
Fig. 8. Integrated downregulation of SIRT1 and DEPTOR contributes to the pathogenesis in patients with ALD.
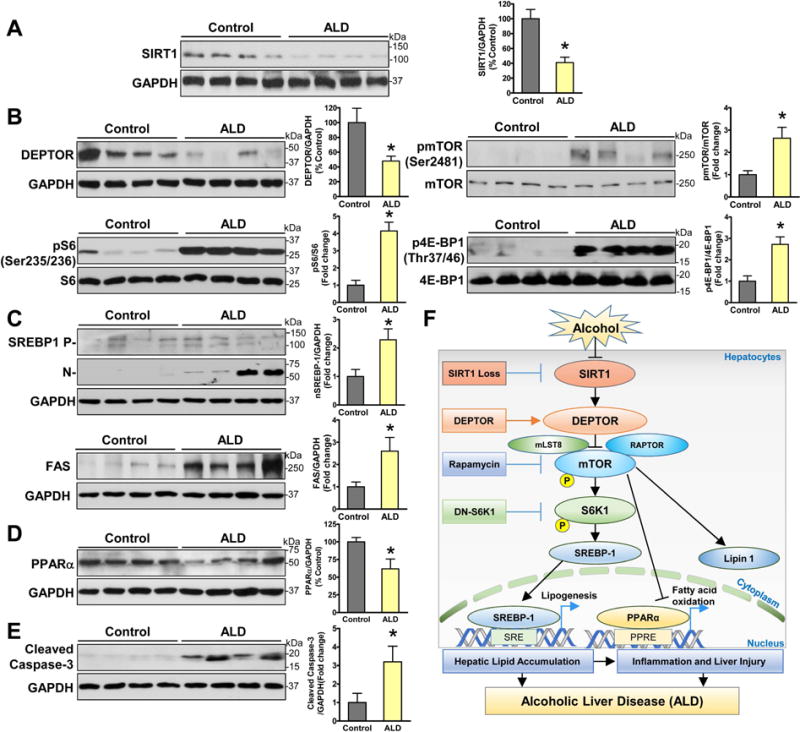
A–E. Representative immunoblots and densitometric quantification for SIRT1, mTORC1 signaling, lipid metabolic regulators, and apoptosis in normal liver tissues (n= 6) and liver tissues from patients with ALD (n =8). The data are presented as the mean ± SEM. *P < 0.05 vs. normal liver tissue. F. The proposed model for the deregulation of the SIRT1-DEPTOR-mTORC1 axis in the pathogenesis of ALD in mice and humans. Chronic alcohol consumption causes SIRT1 suppression in hepatocytes, which is coupled to the downregulation of DEPTOR and activation of mTORC1 and S6K1. Aberrant activation of mTORC1 by alcohol stimulates the proteolytic processing, nuclear translocation, and transcriptional activity of SREBP-1, promotes the cytoplasmic translocation of lipin 1, and inhibits the transcriptional activity of PPARα, which in turn increases fatty acid synthesis and downregulates fatty acid oxidation. Alcohol-induces hepatic lipogenic process acts through parallel S6K1-dependent and independent pathway. Alcohol feeding acts largely via SIRT1 inhibition and mTORC1 activation to induce excess fat accumulation and apoptosis in hepatocytes. Hepatic lipotoxicity and inflammation likely contribute to the development of acute-on-chronic alcoholic liver injury. Hepatic loss of SIRT1 impairs DEPTOR function, stimulates mTORC1 and lipogenesis, and promotes triglyceride overproduction, thereby leading to inflammation and liver injury in ALD. DEPTOR-dependent inhibition of mTORC1 provides a potential druggable target for treating ALD in humans.