

Abstract
The ability of ketamine administration to activate prefrontal glutamate neurotransmission is thought to be a key mechanism contributing to its transient psychotomimetic effects and its delayed and sustained antidepressant effects. Rodent studies employing carbon-13 magnetic resonance spectroscopy (13C MRS) methods have shown ketamine and other N-methyl-D-aspartate (NMDA) receptor antagonists to transiently increase measures reflecting glutamate–glutamine cycling and glutamate neurotransmission in the frontal cortex. However, there are not yet direct measures of glutamate neurotransmission in vivo in humans to support these hypotheses. The current first-level pilot study employed a novel prefrontal 13C MRS approach similar to that used in the rodent studies for direct measurement of ketamine effects on glutamate–glutamine cycling. Twenty-one participants (14 healthy and 7 depressed) completed two 13C MRS scans during infusion of normal saline or subanesthetic doses of ketamine. Compared to placebo, ketamine increased prefrontal glutamate–glutamine cycling, as indicated by a 13% increase in 13C glutamine enrichment (t = 2.4, p = 0.02). We found no evidence of ketamine effects on oxidative energy production, as reflected by 13C glutamate enrichment. During ketamine infusion, the ratio of 13C glutamate/glutamine enrichments, a putative measure of neurotransmission strength, was correlated with the Clinician-Administered Dissociative States Scale (r = −0.54, p = 0.048). These findings provide the most direct evidence in humans to date that ketamine increases glutamate release in the prefrontal cortex, a mechanism previously linked to schizophrenia pathophysiology and implicated in the induction of rapid antidepressant effects.
Introduction
The discovery [1] and replication [2] of the antidepressant effects of ketamine, and the implication of N-methyl-D-aspartate receptor (NMDAR) hypofunction in the pathophysiology of schizophrenia [3] have generated considerable excitement about the potential of targeting glutamate neurotransmission to develop safe, effective, and rapid acting antidepressants (RAADs) [4], as well as to develop glutamate-based “antipsychotics” to treat the often resistant cognitive deficits and negative symptoms in schizophrenia [5, 6].
However, to date not a single RAAD or glutamate-based antipsychotic has been approved to treat depression or schizophrenia, although two decades have passed since the RAAD and psychotomimetic effects of ketamine were first reported in the 1990s [3, 7]. A major obstacle in this field is the lack of biomarkers that directly reflect synaptic glutamate neurotransmission. Such markers would serve to (1) test ketamine’s RAAD and psychotomimetic mechanisms in humans and (2) permit expedited screening and optimization of putative novel glutamate-based RAAD and antipsychotic agents.
Carbon-13 magnetic resonance spectroscopy (13C MRS) is a unique, noninvasive method to measure neurotransmitter cycling and cell-specific neuroenergetics [8–10]. In the current study, we implemented a 13C MRS pharmacoimaging paradigm, using a novel method [11–13] to conduct quantitative 13C MRS acquisition in the human frontal lobe—a region closely related to psychopathology that was not initially accessible in 13C MRS studies. We aimed to determine ketamine’s effect on glutamate–glutamine cycling and to investigate the association between changes in neurotransmission and the psychotomimetic effects of ketamine.
13C MRS is presently the only method that provides direct dynamic measurements of glutamate–glutamine cycling in the human brain [10]. Briefly, labeled 13C-glucose is infused intravenously over 120 min during the MRS acquisition. The incorporation of the 13C in glutamate and glutamine generates unique signals on the 13C spectrum (Fig. 1). Metabolites of 13C-glucose enter the tricarboxylic acid (TCA; also known as Krebs) cycle, which in turn label glutamate. Then, as 13C-glutamate is released into the synaptic cleft, astrocytes take it up and convert it to 13C-glutamine (Fig. 2). The glutamate and glutamine 13C enrichments rise rapidly in the first 20–30 min of 13C-glucose infusion and reach a steady-state in the last 40–60 min (Fig. S1). Accordingly, the glutamate enrichment primarily reflects oxidative energy production through the TCA cycle, while the rate of glutamine enrichment primarily reflects glutamate neurotransmitter cycling [8, 10].
Fig. 1.

Prefrontal 13C magnetic resonance spectroscopy (MRS) acquisition and 13C spectrum. Sagittal (a) and axial (b) view of the region of interest—based on the radius of the carbon coil—primarily rostral Brodmann Area 10. c 13C magnetic resonance spectrum acquired at 4 T from the prefrontal region of a study participant during infusion of [U 13C]-glucose. Color code: blue—raw; red—fitted; green—residual. Abbreviations: GluC45 13C-Glutamate C45, GlnC45 13C-Glutamine C45, and AspC34 13C-Aspartate C34
Fig. 2.
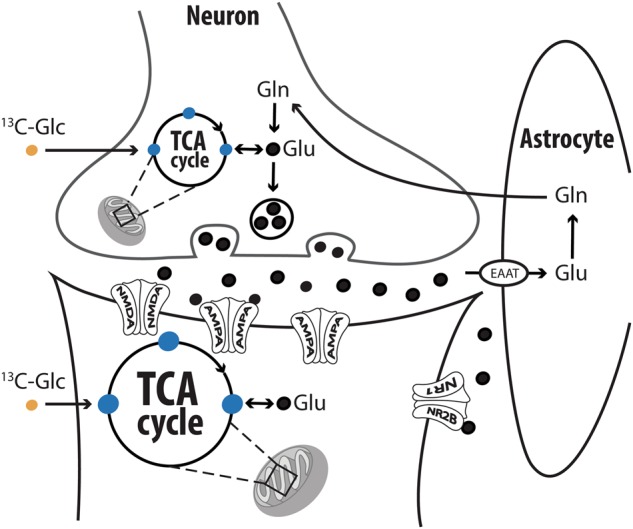
Carbon-13 (13C) labeling of glutamate and glutamine via the TCA and glutamate–glutamine cycle. Following glycolysis, 13C-Glucose (13C-Glc) metabolites (i.e., acetyl-CoA) enter the mitochondrial tricarboxylic acid (TCA) cycle (also known as Krebs cycle) and subsequently label glutamate through exchange with α-ketoglutarate. Next, 13C-glutamate is released into the synaptic cleft and taken up by astrocytes, where the 13C-glutamate is converted to 13C-glutamine and transferred to neurons. Hence, the rate of 13C-glutamate enrichment is primarily affected by neuroenergetics (the neuronal TCA cycle) and the rate of 13C-glutamine enrichment primarily reflects the rate of glutamate–glutamine cycling. The development of this metabolic model and potential impact of other metabolic pathways on the measured labeling are discussed in reference [10]. The figure was adapted with permission from the Emerge Research Program (emerge.care)
A main limitation of previous methods is the lack of distinction between presynaptic glutamate release and postsynaptic activation, the latter has been related to the RAAD effects of ketamine [14, 15]. While these processes are highly coupled under normal conditions, preclinical 13C MRS data suggest disproportionate changes following ketamine administration [16, 17]. In particular, approximately 9 min after intraperitoneal injection in rodents, ketamine induces a rise in prefrontal 13C glutamine enrichment (~20%) with no changes in 13C glutamate (~1%) [17], suggesting a disproportionate change in neuroenergetics (VTCA) vs. glutamate cycling (Vcycle)—i.e., an alteration in neuronal energy-per-cycle (EPC) ratio. By 18 min after the injection, both prefrontal 13C glutamate (~13%) and glutamine (~35%) enrichment increased [17]. Under normal conditions, neuronal energy largely supports postsynaptic neuronal activation and functional increments are consistently proportional to changes in glutamate–glutamine cycling, resulting in a largely constant EPC ratio across species and functional conditions [18, 19]. The alteration of EPC acutely post injection may reflect a transient reduction in postsynaptic activation [9] due to blockade of postsynaptic NMDAR by ketamine and its metabolite, norketamine. Thus, an alteration of EPC immediately post ketamine injection (i.e., an increase in presynaptic release without proportionate increase in postsynaptic activation) would be consistent with the NMDAR hypofunction model of schizophrenia, raising the question whether EPC, as reflected by 13C glutamate/glutamine ratio, would predict the psychotomimetic effects of ketamine during infusion in humans. Consistent with this hypothesis, the increase in 13C glutamate/glutamine ratio in rodents is more evident following the administration of low doses of ketamine (3 and 10 mg/kg), selective NMDAR subtype 2B antagonist, and scopolamine—all of which are expected to have lower psychotomimetic effects compared to 30 mg/kg of ketamine. This possibility, if confirmed, would signify a paradigm shift suggesting that the behavioral disturbances during ketamine infusion may be driven by a loss of communication fidelity between presynaptic neurotransmission release and postsynaptic activation, rather than a correlate only of increased glutamate neurotransmission. Considering that the RAAD effects are associated with postsynaptic glutamate activation, the EPC biomarker will also provide a mechanism through which novel drugs may induce RAAD effects without psychotomimetic symptoms, provided that these new agents equally increase presynaptic release and postsynaptic activation.
In the current study, we primarily aimed to implement a novel, complex yet unique, pharmacoimaging paradigm to translate the preclinical 13C MRS ketamine findings into a biomarker to apply in humans in vivo, investigating its potential utility in rational drug development for mood and psychotic disorders, as well as for a variety of other potential neuropsychiatric indications. Additionally, considering recent preclinical evidence, we tested the hypothesis that ketamine in a cohort of healthy and depressed subjects would induce a large increase in prefrontal 13C glutamine enrichment, with a modest increase in 13C glutamate enrichment comparable to recent preclinical findings [17]. Increased 13C-glutamine enrichment could be interpreted as a ketamine-induced surge in glutamate–glutamine cycling, and a rise in 13C glutamate enrichment would suggest an acute increase in prefrontal oxidative energy. Based on findings from glutamate inhibitor studies [20–23], we predicted a positive association between glutamate–glutamine cycling and the behavioral psychotomimetic symptoms following ketamine administration. We also examined whether variability in 13C glutamate/glutamine ratio, a putative indicator of EPC, would be associated with the psychotomimetic effects of ketamine as suggested by preclinical data.
Methods
Study population
Twenty-four participants [17 healthy and 7 major depressive disorder (MDD)] between the age of 21 and 65 years were recruited for this study. Of the healthy subjects, one subject did not complete the second scan, and two subjects had unsuccessful scans. Subjects completed an informed consent process and were screened for study inclusion and exclusion criteria. For all participants, exclusion criteria consisted of medical conditions that contraindicate MRS or increase the risks of ketamine administration. A negative drug test, and for women, a negative pregnancy test and use of a medically accepted means of contraception were required. Healthy participants were excluded if they had a lifetime history of any psychiatric disorder, or if they had a first-degree relative with mood, anxiety, or psychotic disorder. MDD participants were included if they met the following study criteria: (1) current MDD as determined by a structured interview; (2) Montgomery-Åsberg Depression Rating Scale (MADRS) severity of 18 or more at baseline; (3) no substance abuse or dependence in the previous 12 months; (4) no lifetime history of psychosis or bipolar disorder; and (5) medication-free or on a stable (>4 weeks) antidepressant treatment. The MDD group was included in the current report to explore the possibility of differences between the two groups and to test the feasibility of conducting the 13C MRS pharmacoimaging paradigm in this population. The target sample for the MDD group is 18 subjects, which will be reported in the future once enrollment is completed.
Overview of study design
Two 13C MRS scans, separated by at least 1 week, were performed using a single-blind, placebo-controlled, fixed-order within-subject design (Fig. S2). On day 1, participants received normal saline and uniformly 13C-labeled glucose infused over 120 min as described previously [24]. Concurrently, 13C MRS of the frontal brain region was used to observe glutamate and glutamine 13C enrichments during the placebo infusion. On day 8+, healthy participants received a subanesthetic dose that has been commonly used in studies examining the psychotomimetic effects of the drug (0.23 mg/kg bolus followed by 0.58 mg/kg infusion over approximately 75 min). This dose was administered to ensure rapid and robust psychotomimetic effect [25, 26], which we believed would enhance statistical power to ascertain the relationship between glutamate cycling and the psychotomimetic symptoms during ketamine infusion. MDD patients received the dose most commonly used in depression clinical trials (0.5 mg/kg infused over 40 min) [1, 2], concurrently with otherwise identical 13C-glucose infusion and 13C MRS scans. The psychotomimetic effects of ketamine were assessed using the Clinician-Administered Dissociative States Scale (CADSS) and Brief Psychiatric Rating Scale (BPRS) (Fig. S2). The behavioral assessments in the MDD participants will be investigated in future reports once a larger cohort is enrolled.
13C MRS acquisition and processing
MRS data were acquired on a 4.0 T whole-body magnet interfaced to a Bruker AVANCE spectrometer (Bruker Instruments, Billerica, MA, USA). Subjects were placed supine in the magnet, with their head immobilized with foam. An RF probe consisting of one circular 13C coil (8.5 cm ∅) and two circular, quadrature-driven 1H RF coils (12.5 cm ∅) were used for acquisition of 13C MR spectra from the frontal lobe (Fig. 1a, b). Following tuning and acquisition of scout images, second-order shimming of the region of interest (ROI) was performed using phase mapping provided by Bruker.
13C MR spectra were acquired with a pulse-acquire sequence using an adiabatic 90° excitation pulse and optimized repetition time. Nuclear Overhauser enhancement (nOe) was achieved by applying 1H block pulses before the 13C excitation pulse. 1H decoupling during acquisition consists of pseudo noise decoupling as described by Li et al. [12], to decouple the long-range 1H–13C coupling of the carboxylic carbon positions. The pseudo noise decoupling pulse has a constant amplitude and the phase of each 1.2-ms unit pulse is randomly assigned to either 0° or 180°. Following the start of [U 13C]-glucose infusion, 6.5-min blocks of MR spectra were acquired for 120 min (Figs. 1c and S1). In the last 60 min of acquisition, we alternated 6.5-min blocks of acquisition using our standard parameters with acquisition without nOe and a 30 s delay in order to obtain a correction for nOe efficiency and saturation effects.
Spectral data were analyzed with −2/6 Hz Lorentzian-to-Gaussian conversion and 16-fold zero-filing followed by Fourier transformation. An Linear Combination (LC) model approach was used to fit the peak areas of the labeled carbon positions of glutamate C45 and glutamine C45 (Fig. 1c), which overlapped with aspartate C34. Due to the overlap, we analyzed the combined aspartate and glutamate peaks. However, it is unlikely that the surge in glutamine enrichment was affected by changes in aspartate during ketamine considering that the kinetics of aspartate labeling closely track that of its glutamate precursor [27], which was not shown to be affected by ketamine infusion. The amplitude of 13C-labeling over early time points (approximately 20 min) was averaged and normalized by labeling over the steady-state amplitude of the 13C enrichment time course (post-90 min). This procedure provided a normalized measure of 13C glutamate and 13C glutamine enrichment, which reflects the enrichment rate of these metabolites early during the infusion of placebo and ketamine. Cramer–Rao Lower Bounds were used to estimate the quality of the individual measurements, averaging 3% for glutamate and 11% for glutamine. To ensure that the observed 13C enrichment values are not affected by variability in input glucose enrichment, plasma fractional enrichment (FE) of 13C glucose was determined during placebo (early FE = 51.2%; steady state FE = 61.1%) and ketamine (early FE = 51.6%; steady state FE = 61.8%), which did not differ between sessions (p > 0.2). To provide an estimate of the variability of the 13C MRS measures between sessions, we computed the ratio of the mean over the standard deviation of each of the measures across all 21 subjects during the placebo scans. We found relatively low variability, with ratios of 7.3 for 13C glutamine enrichment and 11 for 13C glutamate enrichment. The ratios were comparable across healthy (7 for 13C glutamine and 11 for 13C glutamate) and depressed subjects (8 for 13C glutamine and 10 for 13C glutamate).
Statistical analyses
Before conducting each analysis, the distributions of outcome measure were examined. Estimates of variation are provided as standard error of mean (SEM). Considering that this is a first-in-human study, this study should be considered a first-level pilot study implementing a novel technique, rather than a confirmatory study. Effect sizes are provided as Cohen’s d. All tests are 2-tailed with significance set at p ≤ 0.05.
The percent change of 13C glutamate and 13C glutamine enrichment during ketamine compared to placebo was computed and represented the primary outcome measures. One sample t-tests examined whether the percent changes of each metabolite were statistically significant. Follow-up analysis used independent t-tests to determine whether the percent changes of each metabolite differed between the study groups. Spearman’s correlations examined the relationship between the metabolites enrichment and psychotomimetic effects of ketamine as determined by CADSS and BPRS in healthy participants.
Results
A total of 21 participants (14 healthy and 7 MDD) successfully completed both study sessions [mean (SEM) age = 30 (1.2) years; 15 males]. In the MDD group, the baseline MADRS was 27 ± 1.8 (mean ± SEM), and three subjects were on a stable antidepressant treatment. As described in “Methods”, the full assessment of the behavioral (antidepressant/psychotomimetic) effects of ketamine in the MDD group, and their association with 13C enrichment, will be investigated in future reports with a larger cohort currently being recruited and studied.
Ketamine effects on glutamine and glutamate enrichment
To assess the presence of a glutamate surge, we compared early time points of the enrichment of glutamate and glutamine between the placebo and ketamine groups. This period was comparable to that studied previously by 13C MRS in rodent models [16, 17]. In the current study, the 13C glutamine enrichment was 13% greater during ketamine infusion than with placebo (t = 2.4, df = 20, p = 0.02; Fig. 3), but no significant effect of ketamine on 13C glutamate enrichment was seen (t = 0.2, df = 20, p = 0.81; Fig. 3). To examine whether the cycling surge differed in MDD, we conducted an exploratory analysis, which showed a comparable effect size in healthy (d = 0.48) and MDD (d = 0.78), with no statistically significant differences between groups (p = 0.85; Fig. 4).
Fig. 3.
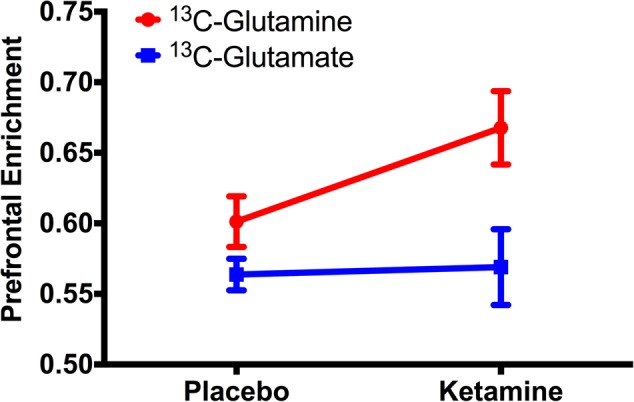
Effects of ketamine on prefrontal glutamate–glutamine cycling and neuroenergetics. In the first 20 min post infusion, ketamine induced a rapid increase in 13C-glutamine enrichment, indicating an acute surge in prefrontal glutamate–glutamine cycling. The enrichment of 13C-glutamate remained stable, suggesting no changes in oxidative energy production early during ketamine infusion
Fig. 4.

Separate effects of ketamine on prefrontal glutamate–glutamine cycling and neuroenergetics in healthy and major depressive disorder (MDD) subjects. a Mean and SEM of 13C-glutamate and 13C-glutamine during placebo (Plc) and during ketamine (Ket). b Individual values, as well as mean and SEM, of percent change in enrichment of 13C-glutamine during ketamine compared to placebo in each group. The pattern of changes in 13C-glutamine and 13C-glutamate enrichments was comparable between healthy and depressed subjects, with no statistically significant differences between the two groups. The similarities indicate that while the impacts of ketamine on glutamate–glutamine cycling were significant, both groups were affected to about the same extent
Exploratory analyses: psychotomimetic effects of ketamine
Compared to placebo, ketamine in the healthy group induced significant increases in CADSS [mean difference (SEM) = 28 (4.1), t = 6.9, df = 13, p < 0.001] and BPRS Positive symptoms [mean difference (SEM) = 10 (1.8), t = 5.5, df = 13, p < 0.001], but not BPRS Negative symptoms [mean difference (SEM) = 1 (0.6), t = 1.7, df = 13, p = 0.10].
During the ketamine infusion, the 13C glutamate/glutamine enrichment ratio was negatively correlated with CADSS (r = −0.54, n = 14, p = 0.048; Cohen’s d = 1.3; Fig. 5a), but not with BPRS Positive (r = −0.43, n = 14, p = 0.13; Cohen’s d = 1.0; Fig. 5b) or BPRS Negative symptoms (r = 0.02, n = 14, p = 0.96; Cohen’s d = 0.03). Otherwise, there were no significant correlations between 13C glutamine or 13C glutamate enrichment and any of CADSS, BPRS Positive, or BPRS Negative scores (p > 0.1). During placebo infusion, there were no significant correlations between any of the metabolites and psychotomimetic measures (p > 0.1).
Fig. 5.

Association between glutamate neurotransmission and the psychotomimetic effects of ketamine in healthy participants. a Reduced neuroenergetics relative to neurotransmitter cycling, as measured by the 13C-glutamate/13C-glutamine (13C Glu/Gln) ratio, is associated with the psychotomimetic effects of ketamine, as measured by the Clinician-Administered Dissociative States Scale (CADSS). b The association with the Brief Psychiatric Rating Scale (BPRS) positive symptoms was comparable in effect size, but did not reach statistical significance in this sample of 14 subjects
Discussion
The study results provide the most direct evidence yet in humans of a ketamine-induced increase in prefrontal glutamate release, based on the detection of increased 13C labeling of glutamine. The rapid initial labeling of glutamine is consistent with previous findings in preclinical models. Comparable to preclinical findings with 13C MRS [17], the changes in 13C-glutamine (13%) were more prominent than 13C-glutamate (1%). Given the exponential decrease in ketamine levels after injection, ketamine and norketamine brain levels during infusions in humans are likely to resemble the immediate time period post-injection in rodents, in which there was no significant increase in 13C glutamate enrichment (also ~1%). The previous 13C MRS work with rodents also examined a set of animals in which 13C administration was begun with an 18-min delay after ketamine administration, and in that condition the increase in 13C glutamate was more apparent, though still less than glutamine.
The ketamine-induced acute glutamate surge also plays an important role in the psychotomimetic effects of the drug, behavioral alterations that are believed to capture core phenomenology of the pathophysiology of schizophrenia [28]. Lacking a direct measure of glutamate neurotransmission in vivo, investigators over the past two decades have employed several elegant approaches to infer the presence of a ketamine-induced glutamate surge in humans and to relate this surge to its psychotomimetic effects. Early studies have shown that inhibitors of glutamate release attenuate the ketamine-induced psychotomimetic symptoms [20–23]. Other studies have employed pharmacoimaging paradigms to demonstrate a relationship between ketamine-induced prefrontal activation/connectivity and its transient behavioral disturbances [29–36], or to block prefrontal activation/connectivity using glutamate inhibitors [20, 37]. More recently, a metabotropic glutamatergic receptor tracer was also used to explore the acute effects of ketamine [38–40]. 1H MRS, a method capable of estimating total—but not dynamic cycling of—prefrontal glutamate and glutamine levels show changes in tissue content of glutamate and/or glutamine during ketamine infusion [32, 41–43], but also see [44]. Together, the data provided indirect evidence supporting the presence of a ketamine-induced surge in glutamate neurotransmission in humans and that this surge may be associated with the psychotomimetic effects of the drug.
While underscoring the pilot nature of the correlation analyses in the current report, it is intriguing that CADSS scores significantly correlated with glutamate/glutamine ratio (r = −0.54, n = 14, p = 0.048; Cohen’s d = 1.3), but not with glutamine enrichment (r = −0.38, n = 14, p = 0.19; Cohen’s d = 0.8). A possible explanation is that the low glutamate/glutamine enrichment ratio reflects low EPC. In this model, healthy individuals with high sensitivity to ketamine (i.e., NMDAR hypofunction) would have higher blockade of synaptic NMDAR, leading to reduced postsynaptic activation per glutamate cycle, and subsequent increase in psychotomimetic symptoms. This hypothesis could be assessed in future 13C MRS studies enrolling larger sample of healthy individuals with variable sensitivity to ketamine (e.g., with/without family history of alcoholism [45, 46]), as well as by testing the ability of NMDAR agonists to affect the 13C glutamate/glutamine ratio. The approach parallels extent preclinical data [16, 17], which facilitates the translatability of the biomarker between species.
Limitations
Thus far, this study provided first-level pilot evidence about the potential utility of the 13C MRS pharmacoimaging paradigm in human models of schizophrenia, demonstrating the long-hypothesized ketamine-induced glutamate surge and presenting pilot evidence associating the psychotomimetic effects of ketamine with reduced EPC, as estimated by the 13C glutamate/glutamine ratio. However, considering the pilot nature of the association findings, and the lack of direct modulation implicating NMDAR hypofunction in the variability of 13C glutamate/glutamine ratio, future replications and laboratory testing are required. To assess the psychotomimetic effects in the healthy group, we administered a larger dose along with a bolus. Future studies aiming to compare neurotransmission changes between healthy and depressed groups would require administering the same doses in both population. Other limitations include the small sample size, which requires replication in larger studies, and the fixed-order acquisition, which requires future test–retest reliability data or randomized parallel groups design. Moreover, the overlap between glutamine and aspartate peaks prevented the individual fitting of aspartate and glutamine. Finally, due to limitations that may include biological variability and low spatial resolution of 13C MRS, the application of the 13C-glutamate/glutamine ratio may be limited to alterations in glutamate neurotransmission in groups of individuals or within subjects. However, new higher spatial resolution 1H–13C MRS methods that can measure glutamate and glutamine, without aspartate, may allow application to evaluate individual subject responses in the future [47].
Preclinical studies have found that the NMDAR antagonist ketamine blocks glutamate neurotransmission, but only at high anesthetic doses [48]. At subanesthetic doses, ketamine induces a paradoxical increase in glutamate neurotransmission [14, 49]. The acute surge in glutamate transmission increases prefrontal synaptic connectivity, through enhanced activity-dependent brain-derived neurotrophic factor (BDNF) and related downstream signaling [14]. Other putative RAADs (e.g., scopolamine and (2R,6R)-Hydroxynorketamine) show a comparable acute surge in prefrontal glutamate [17, 50–53] and enhanced BDNF signaling [54, 55]. Together, the preclinical evidence to date underscores the critical role of acute prefrontal glutamate neurotransmission in the mechanism of action of various RAADs. It also highlights the role of prefrontal synaptic plasticity—i.e., the increased density and connectivity evident at 24 h post treatment and lasting for a week [4, 56].
In humans, functional global brain connectivity was previously used to provide a well-replicated evidence of reduced prefrontal connectivity in individuals with MDD [37, 57–59], a dysconnectivity that is normalized 24 h post-ketamine treatment [37, 57]. The ketamine-induced subacute increases in prefrontal connectivity were positively associated with the antidepressant response [57]. In healthy subjects, a direct link between prefrontal connectivity and glutamate neurotransmission was demonstrated, while showing an acute ketamine-induced surge in prefrontal connectivity, which is attenuated by a glutamate inhibitor [37]. Together, the human evidence is consistent with the findings from preclinical models, with particular focus on the essential role of acute and subacute prefrontal synaptic neurotransmission and connectivity in the mechanism of ketamine and perhaps other RAADs.
In this context, the 13C MRS pharmacoimaging paradigm and findings offer the following evidence and possibilities: (1) It is consistent with the occurrence of an acute ketamine-induced glutamate surge in humans, using a paradigm that is readily translated between animals and humans. (2) The similar time frames between preclinical observations with human connectivity and neurotransmitter cycling data raises the question whether acute increases in glutamate–glutamine cycling would predict the antidepressant effects of ketamine, comparable to findings from preclinical and connectivity data. (3) The findings, taken together with published observations, suggest the possibility of a positive relationship between prefrontal glutamate cycling and global connectivity. Such data would not only inform us about the mechanisms of RAADs, but also provide important information about the interplay between molecular neurotransmission and large-scale intrinsic networks in the human brain. (4) It may be that cycling and connectivity measures could facilitate the optimization of antipsychotics and RAADs doses prior to testing in costly clinical trials. Taken together, these points raise the possibility of application as rigorous and reproducible biomarkers of treatment target engagement and validation to improve the development of novel antipsychotics and RAADs.
Electronic supplementary material
Acknowledgments
The authors would like to thank the individuals who participated in this study for their invaluable contribution. Funding and research support were provided by NIMH (K23MH101498), NIAAA (R01AA021984), the VA National Center for PTSD, Brain & Behavior Foundation (NARSAD), Patterson Trust Award, American Psychiatric Foundation, Clinical Neuroscience Research Unit (CNRU) at Connecticut Mental Health Center, and Yale Center for Clinical Investigation (YCCI UL1 RR024139), an NIH Clinical and Translational Science Award (CTSA). The content is solely the responsibility of the authors and does not necessarily represent the official views of the sponsors, the Department of Veterans Affairs, NIH, or the U.S. Government.
Competing interests
CGA has served as a consultant and/or on advisory boards for Genentech and Janssen, and editor of “Chronic Stress” for Sage Publications, Inc. JHK is a consultant for AbbVie, Inc., Amgen, Astellas Pharma Global Development, Inc., AstraZeneca Pharmaceuticals, Biomedisyn Corporation, Bristol–Myers Squibb, Eli Lilly and Company, Euthymics Bioscience, Inc., Neurovance, Inc., FORUM Pharmaceuticals, Janssen Research & Development, Lundbeck Research USA, Novartis Pharma AG, Otsuka America Pharmaceutical, Inc., Sage Therapeutics, Inc., Sunovion Pharmaceuticals, Inc., and Takeda Industries. JHK is on the Scientific Advisory Board for Lohocla Research Corporation, Mnemosyne Pharmaceuticals, Inc., Naurex, Inc., and Pfizer. JHK is a stockholder in Biohaven Pharmaceuticals, holds stock options in Mnemosyne Pharmaceuticals, Inc., holds patents for Dopamine and Noradrenergic Reuptake Inhibitors in Treatment of Schizophrenia, U.S. Patent No. 5,447,948 (issued September 5, 1995), and Glutamate Modulating Agents in the Treatment of Mental Disorders, U.S. Patent No. 8,778,979 (issued July 15, 2014); and filed a patent for Intranasal Administration of Ketamine to Treat Depression. U.S. Application No. 14/197,767 (filed on March 5, 2014); U.S. application or Patent Cooperation Treaty international application No. 14/306,382 (filed on June 17, 2014). GS reports personal consulting fees from Alkermes, Allergan, Biohaven Pharmaceuticals, Eli Lilly and Co., Genentech, Intra-Cellular Therapies, Janssen Pharmaceuticals, Lundbeck Research USA, Merck & Co., Naurex, Navitor Pharmaceuticals, Noven Pharmaceuticals, Teva Pharmaceuticals Industries, Taisho Pharmaceutical Co., Takeda Pharmaceutical Co., Sage Pharmaceuticals, Inc., Sevier, Valeant Pharmaceuticals, and Vistagen Therapeutics, Inc. GS has grants and research contracts from Eli Lilly and Co., Janssen Pharmaceuticals, Merck & Co., and Sevier; and support from Sanofi-Aventis, in the form of free medication for an NIH sponsored study over the last 36 months. In addition, Dr. GS is a stockholder and holds stock options in Biohaven Pharmaceuticals; and has a patent for Glutamate Modulating Agents in the Treatment of Mental Disorders, U.S. Patent No. 8,778,979 (issued July 15, 2014) with royalties paid from Biohaven Pharmaceuticals. GFM is a consultant for Sumitomo Dainippon Pharma Co. Ltd. and UCB Pharma SA, and serves on the Scientific Advisory Board of Elucidata, Inc. All other authors report no competing interests.
Ethics approval
All study procedures were approved by an institutional review board (ClinicalTrials.gov NCT02037035).
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Douglas L. Rothman, Gerard Sanacora, Graeme F. Mason
Electronic supplementary material
Supplementary Information accompanies this paper at (10.1038/s41386-018-0136-3).
References
- 1.Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351–4. doi: 10.1016/S0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- 2.Zarate CA, Jr., Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63:856–64. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- 3.Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51:199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- 4.Murrough JW, Abdallah CG, Mathew SJ. Targeting glutamate signalling in depression: progress and prospects. Nat Rev Drug Discov. 2017;16:472–86. doi: 10.1038/nrd.2017.16. [DOI] [PubMed] [Google Scholar]
- 5.Javitt DC, Zukin SR, Heresco-Levy U, Umbricht D. Has an angel shown the way? Etiological and therapeutic implications of the PCP/NMDA model of schizophrenia. Schizophr Bull. 2012;38:958–66. doi: 10.1093/schbul/sbs069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moghaddam B, Krystal JH. Capturing the angel in “angel dust”: twenty years of translational neuroscience studies of NMDA receptor antagonists in animals and humans. Schizophr Bull. 2012;38:942–9. doi: 10.1093/schbul/sbs075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krystal JH, Berman RM, Skolnick P, Paul IA, Sanacora G. Glutamate and the pathophysiology and treatment of depression. Biol Psychiatry. 1999;45:1S–47S. doi: 10.1016/S0006-3223(99)80008-0. [DOI] [Google Scholar]
- 8.de Graaf RA, Rothman DL, Behar KL. State of the art direct 13C and indirect 1H-[13C] NMR spectroscopy in vivo. A practical guide. NMR Biomed. 2011;24:958–72. doi: 10.1002/nbm.1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abdallah CG, Jiang L, De Feyter HM, Fasula M, Krystal JH, Rothman DL, et al. Glutamate metabolism in major depressive disorder. Am J Psychiatry. 2014;171:1320–7. doi: 10.1176/appi.ajp.2014.14010067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rothman DL, De Feyter HM, de Graaf RA, Mason GF, Behar KL. 13C MRS studies of neuroenergetics and neurotransmitter cycling in humans. NMR Biomed. 2011;24:943–57. doi: 10.1002/nbm.1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li S, Zhang Y, Wang S, Araneta MF, Johnson CS, Xiang Y, et al. 13C MRS of occipital and frontal lobes at 3 T using a volume coil for stochastic proton decoupling. NMR Biomed. 2010;23:977–85. doi: 10.1002/nbm.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li S, Zhang Y, Wang S, Yang J, Ferraris Araneta M, Farris A, et al. In vivo 13C magnetic resonance spectroscopy of human brain on a clinical 3 T scanner using [2-13C]glucose infusion and low-power stochastic decoupling. Magn Reson Med. 2009;62:565–73. doi: 10.1002/mrm.22044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sailasuta N, Robertson LW, Harris KC, Gropman AL, Allen PS, Ross BD. Clinical NOE 13C MRS for neuropsychiatric disorders of the frontal lobe. J Magn Reson. 2008;195:219–25. doi: 10.1016/j.jmr.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hare B, Ghosal S, Duman R. Rapid acting antidepressants in chronic stress models: molecular and cellular mechanisms. Chronic Stress. 2017;1:1–16. doi: 10.1177/2470547017697317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abdallah CG, Sanacora G, Duman RS, Krystal JH. The neurobiology of depression, ketamine and rapid-acting antidepressants: is it glutamate inhibition or activation? Pharmacol Ther. 2018. 10.1016/j.pharmthera.2018.05.010. [DOI] [PMC free article] [PubMed]
- 16.Chowdhury GM, Behar KL, Cho W, Thomas MA, Rothman DL, Sanacora G. 1H-[13C]-nuclear magnetic resonance spectroscopy measures of ketamine’s effect on amino acid neurotransmitter metabolism. Biol Psychiatry. 2012;71:1022–5. doi: 10.1016/j.biopsych.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chowdhury GM, Zhang J, Thomas M, Banasr M, Ma X, Pittman B, et al. Transiently increased glutamate cycling in rat PFC is associated with rapid onset of antidepressant-like effects. Mol Psychiatry. 2017;22:120–6. doi: 10.1038/mp.2016.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hyder F, Rothman DL, Bennett MR. Cortical energy demands of signaling and nonsignaling components in brain are conserved across mammalian species and activity levels. Proc Natl Acad Sci USA. 2013;110:3549–54. doi: 10.1073/pnas.1214912110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shulman RG, Rothman DL, Behar KL, Hyder F. Energetic basis of brain activity: implications for neuroimaging. Trends Neurosci. 2004;27:489–95. doi: 10.1016/j.tins.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 20.Deakin JF, Lees J, McKie S, Hallak JE, Williams SR, Dursun SM. Glutamate and the neural basis of the subjective effects of ketamine: a pharmaco-magnetic resonance imaging study. Arch Gen Psychiatry. 2008;65:154–64. doi: 10.1001/archgenpsychiatry.2007.37. [DOI] [PubMed] [Google Scholar]
- 21.Krystal JH, Abi-Saab W, Perry E, D’Souza DC, Liu N, Gueorguieva R, et al. Preliminary evidence of attenuation of the disruptive effects of the NMDA glutamate receptor antagonist, ketamine, on working memory by pretreatment with the group II metabotropic glutamate receptor agonist, LY354740, in healthy human subjects. Psychopharmacology (Berl) 2005;179:303–9. doi: 10.1007/s00213-004-1982-8. [DOI] [PubMed] [Google Scholar]
- 22.Krystal JH, Mathew SJ, D’Souza DC, Garakani A, Gunduz-Bruce H, Charney DS. Potential psychiatric applications of metabotropic glutamate receptor agonists and antagonists. CNS Drugs. 2010;24:669–93. doi: 10.2165/11533230-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 23.Anand A, Charney DS, Oren DA, Berman RM, Hu XS, Cappiello A, et al. Attenuation of the neuropsychiatric effects of ketamine with lamotrigine: support for hyperglutamatergic effects of N-methyl-D-aspartate receptor antagonists. Arch Gen Psychiatry. 2000;57:270–6. doi: 10.1001/archpsyc.57.3.270. [DOI] [PubMed] [Google Scholar]
- 24.Chen H, De Feyter HM, Brown PB, Rothman DL, Cai S, de Graaf RA. Comparison of direct (13)C and indirect (1)H-[(13)C] MR detection methods for the study of dynamic metabolic turnover in the human brain. J Magn Reson. 2017;283:33–44. doi: 10.1016/j.jmr.2017.08.004. [DOI] [PubMed] [Google Scholar]
- 25.D’Souza DC, Singh N, Elander J, Carbuto M, Pittman B, de Haes JU, et al. Glycine transporter inhibitor attenuates the psychotomimetic effects of ketamine in healthy males: preliminary evidence. Neuropsychopharmacology. 2012;37:1036–46. doi: 10.1038/npp.2011.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gunduz-Bruce H, Reinhart RM, Roach BJ, Gueorguieva R, Oliver S, D’Souza DC, et al. Glutamatergic modulation of auditory information processing in the human brain. Biol Psychiatry. 2012;71:969–77. [DOI] [PMC free article] [PubMed]
- 27.Gruetter R, Seaquist ER, Ugurbil K. A mathematical model of compartmentalized neurotransmitter metabolism in the human brain. Am J Physiol Endocrinol Metab. 2001;281:E100–12. doi: 10.1152/ajpendo.2001.281.1.E100. [DOI] [PubMed] [Google Scholar]
- 28.Moghaddam B, Javitt D. From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology. 2012;37:4–5. doi: 10.1038/npp.2011.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Breier A, Malhotra AK, Pinals DA, Weisenfeld NI, Pickar D. Association of ketamine-induced psychosis with focal activation of the prefrontal cortex in healthy volunteers. Am J Psychiatry. 1997;154:805–11. doi: 10.1176/ajp.154.6.805. [DOI] [PubMed] [Google Scholar]
- 30.Driesen NR, McCarthy G, Bhagwagar Z, Bloch M, Calhoun V, D’Souza DC, et al. Relationship of resting brain hyperconnectivity and schizophrenia-like symptoms produced by the NMDA receptor antagonist ketamine in humans. Mol Psychiatry. 2013;18:1199–204. doi: 10.1038/mp.2012.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Driesen NR, McCarthy G, Bhagwagar Z, Bloch MH, Calhoun VD, D’Souza DC, et al. The impact of NMDA receptor blockade on human working memory-related prefrontal function and connectivity. Neuropsychopharmacology. 2013;38:2613–22. doi: 10.1038/npp.2013.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Javitt DC, Carter CS, Krystal JH, Kantrowitz JT, Girgis RR, Kegeles LS, et al. Utility of imaging-based biomarkers for glutamate-targeted drug development in psychotic disorders: a randomized clinical trial. JAMA Psychiatry. 2018;75:11–9. doi: 10.1001/jamapsychiatry.2017.3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vollenweider FX, Leenders KL, Oye I, Hell D, Angst J. Differential psychopathology and patterns of cerebral glucose utilisation produced by (S)- and (R)-ketamine in healthy volunteers using positron emission tomography (PET) Eur Neuropsychopharmacol. 1997;7:25–38. doi: 10.1016/S0924-977X(96)00042-9. [DOI] [PubMed] [Google Scholar]
- 34.Vollenweider FX, Leenders KL, Scharfetter C, Antonini A, Maguire P, Missimer J, et al. Metabolic hyperfrontality and psychopathology in the ketamine model of psychosis using positron emission tomography (PET) and [18F]fluorodeoxyglucose (FDG) Eur Neuropsychopharmacol. 1997;7:9–24. doi: 10.1016/S0924-977X(96)00039-9. [DOI] [PubMed] [Google Scholar]
- 35.Anticevic A, Corlett PR, Cole MW, Savic A, Gancsos M, Tang Y, et al. N-methyl-D-aspartate receptor antagonist effects on prefrontal cortical connectivity better model early than chronic schizophrenia. Biol Psychiatry. 2015;77:569–80. doi: 10.1016/j.biopsych.2014.07.022. [DOI] [PubMed] [Google Scholar]
- 36.Anticevic A, Gancsos M, Murray JD, Repovs G, Driesen NR, Ennis DJ, et al. NMDA receptor function in large-scale anticorrelated neural systems with implications for cognition and schizophrenia. Proc Natl Acad Sci USA. 2012;109:16720–5. doi: 10.1073/pnas.1208494109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abdallah CG, Averill CL, Salas R, Averill LA, Baldwin PR, Krystal JH, et al. Prefrontal connectivity and glutamate transmission: relevance to depression pathophysiology and ketamine treatment. Biol Psychiatry Cogn Neurosci Neuroimaging. 2017;2:566–74. doi: 10.1016/j.bpsc.2017.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davis MT, Holmes SE, Pietrzak RH, Esterlis I. Neurobiology of chronic stress-related psychiatric disorders: evidence from molecular imaging studies. Chronic Stress. 2017;1. 10.1177/2470547017710916. [DOI] [PMC free article] [PubMed]
- 39.DeLorenzo C, DellaGioia N, Bloch M, Sanacora G, Nabulsi N, Abdallah C, et al. In vivo ketamine-induced changes in [(11)C]ABP688 binding to metabotropic glutamate receptor subtype 5. Biol Psychiatry. 2015;77:266–75. doi: 10.1016/j.biopsych.2014.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Esterlis I, DellaGioia N, Pietrzak RH, Matuskey D, Nabulsi N, Abdallah CG, et al. Ketamine-induced reduction in mGluR5 availability is associated with an antidepressant response: an [(11)C]ABP688 and PET imaging study in depression. Mol Psychiatry. 2018;23:824–32. doi: 10.1038/mp.2017.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Milak MS, Proper CJ, Mulhern ST, Parter AL, Kegeles LS, Ogden RT, et al. A pilot in vivo proton magnetic resonance spectroscopy study of amino acid neurotransmitter response to ketamine treatment of major depressive disorder. Mol Psychiatry. 2016;21:320–7. doi: 10.1038/mp.2015.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rowland LM, Bustillo JR, Mullins PG, Jung RE, Lenroot R, Landgraf E, et al. Effects of ketamine on anterior cingulate glutamate metabolism in healthy humans: a 4-T proton MRS study. Am J Psychiatry. 2005;162:394–6. doi: 10.1176/appi.ajp.162.2.394. [DOI] [PubMed] [Google Scholar]
- 43.Stone JM, Dietrich C, Edden R, Mehta MA, De Simoni S, Reed LJ, et al. Ketamine effects on brain GABA and glutamate levels with 1H-MRS: relationship to ketamine-induced psychopathology. Mol Psychiatry. 2012;17:664–5. doi: 10.1038/mp.2011.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taylor MJ, Tiangga ER, Mhuircheartaigh RN, Cowen PJ. Lack of effect of ketamine on cortical glutamate and glutamine in healthy volunteers: a proton magnetic resonance spectroscopy study. J Psychopharmacol. 2012;26:733–7. doi: 10.1177/0269881111405359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krystal JH, Petrakis IL, Limoncelli D, Webb E, Gueorgueva R, D’Souza DC, et al. Altered NMDA glutamate receptor antagonist response in recovering ethanol-dependent patients. Neuropsychopharmacology. 2003;28:2020–8. doi: 10.1038/sj.npp.1300252. [DOI] [PubMed] [Google Scholar]
- 46.Petrakis IL, Limoncelli D, Gueorguieva R, Jatlow P, Boutros NN, Trevisan L, et al. Altered NMDA glutamate receptor antagonist response in individuals with a family vulnerability to alcoholism. Am J Psychiatry. 2004;161:1776–82. doi: 10.1176/ajp.161.10.1776. [DOI] [PubMed] [Google Scholar]
- 47.De Feyter HM, Herzog RI, Steensma BR, Klomp DWJ, Brown PB, Mason GF, et al. Selective proton-observed, carbon-edited (selPOCE) MRS method for measurement of glutamate and glutamine (13) C-labeling in the human frontal cortex. Magn Reson Med. 2018;80:11–20. [DOI] [PMC free article] [PubMed]
- 48.Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17:2921–7. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abdallah CG, Adams TG, Kelmendi B, Esterlis I, Sanacora G, Krystal JH. Ketamine’s mechanism of action: a path to rapid-acting antidepressants. Depress Anxiety. 2016;33:689–97. doi: 10.1002/da.22501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pham TH, Defaix C, Xu X, Deng SX, Fabresse N, Alvarez JC, et al. Common neurotransmission recruited in (R,S)-ketamine and (2R,6R)-hydroxynorketamine-induced sustained antidepressant-like effects. Biol Psychiatry. 2018;84:e3–6. [DOI] [PubMed]
- 51.Schwarcz R, Wu HQ, Zanos P, Gould T. Endogenous kynurenic acid mediates ketamine- and (2R,6R)-hydroxynorketamine-induced increases in extracellular glutamate and antidepressant actions. Neuropsychopharmacology. 2017;43:S111. doi: 10.1038/npp.2017.264. [DOI] [Google Scholar]
- 52.Voleti B, Navarria A, Liu RJ, Banasr M, Li N, Terwilliger R, et al. Scopolamine rapidly increases mammalian target of rapamycin complex 1 signaling, synaptogenesis, and antidepressant behavioral responses. Biol Psychiatry. 2013;74:742–9. doi: 10.1016/j.biopsych.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wohleb ES, Wu M, Gerhard DM, Taylor SR, Picciotto MR, Alreja M, et al. GABA interneurons mediate the rapid antidepressant-like effects of scopolamine. J Clin Invest. 2016;126:2482–94. doi: 10.1172/JCI85033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fukumoto K, Kato T, Li XY, Duman R. Role of BDNF/mTORC1 signaling in the long-lasting antidepressant effects of (2R,6R)-hydroxynorketamine. Neuropsychopharmacology. 2017;43:S111. doi: 10.1038/npp.2017.264. [DOI] [Google Scholar]
- 55.Ghosal S, Bang E, Yue W, Hare BD, Lepack AE, Girgenti MJ, et al. Activity-dependent brain-derived neurotrophic factor release is required for the rapid antidepressant actions of scopolamine. Biol Psychiatry. 2018;83:29–37. doi: 10.1016/j.biopsych.2017.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zanos P, Gould TD. Intracellular signaling pathways involved in (S)- and (R)-ketamine antidepressant actions. Biol Psychiatry. 2018;83:2–4. doi: 10.1016/j.biopsych.2017.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abdallah CG, Averill LA, Collins KA, Geha P, Schwartz J, Averill C, et al. Ketamine treatment and global brain connectivity in major depression. Neuropsychopharmacology. 2017;42:1210–9. doi: 10.1038/npp.2016.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Murrough JW, Abdallah CG, Anticevic A, Collins KA, Geha P, Averill LA, et al. Reduced global functional connectivity of the medial prefrontal cortex in major depressive disorder. Hum Brain Mapp. 2016;37:3214–23. doi: 10.1002/hbm.23235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scheinost D, Holmes SE, DellaGioia N, Schleifer C, Matuskey D, Abdallah CG, et al. Multimodal investigation of network level effects using intrinsic functional connectivity, anatomical covariance, and structure-to-function correlations in unmedicated major depressive disorder. Neuropsychopharmacology. 2018;43:1119–27. doi: 10.1038/npp.2017.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.