313::AID-AJMG11>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 8.Tuchman M. Mutations and polymorphisms in the human ornithine transcarbamylase gene. Hum. Mutat. 1993;2:174–178. doi: 10.1002/humu.1380020304. [DOI] [PubMed] [Google Scholar]
- 9.Tuchman M, Morizono H, Rajagopal BS, Plante RJ, Allewell NM. Identification of ‘private’ mutations in patients with ornithine transcarbamylase deficiency. J. Inherit. Metab. Dis. 1997;20:525–527. doi: 10.1023/A:1005301513465. [DOI] [PubMed] [Google Scholar]
- 10.Tuchman M, Jaleel N, Morizono H, Sheehy L, Lynch MG. Mutations and polymorphisms in the human ornithine transcarbamulase gene. Hum. Mutat. 2002;19:93–107. doi: 10.1002/humu.10035. [DOI] [PubMed] [Google Scholar]
- 11.Maddalena A, Sosnoski DM, Berry GT, Nussbaum RL. Mosaicism for an intragenic deletion in a boy with mild ornithine transcarbamylase deficiency. N. Engl. J. Med. 1988;319:999–1003. doi: 10.1056/NEJM198810133191507. [DOI] [PubMed] [Google Scholar]
- 12.Legius E, Baten E, Stul M, Marynen P, Cassiman JJ. Sporadic late onset ornithine transcarbamylase deficiency in a boy with somatic mosaicism for an intragenic deletion. Clin. Genet. 1990;38:155–159. doi: 10.1111/j.1399-0004.1990.tb03565.x. [DOI] [PubMed] [Google Scholar]
- 13.Komaki S, et al. Familial lethal inheritance of a mutated paternal gene in females causing X-linked ornithinetranscarbamylase (OTC) deficiency. Am. J. Med. Genet. 1997;69:177–181. doi: 10.1002/(SICI)1096-8628(19970317)69:2<177::AID-AJMG12>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 14.Qin L, et al. Detection and quantification of mosaic mutations in disease genes by next-generation sequencing. J. Mol. Diagn. 2016;18:446–453. doi: 10.1016/j.jmoldx.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 15.Ogino W, et al. Mutation analysis of the transcarbamylase (OTC) gene in five Japanese OTC deficiency patients revealed two known and three novel mutations including a deep intronic mutation. Kobe J. Med. Sci. 2007;53:229–240. [PubMed] [Google Scholar]
- 16.Caldovic L, Abdikarim I, Narain S, Tuchman M, Morizono H. Genotype-phenotype correlations in ornithine transcarbamylase deficiency: a mutation update. J. Genet. Genom. 2015;42:181–194. doi: 10.1016/j.jgg.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hall JG. Review and hypotheses: somatic mosaicism: observations related to clinical genetics. Am. J. Hum. Genet. 1988;43:355–363. [PMC free article] [PubMed] [Google Scholar]
- 18.Girgis N, McGravey V, Shah BL, Herrin J, Shih VE. Lethal ornithine transcarbamylase deficiency in a female neonate. J. Inherit. Metab. Dis. 1987;10:274–275. doi: 10.1007/BF01800079. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The relevant data from this Data Report are hosted at the Human Genome Variation Database at 10.6084/m9.figshare.hgv.2363