

Abstract
Homoisoflavanone-1 is a natural compound that may be extracted from the Chinese medicinal herb Polygonatum odoratum, which has pronounced antioxidant activities. The present study reports that homoisoflavanone-1 significantly inhibited tumor cell growth and induced apoptosis in A549 non-small cell lung cancer (NSCLC) cells in a dose-dependent manner. Homoisoflavanone-1 arrested the cell cycle at the G2/M stage, which was associated with an increase in the accumulation of phosphorylated (p-)p38, p38, p53, and p-cyclin dependent kinase (Cdc)2 proteins, as well as a decrease in Cdc2 expression. In addition, treatment with homoisoflavanone-1 increased the levels of active caspase-3 and decreased Poly ADP-ribose polymerase, which was accompanied by a reduction in the B-cell lymphoma-2/Bak ratio and consequently, apoptosis. Furthermore, homoisoflavanone-1 increased the expression of endoplasmic reticulum (ER) stress-related proteins, including PERK, ATF4 and GADD34 in a dose-dependent manner. In conclusion, homoisoflavanone-1 induced apoptosis in A549 cells by regulating the mitochondria-caspase-dependent and ER stress pathways and resulted in G2/M arrest by activating the p38/p53 signaling pathway. These findings suggest that homoisoflavanone-1 extracted from Polygonatum odoratum may function as a cancer-suppressing agent and has potential as a novel therapeutic method against NSCLC.
Keywords: homoisoflavanone-1, A549 cells, apoptosis, immunoblot
Introduction
Lung cancer is the leading cause of death for both men and women in many countries (1). Νon-small cell lung cancer (NSCLC) accounts for approximately 85% of lung cancer cases (2). Surgery is the most effective treatment for NSCLC if the cancer is diagnosed early. However, up to 75% of patients are diagnosed with NSCLC at an advanced disease (3,4). Although there has been some progress in methods for diagnosis and treatment, the prognosis of the patients with NSCLC has not improved much, and the 5-year survival rate is currently less than 15% (5). Therefore, there is a need to develop new and safe therapeutic agents against NSCLC.
Conventional chemotherapies kill normal cells in addition to cancer cells by inducing cell cycle checkpoint arrest or apoptosis. The initiation of apoptosis is tightly regulated by intrinsic caspase-dependent pathways (including one that is mitochondrion-dependent) and extrinsic caspase-dependent pathways (such as the death receptor-induced caspase activation pathway) (6,7). A shared a terminal execution pathway begins with caspase-3 cleavage and ultimately leads to phagocytosis (8). The best characterized regulators of apoptosis are the Bcl-2 family of proteins, which promote or repress apoptosis by directly regulating a mitochondrial apoptosis-induced channel. The multidomain pro-apoptotic protein Bax/Bak is responsible for forming this channel, while Bcl-2, Bcl-xL, or Mcl-1 inhibit its formation (9,10). Additionally, the intrinsic ER stress pathway is triggered by conditions that disturb protein folding in the endoplasmic reticulum (ER). The induction of the ER stress pathway may contribute to cell death by activating the IRE1 kinase via activation of APOPTOTIC-SIGNALING KINASE (ASK) and p38 MAPK, which itself activates CHOP by phosphorylating the CHOP transactivation domain (11). Additionally, both p38 MAPK and the Jun-N-terminal kinase (JNK) were reported to promote phosphorylation and activation of Bak (12). In addition, Bax and Bak induce cell death by binding and activating INOSITOL-REQUIRING PROTEIN-1α (IRE1α) (13). Intriguingly, accumulating evidence indicates that activated p38 can cause mitotic arrest in the somatic cell cycle at the spindle assembly checkpoint (14).
Traditional herbal medicines with anti-cancer properties that trigger apoptosis in various types of tumor cells with minimal adverse effects have been valuable sources for therapeutic agents. Polygonatum odoratum is widely used as an herbal medicine with procoagulant (15), anti-hyperglycemic (16), anti-herpes simplex virus-II, and apoptosis-inducing (17) activities, that can also improve glucose tolerance (18). However, the active components in P. odoratum for the anti-cancer effects and the underlying mechanisms of these effects remain largely unknown.
Homoisoflavanone-1 is a type of phenolic compound isolated from P. odoratum that has apparent antioxidant activities (19,20). However, the effects of homoisoflavanone-1 on human NSCLC cells, and therefore the mechanism of this effect, have never been elucidated. In the present study, we investigated the effect of homoisoflavanone-1 on NSCLC A549 cell proliferation and cell cycle progression. Our result shows that homoisoflavanone-1 has potential as a new natural anti-tumor medicine for treatment of NSCLC.
Materials and methods
Cell culture and reagents
The human NSCLC cell line A549 was purchased from Basic medical cell center of Peking Union Medical College (Peking, China). A549 cells were cultured in DMEM containing 10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA), 1% penicillin, and 1% streptomycin in 5% CO2 at 37°C. Cells in the exponential phase were used in the experiments.
Dimethyl sulfoxide (DMSO) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Amresco, LLC, (Solon, OH, USA) and Propidium iodide (PI)/Annexin V-FITC was obtained from Sigma-Aldrich; Merck KGaA, (Darmstadt, Germany). Polyclonal antibodies against Caspase 3, Active caspase 3, Bak, Bcl-2, p-p38, p38, p53, Cdc2, p-Cdc2, β-actin, and the horseradish peroxidase-conjugated secondary antibody were bought from Cell Signaling Technology, Inc., (Danvers, MA, USA).
Isolation and purification of homoisoflavanone-1
The homoisoflavanone-1 used in this study was extracted and purified from P. odoratum roots (Fig. 1A) according to the methods described previously with appropriate modification (20). Briefly, 10 kg of dried P. odoratum roots was ground and subjected to two 95% ethanol extractions at room temperature. The solvent was removed under reduced pressure, and the concentrate was diluted in water, followed by filtering. The precipitate including insoluble metabolites was dissolved in 90% methanol. The methanol-soluble fraction was collected and subjected to chromatography on a silica gel column, with a gradient of petroleum ether and petroleum ether-ethyl acetate as the eluting solvent, followed by thin layer chromatography to collect cytotoxic fractions. Based on HPLC analysis of the collected components, an elution with a single component was collected. Homoisoflavanone-1 (17.84 mg) was isolated by reverse-phase preparative HPLC, using methanol:H2O (60:40) as the mobile phase, and was identified by comparing ESI-MS/MS and spectroscopic (1H-NMR and 13C-NMR) data.
Figure 1.
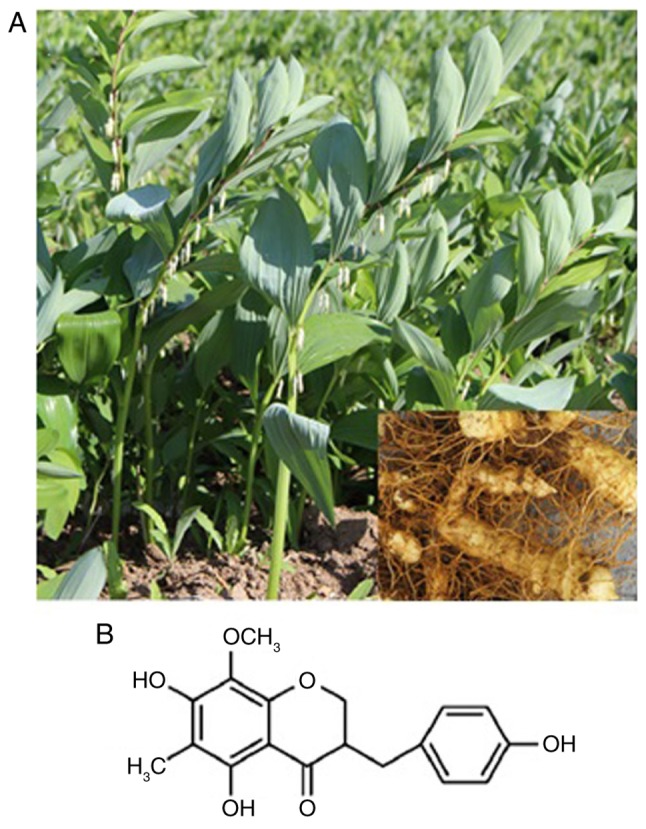
Structure of homoisoflavanone-1 extracted from the roots of Polygonatum odoratum. (A) Image of Polygonatum odoratum. (B) The structure of homoisoflavanone-1 was established via 1H-NMR and 13C-NMR.
The structures of homoisoflavanone-1 were in good agreement with a previous report (20) of it being 5,7-dihydroxyl-6-methyl-8-methoxyl-3-(4′-hydroxylbenzyl)-chroman-4-one (Fig. 1B).
Cell viability assay
The effect of homoisoflavanone-1 on A549 cell viability was determined using the MTT assay. Briefly, cells were incubated in 96-well plates at a density of 10×104 per well for 24 h before treatment with homoisoflavanone-1 or DMSO, as the control. Homoisoflavanone-1 was dissolved in DMSO at serial concentrations (12.5, 25, 50, and 100 µg/ml), and 8 µl of these solutions were added to each well and the samples were incubated for 6, 12, and 24 h. The results were read using a microplate spectrophotometer (BioTek Instruments, Inc., Winooski, VT, USA) at 570 nm. The rate of cell growth inhibition (%) was calculated as: [1-A549 experiment group OD/A549 control OD] ×100%. The half maximal inhibitory concentration (IC50) values were determined by plotting a linear regression curve.
Colony formation assay
A549 cells were seeded in triplicate into 12-well tissue culture plates and cultured for 6 days after treatment with homoisoflavanone-1 at serial concentrations (12.5, 25, 50, and 100 µg/ml). Then, crystal violet was used for staining, and the rate of colony formation was calculated.
Cell cycle analysis
A549 cells were seeded into 6-well plates, incubated for 12 h, and then treated with either homoisoflavanone-1 (12.5, 25, 50, or 100 µg/ml) or DMSO for 24 h. Cells were collected, fixed in 70% ethanol, centrifuged (3,000 g, 5 min) and washed twice with ice-cold PBS. The cells were stained with PI for 20 min at 4°C in the dark. Cell cycle analysis was carried out with a fluorescence-activated cell sorting (FACS) Calibur flow cytometer (BD FACSCalibur; BD Biosciences, Franklin Lakes, NJ, USA USA).
Cell apoptosis analysis
Apoptosis analyses were performed with an Annexin Vfluorescein isothiocyanate (FITC) apoptosis detection kit (BioVision, Inc., Milpitas, CA, USA). A549 cells were seeded in 96-well plates, incubated for 12 h, and then treated with homoisoflavanone-1 (12.5, 25, 50, or 100 µg/ml) for 24 h. Cells were collected and washed twice with ice-cold PBS, followed by incubation with Annexin-V for 10 min in the dark. Next, the cells were incubated with PI for 5 min and evaluated for apoptosis using a FACS Calibur flow cytometer (BD FACSCalibur; BD Biosciences).
Immunoblot analysis
A549 cells were seeded in 6-well plates for 12 h, and then treated with either homoisoflavanone-1 (12.5, 25, 50, or 100 µg/ml) or DMSO for 24 h. Cellular proteins were harvested and immunoblotting was carried out as previously described (21) with appropriate modifications. Cells were resuspended in lysis buffer (1% Triton-X100, 50 mM Hepes pH 7.4, 2 mM sodium orthovanadate, 1 mM edetic acid, 100 mM sodium fluoride, 1 mM PMSF, 10 µg/ml of leupeptin and 10 µg/ml of aprotinin), and the supernatants of the lysates were collected after centrifugation at 14,000 g for 10 min at 4°C. The protein concentration was determined using a protein concentration kit. Proteins were separated with 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinyldene fluoride membranes. The membranes were blocked in 5% skimmed milk for 2 h at room temperature. Protein was detected with antibodies against active caspase 3, caspase 3, PARP, Bcl-2, Bak, p-Cdc2, Cdc2, p-P38, P38, P53 or β-actin at 4°C. The membranes were washed three times with TBST before incubation with the horseradish peroxidase-conjugated secondary antibody at room temperature for 1 h. Immunoreactive bands were visualized using an ECL kit (Sigma-Aldrich; Merck KGaA). Protein levels were quantified relative to the control group.
Statistical analysis
All experiments were performed at least three independent times (n=3), and data are presented as the mean ± standard deviation (SD). The comparisons of multiple groups were performed using the one-way analysis of variance and the group differences were analyzed using Dunnett's post hoc tests. Statistical analyses were performed in SPSS v.13.0 (SPSS, Inc., Chicago, IL, USA) and P<0.05 was considered to indicate a statistically significant difference.
Results
Homoisoflavanone-1 inhibits cell proliferation and colony formation of A549 Cells
An MTT assay was performed to determine the effect of homoisoflavanone-1 on the proliferation of A549 cells. As shown in Table I and Fig. 2A, homoisoflavanone-1 inhibited cell growth in a dose- and time-dependent manner. For example, a remarkable inhibitory effect was observed in cells treated with higher concentrations of homoisoflavanone-1 (50, 100 µg/ml) for 6, 12, and 24 h vs. controls at the same time points (P<0.01), and a significant effect on the growth of A549 cells was still observed at a lower concentration of homoisoflavanone-1 (25 µg/ml) for 12, 24 h (P<0.05). The IC50 values were 49.11, 37.87 and 37.11 µg/ml for 6, 12 and 24 h incubation with homoisoflavanone-1, respectively. In addition, the colony formation rate of A549 cells in homoisoflavanone-1-treated groups was obviously lower than that of the control group (Fig. 2B and C).
Table I.
Effects of homoisoflavanone-1 on cell proliferation in lung cancer A549 cells.
| Concentration (µg/ml) | 6 h | 12 h | 24 h |
|---|---|---|---|
| 0 | 99.04±4.47 | 123.15±9.50 | 114.18±15.21 |
| DMSO | 100.00±0.00 | 100.00±0.00 | 100.00±0.00 |
| 12.5 | 98.80±0.57 | 93.54±26.32 | 94.15±21.33 |
| 25 | 71.70±0.05 | 67.47±22.90a | 67.14±25.07 a |
| 50 | 53.66±4.94b | 38.11±5.94a | 39.27±27.99 b |
| 100 | 22.86±4.84 b | 11.78±5.61a | 7.51±1.22b |
P<0.05
P<0.01 vs. controls at the same time point. Data are shown as the mean ± standard deviation from three independent experiments.
Figure 2.
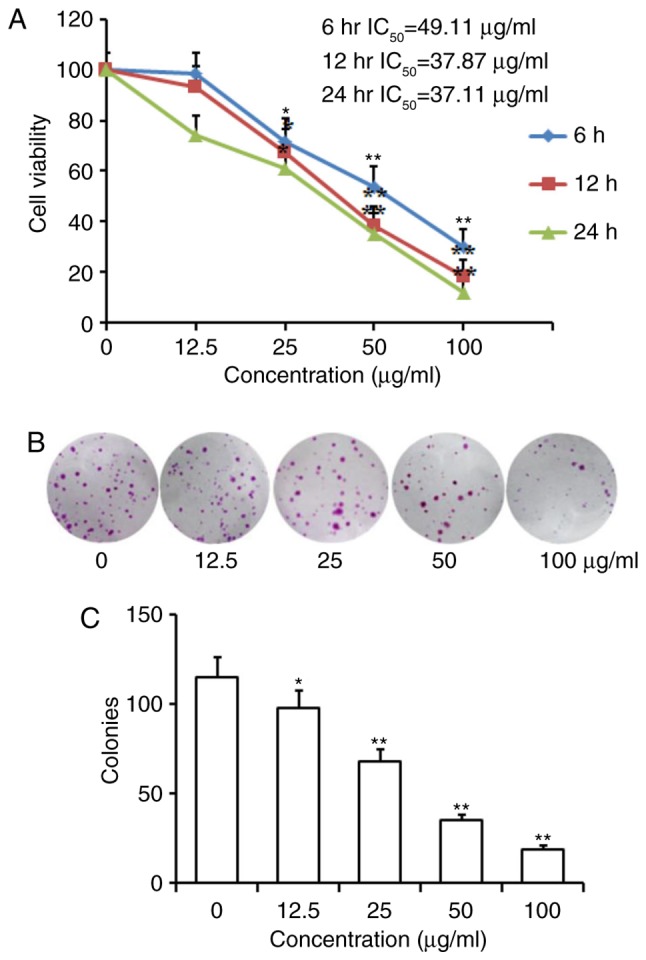
Homoisoflavanone-1 inhibits A549 cells proliferation and colony formation. (A) A549 cells were treated with various concentrations of homoisoflavanone-1 for 6, 12 and 24 h. Cell viability was assessed using MTT assay. (B and C) Homoisoflavanone-1 suppressed colony formation of A549 cells. Cells were treated with homoisoflavanone-1 and then cultured in a fresh medium for 6 days to form colonies. Data are presented as the mean ± standard deviation from three independent experiments. *P<0.05 and **P<0.01 vs. control group.
Homoisoflavanone-1 arrests the cell cycle between the G2/M phases in A549 cells
To address the precise action responsible for the anti-proliferative effect mediated by homoisoflavanone-1, the cell cycle distribution profile was examined. As indicated in Table II, A549 cells were exposed to 0, 12.5, 25, 50, or 100 µg/ml homoisoflavanone-1 for 24 h. The ratio of the G2/M phase was 1.07, 7.34, 10.13, 12.75 and 17.76 for 0, 12.5, 25, 50 and 100 µg/ml, respectively. There was no difference in the proportions of cells in the G0/G1 phases, however, the proportion of cells in the S phase was also reduced in response to homoisoflavanone-1 treatment (Fig. 3A and B).
Table II.
Effects of homoisoflavanone-1 on the cell cycle of A549 cells.
| Concentration (µg/ml) | G0/G1 phase | S phase | G2/M phase |
|---|---|---|---|
| 0 | 55.39±2.37 | 43.99±1.50 | 1.07±1.11 |
| 12.5 | 57.89±2.57 | 36.39±2.09 | 7.34±1.31a |
| 25 | 53.48±3.05 | 36.92±1.98 | 10.13±2.07a |
| 50 | 48.54±2.97 | 27.73±1.71* | 12.75±1.89b |
| 100 | 54.52±2.86 | 26.21±1.42* | 17.76±1.43b |
P<0.05
P<0.01 vs. controls at the same time point. Data are shown as the mean ± standard deviation from three independent experiments.
Figure 3.

Homoisoflavanone-1 induces cell cycle arrest in the G2/M phase in A549 cells. (A) Flow cytometry was performed to investigate, (B) the percentage of cells at each stage of the cell cycle. This revealed the arresting effect of homoisoflavanone-1 on A549 cells. A549 cells were exposed to 0, 12.5, 25, 50 or 100 µg/ml homoisoflavanone-1 for 24 h as indicated (n=3).
Homoisoflavanone-1 induces apoptosis in A549 cells
We conducted flow cytometry to evaluate the effect of homoisoflavanone-1 on A549 cell apoptosis. The results demonstrate that A549 cells treated with increasing concentrations (12.5, 25, 50 and 100 µg/ml) of homoisoflavanone-1 over 24 h showed an increase in the proportion of apoptotic cells (17.22, 18.23, 25.53, and 47.12%, respectively) (Fig. 4A and B). These data showed that homoisoflavanone-1 induced dose-dependent apoptosis in the cells and significantly increased the rate of apoptosis compared with untreated control cells (5.44%).
Figure 4.

Homoisoflavanone-1 induces apoptosis in A549 cells. (A) A549 cells were exposed to 0, 12.5, 25, 50 or 100 µg/ml homoisoflavanone-1 for 24 h, prior to analysis by (A) flow cytometry. (B) Histogram of the rate of apoptosis as determined by FACS analysis for three separate treatments. *P<0.05; **P<0.01 vs. the control. FACS, fluorescence-activated cell sorting.
Homoisoflavanone-1 regulates cell cycle-related proteins in A549 Cells
Activated p38, which is involved in various types cell differentiation, can cause mitotic arrest at the spindle assembly checkpoint in somatic cell cycle (14,22). Additionally, p53 is a key regulator mediating cell cycle arrest induced by a range of factors, including DNA damage and exposure to chemotherapeutic compounds (23). To investigate the molecular events governing homoisoflavanone-1-mediated cell cycle retardation, we used immunoblots to detect the protein levels of p-p38, p38 and p53 in A549 cells after 24 h of various doses of homoisoflavanone-1 (0, 12.5, 25, 50 and 100 µg/ml) treatment. As shown in Fig. 5A-D, there was a dose-dependent increase in the active forms of p-p38, p38 and p53 in response to homoisoflavanone-1 treatment.
Figure 5.

Accumulation of cell-cycle associated proteins in response to homoisoflavanone-1 treatment. (A) A549 cells were treated with 0, 12.5, 25, 50 and 100 µg/ml homoisoflavanone-1 for 24 h and analyzed by western blot analysis. Immunoblotting detected the protein expression of (B) p-p38, (C) p38, (D) p53, (E) p-Cdc2 and (F) Cdc2. β-actin was the loading control. Data are presented as the mean ± standard deviation from three independent experiments. *P<0.05 and **P<0.01 vs. the control group. Cdc, cyclin dependent kinase.
Cdc 2/cyclin B complexes initiate mitosis (M) by phosphorylating both regulatory and structural proteins involved in mitosis (24). CDK activity is itself regulated by phosphorylation and dephosphorylation, as simply binding to cyclins is not sufficient for activation of the complexes (25). As shown in Fig. 5E and F, p-Cdc2 levels increased in a dose-dependent manner in cells that were treated with homoisoflavanone-1 (0, 12.5, 25, 50, and 100 µg/ml), while Cdc2 levels decreased.
Homoisoflavanone-1 induces intrinsic mitochondria-mediated apoptosis
As an effector caspase, caspase-3 remains inactive until apoptotic signaling leads to its cleavage by an initiator caspase (26). Poly ADP-ribose polymerase (PARP) is cleaved between Asp214 and Gly215 by the activated caspase-3, which leads to PARP inactivation and further facilitates apoptotic cell death. As shown in Fig. 6A-D, the amount of caspase 3 and active caspase-3 proteins increased dramatically in a dose-dependent manner after cells were treated with high concentrations of homoisoflavanone-1 (25, 50 and 100 µg/ml), while the amount of PARP protein decreased.
Figure 6.

Accumulation of proteins associated with the mitochondria-caspase pathway for apoptosis in response to homoisoflavanone-1 treatment. (A) A549 cells were treated with 0, 12.5, 25, 50 and 100 µg/ml of homoisoflavanone-1 for 24 and analyzed by western blotting. Immunoblotting detected the protein expression of (B) caspase 3, (C) active caspase 3, (D) PARP, (E) Bcl-2 and (F) Bak. β-actin was the loading control. Data are presented as the mean ± standard deviation from three independent experiments. *P<0.05 and **P<0.01 vs. the control group. PARP, poly ADP-ribose polymerase; Bcl-2, B-cell lymphoma 2.
Bcl-2 proteins regulate caspase activity, and function in intrinsic mitochondrion-mediated apoptosis (27). Within the Bcl-2 family, Bcl-2 is anti-apoptotic, whereas Bak is pro-apoptotic (28). After treatment with various concentrations of homoisoflavanone-1, the amount of Bcl-2 protein decreased significantly, whereas the amount of Bak protein increased in a dose-dependent manner (Fig. 6E and F).
Homoisoflavanone-1 treatment increased the abundance of er stress-related proteins
To further examine homoisoflavanone-1 induced apoptosis in A549 cells, we assessed the expressions of the ER stress-related proteins PERK, ATF4, and GADD34 in homoisoflavanone-1 treated cells. As shown in Fig. 7A and B, phomoisoflavanone-1 (100 µg/ml) significantly increased the abundance of ER stress-related proteins in a time-dependent manner. PERK levels peaked 12 h after homoisoflavanone-1 treatment, while ATF4 and GADD34 reached a peak at 24 h with the same treatment. Homoisoflavanone-1 also increased the expression of these three proteins in a dose-dependent manner at both 12 and 24 h (Fig. 7C-F). These results suggest that the ER stress pathway functions in homoisoflavanone-1-induced lung cancer cell apoptosis.
Figure 7.

Accumulation of proteins associated with the ER stress pathway for apoptosis in response to homoisoflavanone-1 treatment. A549 cells were treated with homoisoflavanone-1 (100 µg/ml) for 0, 6, 12 and 24 h. (A) Western blot analysis was performed to detect the protein expression of (B) PERK, ATF4 and GADD34. A549 cells were treated with 0, 12.5, 25, 50 or 100 µg/ml of homoisoflavanone-1 for 12 h and (C) western blot analysis was performed to detect the protein expression of (D) PERK. A549 cells were treated with 0, 12.5, 25, 50 or 100 µg/ml of homoisoflavanone-1 for 24 h and (E) western blot analysis was performed to detect the protein expression of (F) ATF4 and (G) GADD34. β-actin was the loading control. Data are presented as the mean ± standard deviation from three independent experiments. *P<0.05 and **P<0.01 vs. the control group. ER, endoplasmic reticulum.
Discussion
Homoisoflavanone-1 is a natural product that can be purified from P. odoratum, a traditional herbal medicine due to its anti-hyperglycemic effects (16), procoagulant activity (15), and apoptosis inducing activity (17,29). In the present study, we demonstrated that homoisoflavanone-1 exerted anti-cancer activity by reducing the viability of A549 cells, and that this activity was associated with cell cycle arrest and apoptosis. Moreover, homoisoflavanone-1 activated the mitochondrion and ER stress apoptotic pathways in A549 cells, concomitant with changes in the cell cycle and the expression of apoptosis-related proteins.
Cell cycle progression from G2 to M is regulated by cyclin B/Cdc2, which is a component of the mitosis-promoting factor (MPF) (30). To move past the M phase, the cyclin B/Cdc2 must be inactivated by the cyclin-CDK complex bound to p21 (31). Interestingly, p53 is upstream of p21 in this cascade, and regulates it at the transcriptional level (32). Then, active p38 can induce p53 and p21 activation, at least in part by increasing ROS levels. Upregulated p38 expression can also promote Cdc2 phosphorylation, which inhibits the transcription of cyclin B1 and Cdc2 and therefore reduces the levels of the Cdc2/cyclin B1 complex required for progression from G2 to M (33). Our findings of increased levels of P-P38, p38, p53 and p-Cdc2, and decreased levels of Cdc2, suggest that these proteins are involved in the homoisoflavanone-1-induced G2/M arrest by activating the p38-p53 signaling pathway.
As a process in programmed cell death, apoptosis is necessary for cell growth, development, and homeostasis in metazoans associated with G2/M arrest (34,35). Three well-studied pathways initiate apoptosis: the mitochondrion-mediated intrinsic pathway, the ER stress-induced pathway, and the death receptor-induced extrinsic pathway (36). In the mitochondria-mediated intrinsic pathway, apoptosis is mediated primarily by Bcl-2 family proteins including anti-apoptotic proteins such as Bcl-2 and pro-apoptotic proteins such as Bak. Altering the balance between Bcl-2 and Bak can increase permeability of the mitochondrial outer membrane, leading to cytochrome c release, and ultimately activate caspase cascades (37,38). In addition, ligands in the extrinsic pathway induce the caspase-8 initiator protease, which activates effector proteases such as caspase-3 (39,40). Active caspase-3 cleaves PARP, a nuclear DNA repair enzyme involved maintaining genomic integrity, which further facilitates apoptotic cell death (41,42). In the present study, flow cytometric analysis showed homoisoflavanone-1 induced apoptosis in A549 cells. This was further supported by a reduced Bcl-2/Bak ratio, reflecting by increase in Bak and a decrease in Bcl-2. Together, these data indicate that homoisoflavanone-1 affects the response to mitochondrial-mediated apoptosis. Moreover, homoisoflavanone-1 treatment resulted in an increase in active caspase-3 levels and a decrease in PARP levels, which suggests that homoisoflavanone-1 also induced caspase-associated cell apoptosis.
ER stress-induced cancer cell apoptosis is a high-profile signaling target for the development of cancer therapy drugs. When the cell is under reactive oxygen and calcium ion stress, the ER stress pathway is induced by unfolded or misfolded protein accumulation in the ER lumen (43). Activated PERK is an important sensor for ER stress. When ER stress occurs, the downstream signaling pathway is induced and inhibits protein translation, consequently restoring ER homeostasis (44). As ER stress increases and/or time passes, the PERK signaling pathway activates ATF4 (45), which in turn drives growth arrest and transcription of DNA damage-inducible 34 (GADD34). Continued expression of GADD34 will then induce cell death (46). Consistent with the present study, the protein levels of PERK peaked 12 h after homoisoflavanone-1 (100 mg/l) treatment, while ATF4 and GADD34 peaked 24 h after the same treatment. In addition, we observed an increase in abundance of the ER stress-related proteins PERK, ATF4, and GADD34 in a dose-dependent manner, indicating that ER stress was activated in response to homoisoflavanone-1 treatment in A549 cells.
In conclusion, our study presents the first evidence of a role for homoisoflavanone-1 in inducing cell cycle arrest and in promoting apoptosis in NSCLC A549 cells and reveals that homoisoflavanone-1 accomplishes this by regulating the mitochondrion-caspase-dependent and ER stress pathways. These findings suggest that homoisoflavanone-1 extracted from P. odoratum may function as a tumor suppressor and has potential as a therapeutic agent to treat lung cancer.
Acknowledgements
Not applicable.
Funding
The present study was supported by Scientific Research Fund of Heilongjiang Provincial Education Department (grant no. 2016-KYYWF-0859).
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Authors' contributions
DN participated in the study design and all experimental procedures as well as drafting the manuscript. MJ and TX and JS performed the experiments. ML performed protein identification. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Kohler BA, Ward E, McCarthy BJ, Schymura MJ, Ries LA, Eheman C, Jemal A, Anderson RN, Ajani UA, Edwards BK. Annual report to the nation on the status of cancer, 1975–2007, featuring tumors of the brain and other nervous system. J Natl Cancer Inst. 2011;103:714–736. doi: 10.1093/jnci/djr077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wallace WA. The challenge of classifying poorly differentiated tumours in the lung. Histopathology. 2009;54:28–42. doi: 10.1111/j.1365-2559.2008.03181.x. [DOI] [PubMed] [Google Scholar]
- 3.Smith SL, Palma D, Parhar T, Alexander CS, Wai ES. Inoperable early stage non-small cell lung cancer: Comorbidity, patterns of care and survival. Lung Cancer. 2011;72:39–44. doi: 10.1016/j.lungcan.2010.07.015. [DOI] [PubMed] [Google Scholar]
- 4.Zhao D, Yang G, Meng Q, Liu J, Yang S. Linobiflavonoid inhibits human lung adenocarcinoma A549 cells: Effect on tubulin protein. Mol Biol Rep. 2013;40:6019–6025. doi: 10.1007/s11033-013-2711-3. [DOI] [PubMed] [Google Scholar]
- 5.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 6.Elmore S. Apoptosis: A review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fang C, Zhang J, Qi D, Fan X, Luo J, Liu L, Tan Q. Evodiamine induces G2/M arrest and apoptosis via mitochondrial and endoplasmic reticulum pathways in H446 and H1688 human small-cell lung cancer cells. PLoS One. 2014;9:e115204. doi: 10.1371/journal.pone.0115204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martinvalet D, Zhu P, Lieberman J. Granzyme A induces caspase-independent mitochondrial damage, a required first step for apoptosis. Immunity. 2005;22:355–370. doi: 10.1016/j.immuni.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 9.Dejean LM, Martinez-Caballero S, Manon S, Kinnally KW. Regulation of the mitochondrial apoptosis-induced channel, MAC, by BCL-2 family proteins. Biochim Biophys Acta. 2006;1762:191–201. doi: 10.1016/j.bbadis.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 10.Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ. BAX and BAK regulation of endoplasmic reticulum Ca2+: A control point for apoptosis. Science. 2003;300:135–139. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- 11.Wang XZ, Ron D. Stress-induced phosphorylation and activation of the transcription factor CHOP (GADD153) by p38 MAP Kinase. Science. 1996;272:1347–1349. doi: 10.1126/science.272.5266.1347. [DOI] [PubMed] [Google Scholar]
- 12.Kim BJ, Ryu SW, Song BJ. JNK- and p38 kinase-mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. J Biol Chem. 2006;281:21256–21265. doi: 10.1074/jbc.M510644200. [DOI] [PubMed] [Google Scholar]
- 13.Hetz C, Bernasconi P, Fisher J, Lee AH, Bassik MC, Antonsson B, Brandt GS, Iwakoshi NN, Schinzel A, Glimcher LH, Korsmeyer SJ. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science. 2006;312:572–576. doi: 10.1126/science.1123480. [DOI] [PubMed] [Google Scholar]
- 14.Takenaka K, Moriguchi T, Nishida E. Activation of the protein kinase p38 in the spindle assembly checkpoint and mitotic arrest. Science. 1998;280:599–602. doi: 10.1126/science.280.5363.599. [DOI] [PubMed] [Google Scholar]
- 15.Zhang H, Chen L, Kou JP, Zhu DN, Qi J, Yu BY. Steroidal sapogenins and glycosides from the fibrous roots of Polygonatum odoratum with inhibitory effect on tissue factor (TF) procoagulant activity. Steroids. 2014;89:1–10. doi: 10.1016/j.steroids.2013.10.007. [DOI] [PubMed] [Google Scholar]
- 16.Deng Y, He K, Ye X, Chen X, Huang J, Li X, Yuan L, Jin Y, Jin Q, Li P. Saponin rich fractions from Polygonatum odoratum (Mill.) Druce with more potential hypoglycemic effects. J Ethnopharmacol. 2012;141:228–233. doi: 10.1016/j.jep.2012.02.023. [DOI] [PubMed] [Google Scholar]
- 17.Yang Y, Xu HL, Zhang ZT, Liu JJ, Li WW, Ming H, Bao JK. Characterization, molecular cloning, and in silico analysis of a novel mannose-binding lectin from Polygonatum odoratum (Mill.) with anti-HSV-II and apoptosis-inducing activities. Phytomedicine. 2011;18:748–755. doi: 10.1016/j.phymed.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 18.Park S, Hong SM, Ahn IS, Kim YJ, Lee JB. Huang-Lian-Jie-Du-Tang supplemented with Schisandra chinensis Baill. and Polygonatum odoratum Druce improved glucose tolerance by potentiating insulinotropic actions in islets in 90% pancreatectomized diabetic rats. Biosci Biotechnol Biochem. 2009;73:2384–2392. doi: 10.1271/bbb.90276. [DOI] [PubMed] [Google Scholar]
- 19.Wang D, Zeng L, Li D, Pu W. Antioxidant activities of different extracts and homoisoflavanones isolated from the Polygonatum odoratum. Nat Prod Res. 2013;27:1111–1114. doi: 10.1080/14786419.2012.701212. [DOI] [PubMed] [Google Scholar]
- 20.Guo H, Zhao H, Kanno Y, Li W, Mu Y, Kuang X, Inouye Y, Koike K, Jiang H, Bai H. A dihydrochalcone and several homoisoflavonoids from Polygonatum odoratum are activators of adenosine monophosphate-activated protein kinase. Bioorg Med Chem Lett. 2013;23:3137–3139. doi: 10.1016/j.bmcl.2013.04.027. [DOI] [PubMed] [Google Scholar]
- 21.Bloom J, Cross FR. Multiple levels of cyclin specificity in cell-cycle control. Nat Rev Mol Cell Biol. 2007;8:149–160. doi: 10.1038/nrm2105. [DOI] [PubMed] [Google Scholar]
- 22.Nebreda AR, Porras A. p38 MAP kinases: Beyond the stress response. Trends Biochem Sci. 2000;25:257–260. doi: 10.1016/S0968-0004(00)01595-4. [DOI] [PubMed] [Google Scholar]
- 23.El-Deiry WS. The role of p53 in chemosensitivity and radiosensitivity. Oncogene. 2003;22:7486–7495. doi: 10.1038/sj.onc.1206949. [DOI] [PubMed] [Google Scholar]
- 24.Malumbres M. Physiological relevance of cell cycle kinases. Physiol Rev. 2011;91:973–1007. doi: 10.1152/physrev.00025.2010. [DOI] [PubMed] [Google Scholar]
- 25.Morgan DO. Cyclin-dependent kinases: Engines, clocks, and microprocessors. Annu Rev Cell Dev Biol. 1997;13:261–291. doi: 10.1146/annurev.cellbio.13.1.261. [DOI] [PubMed] [Google Scholar]
- 26.Walters J, Pop C, Scott FL, Drag M, Swartz P, Mattos C, Salvesen GS, Clark AC. A constitutively active and uninhibitable caspase-3 zymogen efficiently induces apoptosis. Biochem J. 2009;424:335–345. doi: 10.1042/BJ20090825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Youle RJ, Strasser A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 28.Harris MH, Thompson CB. The role of the Bcl-2 family in the regulation of outer mitochondrial membrane permeability. Cell Death Differ. 2000;7:1182–1191. doi: 10.1038/sj.cdd.4400781. [DOI] [PubMed] [Google Scholar]
- 29.Ouyang L, Chen Y, Wang XY, Lu RF, Zhang SY, Tian M, Xie T, Liu B, He G. Polygonatum odoratum lectin induces apoptosis and autophagy via targeting EGFR-mediated Ras-Raf-MEK-ERK pathway in human MCF-7 breast cancer cells. Phytomedicine. 2014;21:1658–1665. doi: 10.1016/j.phymed.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 30.Arion D, Meijer L, Brizuela L, Beach D. cdc2 is a component of the M phase-specific histone H1 kinase: Evidence for identity with MPF. Cell. 1988;55:371–378. doi: 10.1016/0092-8674(88)90060-8. [DOI] [PubMed] [Google Scholar]
- 31.Bloom J, Pagano M. To be or not to be ubiquitinated? Cell Cycle. 2004;3:138–140. doi: 10.4161/cc.3.2.659. [DOI] [PubMed] [Google Scholar]
- 32.Bates S, Ryan KM, Phillips AC, Vousden KH. Cell cycle arrest and DNA endoreduplication following p21Waf1/Cip1 expression. Oncogene. 1998;17:1691–1703. doi: 10.1038/sj.onc.1202104. [DOI] [PubMed] [Google Scholar]
- 33.Kang N, Jian JF, Cao SJ, Zhang Q, Mao YW, Huang YY, Peng YF, Qiu F, Gao XM. Physalin A induces G2/M phase cell cycle arrest in human non-small cell lung cancer cells: Involvement of the p38 MAPK/ROS pathway. Mol Cell Biochem. 2016;415:145–155. doi: 10.1007/s11010-016-2686-1. [DOI] [PubMed] [Google Scholar]
- 34.Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. 2011;147:742–758. doi: 10.1016/j.cell.2011.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Green DR, Galluzzi L, Kroemer G. Cell biology. Metabolic control of cell death. Science. 2014;345:1250256. doi: 10.1126/science.1250256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beere HM. Death versus survival: Functional interaction between the apoptotic and stress-inducible heat shock protein pathways. J Clin Invest. 2005;115:2633–2639. doi: 10.1172/JCI26471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang C, Youle RJ. The role of mitochondria in apoptosis*. Annu Rev Genet. 2009;43:95–118. doi: 10.1146/annurev-genet-102108-134850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang F, Kong DS, Zhang ZL, Lei N, Zhu XJ, Zhang XP, Chen L, Lu Y, Zheng SZ. Tetramethylpyrazine induces G0/G1 cell cycle arrest and stimulates mitochondrial-mediated and caspase-dependent apoptosis through modulating ERK/p53 signaling in hepatic stellate cells in vitro. Apoptosis. 2013;18:135–149. doi: 10.1007/s10495-012-0791-5. [DOI] [PubMed] [Google Scholar]
- 39.Cardile V, Musumeci G, Sicurezza E, Caggia S, Rusu MC, Leonardi R, Loreto C. Expression of TRAIL and its receptors DR5 and DcR2 in orthodontic tooth movement. Histol Histopathol. 2013;28:933–940. doi: 10.14670/HH-28.933. [DOI] [PubMed] [Google Scholar]
- 40.Musumeci G, Loreto C, Leonardi R, Castorina S, Giunta S, Carnazza ML, Trovato FM, Pichler K, Weinberg AM. The effects of physical activity on apoptosis and lubricin expression in articular cartilage in rats with glucocorticoid-induced osteoporosis. J Bone Miner Metab. 2013;31:274–284. doi: 10.1007/s00774-012-0414-9. [DOI] [PubMed] [Google Scholar]
- 41.Shakibaei M, Csaki C, Nebrich S, Mobasheri A. Resveratrol suppresses interleukin-1beta-induced inflammatory signaling and apoptosis in human articular chondrocytes: Potential for use as a novel nutraceutical for the treatment of osteoarthritis. Biochem Pharmacol. 2008;76:1426–1439. doi: 10.1016/j.bcp.2008.05.029. [DOI] [PubMed] [Google Scholar]
- 42.Virág L, Szabó C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev. 2002;54:375–429. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- 43.Boyce M, Yuan J. Cellular response to endoplasmic reticulum stress: A matter of life or death. Cell Death Differ. 2006;13:363–373. doi: 10.1038/sj.cdd.4401817. [DOI] [PubMed] [Google Scholar]
- 44.Wang J, Hu X, Jiang H. ERS-PERK signaling pathwaymediated Nrf2/ARE-HO-1 axis: A novel therapeutic target for attenuating myocardial ischemia and reperfusion injury. Int J Cardiol. 2016;203:779–780. doi: 10.1016/j.ijcard.2015.11.033. [DOI] [PubMed] [Google Scholar]
- 45.Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6:1099–1108. doi: 10.1016/S1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 46.Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013;1833:3460–3470. doi: 10.1016/j.bbamcr.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.