

Abstract
The clustered regularly interspersed palindromic repeats (CRISPR)-CRISPR-associated protein 9 (Cas9) prokaryotic adaptive immune defense system has been co-opted as a powerful tool for precise eukaryotic genome engineering. Here, we present a rapid and simple method using chimeric single guide RNAs (sgRNA) and CRISPR-Cas9 Ribonucleoproteins (RNPs) for the efficient and precise generation of genomic point mutations in C. elegans. We describe a pipeline for sgRNA target selection, homology-directed repair (HDR) template design, CRISPR-Cas9-RNP complexing and delivery, and a genotyping strategy that enables the robust and rapid identification of correctly edited animals. Our approach not only permits the facile generation and identification of desired genomic point mutant animals, but also facilitates the detection of other complex indel alleles in approximately 4 - 5 days with high efficiency and a reduced screening workload.
Keywords: Genetics, Issue 134, CRISPR, Cas9, Ribonucleoprotein, C. elegans, genome engineering, sod-1
Introduction
Recent technological advances have radically transformed and accelerated the ability to precisely engineer genomes. In particular, the CRISPR-Cas9 system, which relies on the RNA-guided endonuclease Cas9 to induce a double strand break (DSB) near the target sequence of interest, has been extensively used to accurately engineer the genome of the majority of model organisms used in biomedical research1,2,3,4. Significantly, the use of CRISPR-Cas9 has unlocked genome editing even in difficult species like C. elegans5. Regardless of species, generating point mutations with the CRISPR-Cas9 based genome editing system relies on three core components: 1) Cas9 endonuclease, 2) a single guide RNA (sgRNA) that directs the Cas9 endonuclease to a target sequence, and 3) a user designed homology-directed repair (HDR) template containing the desired edit(s) of interest2.
There are several methods that can be used to introduce the targeting sgRNA and Cas9 nuclease into cells including plasmid, RNA, and viral-based delivery methods6. Recently, direct delivery of pre-complexed sgRNA-Cas9 Ribonucleoproteins (RNPs) has emerged as a powerful and efficient tool in CRISPR-Cas9-based genome editing7. The direct delivery of pre-complexed CRISPR-Cas9 RNPs has several distinct advantages, namely: 1) RNPs bypass the need for cellular transcription and translation, 2) RNPs are rapidly cleared, which may increase specificity by reducing available time for off-target cleavage, and 3) RNPs contain no foreign DNA/RNA elements which circumvents the introduction of non-native sequences into the host genome through random integration. Together, these attributes likely provide a short-lived burst of on-target CRISPR editing while minimizing off-target effects.
We describe a simple and efficient protocol for introducing site-specific genomic changes in C. elegans. This protocol includes targeting sgRNA and single stranded oligonucleotide (ssODN) HDR template design, sgRNA-Cas9 RNP complexing and delivery, and a genotyping strategy for the unequivocal identification of properly edited animals. Using this strategy, not only can the desired site-specific changes be recovered, but other non-specific indel mutations may also be recovered. Thus, our strategy permits the generation of an allelic series using a single strategy, where both mono-allelic, bi-allelic, and indel mutants can be generated in the F1 generation.
Protocol
All animal care and experimental procedures followed the guideline from the National Institutes of Health and the Institutional Animal Care and Use Committee (IACUC) at the University of Michigan. Use RNase-free solutions and pipette tips throughout the protocol. Clean the working area, pipettes, tubes, and centrifuge with RNase Decontamination solution following the manufacturer guidelines (see Materials Table).
1. sgRNA Target Selection
- Using a web browser, open the webpage: http://crispor.tefor.net8
- Input ~60 base pairs (bp) of sequence flanking the desired edit of interest (i.e. 30 bp 5' and 3' of desired edit).
- Select the Caenorhabditis elegans genome from the pulldown menu.
- Select the Protospacer Adjacent Motif (20 bp-NGG - SpCas9, SpCas9-HF1, eSPCas9 1.1) from the pulldown menu.
- Click submit. In the results page, choose the top ranked target sequence closest to the edit of interest. If there are no suitable target sites within the flanking 60 bp input sequence, expand the query sequence up to 100 bp (i.e. 50 bp either side of the desired edit).
From an available source, e.g. synthego, obtain a 20-mer target sgRNA with the following specifications: 3 nmol; no modifications.
Upon receipt, centrifuge lyophilized sgRNA containing tube at >12,000 x g for 1 min at room temperature. Add 60 µL of nuclease-free TE to the tube, and gently re-suspend by pipetting up and down 15 - 20 times using a P200 pipette. The final concentration is 50 µM.
2. Homology-directed Repair Template Design
- Design an ssODN HDR template containing: 1) the mutation of interest, 2) a unique in-frame restriction endonuclease site, 3) a silent mutation of one or both of the Gs within the NGG PAM sequence, and 4) 50 bp 5' and 3' homology arms flanking the first and last mutation. (Figure 1C)9,10.
- If mutation of the NGG PAM sequence is not possible, introduce 5 - 6 silent mutations within the sgRNA target recognition sequence to prevent sgRNA-mediated cleavage of the ssODN HDR template.
From an available source, e.g. idtdna, obtain an "Ultramer DNA oligonucleotide" with the following parameters: Scale: 4 nmol; Formulation: None; Purification: Standard Desalting.
Upon receipt, centrifuge lyophilized ssODN containing tube at >12,000 x g for 1 min at room temperature. Gently re-suspend ssODN to a final concentration of 100 µM in nuclease-free ddH2O by pipetting up and down 15 - 20 times using a P200 pipette.
3. Design Genotyping Primers
4. Prepare Injection Mix
Centrifuge Table 1 reagents at maximum speed for 2 min at 4 °C.
In the order listed, add Table 1 reagents to a nuclease-free PCR tube.
Mix thoroughly by gently pipetting up and down 10 times with a P20 pipette.
Incubate injection mix for 10 min at room temperature.
5. Injection Protocol
Load injection micropipette with approximately one-half of the injection mix12. Save remaining injection mix in case the injection needle clogs or breaks.
Break micropipette tip so that ~20 - 30 p.s.i. produces a gradual flowing solution12. NOTE: Larger diameter tips negatively affect animal health and decrease F1 progeny yield.
Inject 10 - 15 young adult worms in both gonadal arms if possible12. If not, injection into one gonadal arm is sufficient.
Allow injected animals (P0) to recover for 1 - 2 h at room temperature on an OP50-seeded 35 mm nematode growth media (NGM) plate. Subsequently, single injected P0 animals to individual OP50-seeded 35 mm NGM plates using a platinum wire worm pick12.
6. Screen P0 Plates and Single mCherry(+) F1s
Two days post-injection, identify P0 plates containing F1 progeny expressing mCherry in the pharynx using a fluorescent stereomicroscope (Figure 1D, day 2 - 3). Choose three P0 plates that contain the most mCherry(+) F1 animals.
From each of the three selected P0 plates, single 8 - 12 mCherry(+) F1 animals for a total of 24-36 mCherry(+) F1s using a worm pick (Figure 1D, day 2 - 3). NOTE: mCherry(-) animals can also be singled from these P0 plates, but the probability of identifying correctly edited animals is much lower (Figure 2A).
Allow individual F1s to self-fertilize and lay eggs for 1 - 2 days at room temperature.
7. Single Worm PCR and Genotyping
After 1 - 2 days of egg-laying, transfer the F1s into individual PCR strip tube caps containing 7 µL of Worm Lysis Buffer using a worm pick (Table 2, Figure 1D, day 4 - 5).
Centrifuge PCR tubes at maximum speed for 1 min at room temperature to bring animals to the tube bottom, and freeze tubes at -80 °C for 1 h. NOTE: Worms can be stored at -80 °C indefinitely.
Lyse frozen worms in a thermocycler using the following program: 60 °C for 60 min, 95 °C for 15 min, 4 °C hold.
Set-up PCR mastermix as shown in Table 3.
Add 21 µL of PCR mastermix into clean PCR tubes, and then add 4 µL of worm lysis from step 3. Mix well using a pipette and run PCR program following the manufacturers guidelines (see Materials Table).
Purify PCR reactions using a DNA Clean and Concentrate kit, following the manufacturer instructions (see Materials Table). Elute DNA by adding 10 µL water to the spin column.
Set-up restriction enzyme mastermix as shown in Table 4.
Add 10 µL of the restriction enzyme mastermix to each cleaned PCR reaction and incubate for 1 - 2 h at 37 °C13.
Separate digested PCR products on a 1.5% agarose gel run at 120 V using 1x Tris-acetate-EDTA (TAE) buffer14.
8. Identification and Sequence Verification of Edited Animals
Examine enzyme digested samples for the presence of bands that indicate potential edited animals14 (Figure 1D, day 4 - 5). NOTE: Load an N2 control to identify potential indel mutations which can be observed as shifts in band size and/or unexpected and complex restriction enzyme digestion banding patterns.
After identifying potential edited animals, use the remaining 3 µL of worm lysis and repeat the PCR reaction. Do not clean or restriction enzyme digest the PCR reaction after the program is complete. Load the PCR reaction on a 1% agarose gel, separate and extract the band using a gel extraction kit15 (see Materials Table).
Sanger sequence16 gel extracted DNA using the F1 primer to identify correctly edited animals (Figure 1A) NOTE: Heterozygote reads will contain overlaid peaks due to the presence of the wild type and engineered allele.
To obtain homozygous animals from verified heterozygous edited animals, single 8 - 12 F2 animals and perform steps 7 - 8.
Representative Results
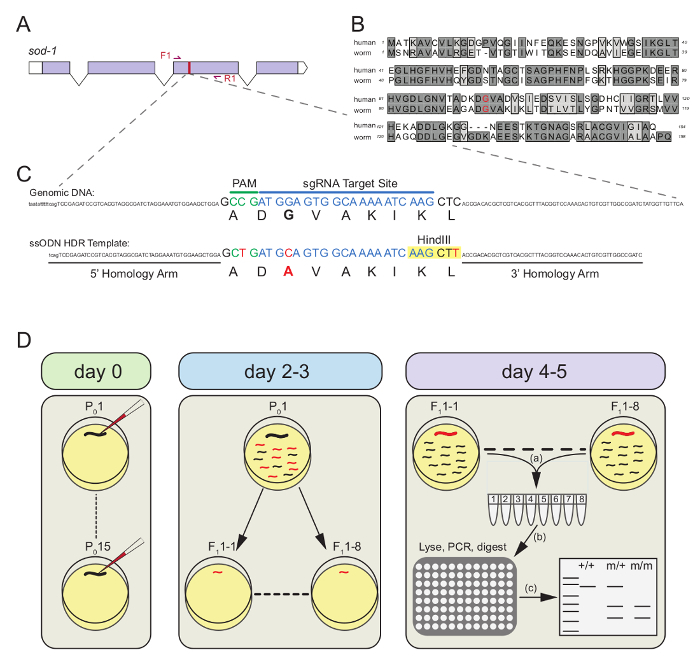
Mutations in human superoxide dismutase 1 (SOD1) account for ~10 - 20% of familial amyotrophic lateral sclerosis, a devastating neurodegenerative disease that invariably leads to paralysis and death17. Human SOD-1 is an evolutionarily conserved protein sharing 55% identity and 70% similarity with the C. elegans SOD-1 protein (Figure 1B). To demonstrate the simplicity, feasibility, and efficiency of the CRISPR-Cas9 RNP-based approach, we targeted the C. elegans sod-1 gene to introduce the disease-associated human G93A variant in the worm genome (Figure 1A).
To engineer the G93A mutation in the C. elegans genome, an sgRNA was chosen whose PAM recognition site sits 3 bp upstream of the G93 codon (Figure 1C). We designed an ssODN HDR repair template containing: 1) nucleotide changes to convert the G93 codon to A93 (GGA GCA), 2) a silent change to the NGG PAM sequence (CGG CAG) to prevent sgRNA-Cas9-mediated cleavage of the HDR template, 3) a silent mutation (CT) that introduces a unique HindIII restriction enzyme site for genotyping, and 4) 50 bp 5' and 3' homology arms (Figure 1C). A PCR primer set (F1-R1) was designed to produce a single 592 bp PCR product in N2 controls (Figure 1A). An injection mix containing the sgRNA, Cas9, ssODN and a fluorescent marker plasmid (pCFJ90) were mixed together, incubated for 10 min at room temperature, and loaded into a microinjection pipette.
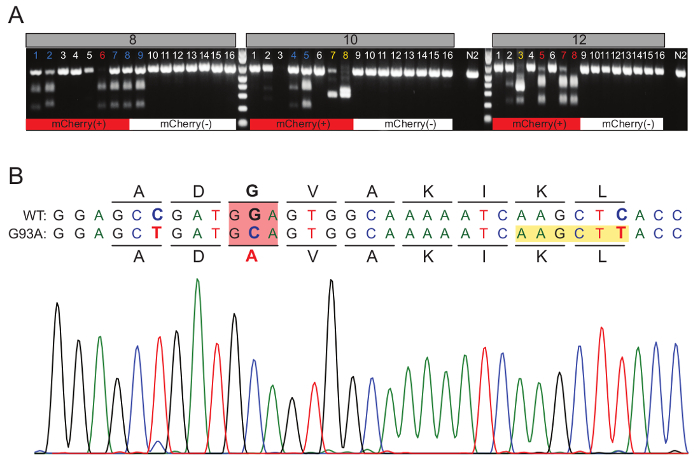
Figure 1D depicts the workflow after the injection mix is loaded into an injection micropipette. On Day 0, 10 - 15 P0 animals were injected in one or both gonad arms using the standard microinjection technique18,19. Injected animals recovered for 1 - 2 h and were then individually plated on OP50-seeded NGM plates. After 2 - 3 days, successful P0 injections were identified by those having mCherry(+) F1 progeny. The top three P0 plates (i.e. those having the greatest number of mCherry(+) F1s) were chosen for subsequent analysis. From each of these three plates (P0-8, P0-10, P0-12), 8 mCherry(+) F1s were singled to individual OP50-seeded 35 mm NGM plates for a total of 24 mCherry(+) F1s. After 2 - 3 days of egg-laying (Day 4 - 5), the individual F1s were transferred into PCR tubes containing worm lysis buffer and frozen for 1 h at -80 °C. Subsequently, the F1s were lysed to liberate genomic DNA, and subjected to PCR. The PCR products were then purified and digested with HindIII for 1 h, and run on a 1.5% agarose gel to identify potential edited animals. In correctly edited animals, HindIII digestion cuts the full-length PCR product (592 bp) into two bands of 370 bp and 222 bp. This genotyping strategy unambiguously differentiates between wild-type (592 bp), heterozygote (592, 370, 222 bp) and homozygote (370, 222 bp) animals. Figure 2A illustrates the genotyping results for the 24 mCherry(+) animals.
To demonstrate that the fluorescent mCherry marker enriches for genome modification, we also picked 8 mCherry(-) animals from each of the three P0 plates. Of the 24 mCherry(+) F1s screened (red bar), 10 contained mono-allelic or bi-allelic modification (42%), 10 were wild type (42%), 3 were potential indels (13%), and there was 1 PCR failure (Figure 2A). In contrast, of the 24 mCherry(-) F1s screened, only 1 animal was correctly modified (4%); whereas, 23 were wild type (96%) (Figure 2A). Figure 2B shows the chromatogram from the full-length gel extracted PCR product (F18-6), demonstrating that the precise nucleotide changes designed in the ssODN HDR template were faithfully incorporated into the genome of this homozygous F1 animal. Notably, F1 animals 10-7, 10-8, and 12-3 exhibited unexpected PCR product sizes and restriction digest banding patterns, suggesting these animals may carry complex indel mutations (Figure 2A). Thus, our method is not only capable of generating desired genome modification(s) with ease, efficiency, and fidelity, but also permits the identification and recovery of unique indel alleles that may shed important and unexpected insight into gene function.
Figure 1. CRISPR-Cas9 engineering of theC. eleganssod-1 locus. (A)Schematic illustration depicting the exon-intron structure of the sod-1 locus in C. elegans. The red bar denotes the location of G93 in exon 3. The primer pair set is noted by the red arrows (F1-R1). (B) Sequence alignment of human and C. elegans (worm) SOD-1 proteins. The targeted G93 residue is highlighted in red. (C) Top, a section of genomic DNA surrounding the G93 codon is shown as a reference, and the PAM (green) and sgRNA target (blue) sites are highlighted. Bottom, the single stranded oligonucleotide (ssODN) homology directed repair (HDR) template is shown containing: the codon edit changing G93 to A93, a silent change to the PAM motif to prevent cleavage by the sgRNA-Cas9 complex, a silent change to introduce a unique HindIII restriction enzyme site (yellow box), and flanking 5' and 3' 50 bp homology arms (black bars). All nucleotide changes designed in the ssODN are highlighted red. (D) Schematic illustration of the pipeline for generating and identifying CRISPR-Cas9 RNP-based point mutants. On day 0, inject 10 - 15 P0 animals with injection mix. On day 2 - 3, identify successfully injected P0 animals by those containing fluorescent F1 progeny, and select three P0 plates with the most fluorescent progeny. From each of the three P0 plates, single 8 fluorescent F1 progeny. On day 4 - 5, (a) step 1, individual F1s are picked and placed into PCR tubes containing Lysis Buffer; (b) step 2, after freezing at -80 °C, PCR tubes containing F1 worms are lysed to release genomic DNA, which is used as a PCR template. The PCR products are then cleaned and digested with the unique restriction enzyme; (c) step 3, enzyme digested products are resolved on an agarose gel where wild type (+/+), heterozygous (m/+), and homozygous (m/m) animals can be identified by restriction digest banding patterns. Please click here to view a larger version of this figure.
Figure 2. Genotyping data forsod-1 (G93A) mutants.(A)Gel image of F1 animals that were individually lysed, PCRed and digested with HindIII. For each P0 plate (P0-8, P0-10, P0-12), 8 mCherry(+) and 8 mCherry(-) F1 animals were analyzed. Both mono-allelic (blue numbers) and bi-allelic (red numbers) edited animals were recovered. Size shifted PCR products or unpredicted HindIII digested bands were also recovered that are indicative of indel mutants (yellow numbers). N2 controls are shown as a reference for PCR product sizing and enzyme specificity. (B) Top, codon spacing and amino acid translations are shown above for the wild type (WT) and below for G93A modified strands. The desired edit is highlighted by a red box, and the silent changes that create an in-frame HindIII restriction site are highlighted by a yellow box. All designed nucleotide changes are shown in bold. Bottom, representative chromatogram from homozygous F1 animal 8-6. Please click here to view a larger version of this figure.
| Reagent | Volume | Final Concentration |
| ddH2O | 6.3 μL | ---- |
| KCl (4M) | 0.94 μL | 300 mM |
| HEPES (0.5M) pH 7.4 | 0.5 μL | 20 mM |
| pCFJ90 (25 ng/μL) | 1.25 μL | 2.5 ng/μL |
| ssODN (500 ng/μL) | 1.25 μL | 50 ng/μL |
| sgRNA (50 μM) | 1.25 μL | 5 μM |
| Cas9 (61 μM) | 1.03 μL | 5 μM |
| Final Volume | 12.5 μL | ---- |
Table 1. CRISPR-Cas9 RNP Injection Mix. All reagents should be re-suspended in nuclease-free ddH2O. RNase free techniques should be used when handling reagents and when making the injection mix.
| Reagent | [Final] |
| KCl | 50 mM |
| Tris-HCl pH 8.3 | 10 mM |
| MgCl2 | 2.5 mM |
| NP-40 | 0.45% |
| Tween-20 | 0.45% |
| Proteinase K | 1 mg/mL |
Table 2. Worm Lysis Buffer Recipe. Proteinase K should be added fresh before each use.
| Reagent | 1x | 24x |
| 2X Q5 Mastermix | 12.5 μL | 300 μL |
| Primer F1 (10 μM) | 1.25 μL | 30 μL |
| Primer R1 (10 μM) | 1.25 μL | 30 μL |
| Worm Lysis | 4 μL | ---- |
| ddH2O | 6 μL | 144 μL |
| Final Volume | 25 μL | 504 μL |
Table 3. PCR Mastermix. The PCR mastermix should be mixed well by pipetting until the solution is homogeneous. 21 µL of the PCR mastermix should be added to clean PCR tubes, and then 4 µL of individual worm lysate should be added to properly labeled tubes.
| Reagent | 1x | 24x |
| 10X Enzyme Buffer | 2 μL | 48 μL |
| Cleaned PCR Reaction | 10 μL | ---- |
| ddH2O | 7 μL | 168 μL |
| Restriction Enzyme (10 U/μL) | 1 μL | 24 μL |
| Final Volume | 20 μL | 240 μL |
Table 4. Restriction Enzyme Mastermix. The restriction enzyme mastermix should be mixed well by pipetting until the solution is homogeneous. 10 µL of the restriction enzyme mastermix should be added to 10 µL of cleaned PCR product
Discussion
The CRISPR-Cas9 system is a powerful and effective tool for precisely modifying the genome of model organisms. Here, we demonstrate that chimeric sgRNAs20 coupled with ssODN HDR templates enable the highly efficient generation of genomic point mutations in C. elegans. Importantly, we demonstrate that RNP delivery produces high editing efficiency when fluorescence is used as a co-selection marker, highlighting the ease and reliability of the technique.
The majority of CRISPR-Cas9 methods for C. elegans genome engineering rely on specific genetic backgrounds or co-CRISPR strategies1. The co-CRISPR method has proven to be an incredibly useful tool that increases the likelihood of obtaining a successful genome edit. However, co-CRISPR methods require introducing a phenotype-selectable edit at a marker locus that enriches for the genome edit of interest. While powerful, introducing selectable phenotype-bearing mutations may be undesirable if the strain to edit or the desired point mutation of interest itself exhibits phenotypes that might be exacerbated or suppressed by the marker phenotype. Moreover, co-CRISPR strategies necessitate creating an additional double strand break at a marker locus that may contribute to off-target effects. Here, we present a method that uses a transient fluorescent marker that not only serves to identify successfully injected P0 animals, but also enriches for proper genome edits. Our data demonstrate this method significantly enriches for successfully edited animals in comparison to non-fluorescent animals, and provides several distinct advantages: 1) only one targeting site-specific sgRNA is required, 2) a 5-fold reduction in Cas9 and sgRNA concentration, 3) strain- and phenotype-selection independence, and 4) a nominal screening workload (~24 F1s). Robust CRISPR-Cas9 genome editing in the F1 generation was observed using this method. Notably, the PCR-restriction enzyme-based detection strategy described enables the unequivocal detection of mono- and bi-allelic edited animals in the F1 generation. Finally, the genotyping strategy facilitates identification of potential indel mutations, which can be observed as PCR band shifts when compared to N2 controls, that are either not susceptible to restriction digest or yield complex bands when digested.
The advent of next generation sequencing coupled with large scale genomic efforts has accelerated the identification of genomic variants that segregate with disease21. However, functional tests of pathogenicity have not been demonstrated for the majority of variants in cellular and organismal model systems. In this study, we targeted the sod-1 gene in C. elegans to introduce the widely studied ALS-associated SOD-1G93A mutation. To date, this mutation has been modeled and studied in C. elegans and mice primarily using transgenic overexpression. Although overexpression of variants in animal models may recapitulate key cellular, molecular and behavioral aspects of disease, it is increasingly clear that 'dose' is an extenuating factor in determining phenotypes22. For example, high copy number SOD-1G93A mice carrying ~24 copies of the mutant human SOD-1 gene exhibit greatly accelerated disease course compared to SOD-1G93Adl mice, which carry only ~8 - 10 copies23,24. Here, we demonstrate that our CRISPR-Cas9 RNP-based method can be utilized to rapidly generate human variant-associated mutations in the orthologous worm gene without requiring transgenic overexpression, in an effort to more precisely replicate the genetic context of disease. Our method provides a feasible platform to produce and test genomic variants of unknown significance in the context of endogenous regulatory control and expression, which precludes the confines of overexpression. Finally, we have also used this method to tag endogenous genes with fluorescent proteins (i.e. GFP), which will provide an additional tool for visualizing how variants affect protein localization, aggregation, and turnover.
Disclosures
We thank members of the Beg laboratory for critical reading of this manuscript. Strains were provided by the Caenorhabditis Genetics Center, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). Research in the Beg laboratory is supported by grants from the NIH (R01 NS094678) and Muscular Dystrophy Association (MDA382300) to A.A.B.
Acknowledgments
There are no conflicts of interest related to this report.
References
- Dickinson DJ, Goldstein B. CRISPR-Based Methods for Caenorhabditis elegans Genome Engineering. Genetics. 2016;202:885–901. doi: 10.1534/genetics.115.182162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- Ma D, Liu F. Genome Editing and Its Applications in Model Organisms. Genomics Proteomics Bioinformatics. 2015;13:336–344. doi: 10.1016/j.gpb.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32:347–355. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HM, Colaiacovo MP. CRISPR-Cas9-Guided Genome Engineering in C. elegans. Curr Protoc Mol Biol. 2016;115:31–31. doi: 10.1002/cpmb.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Kauffman KJ, Anderson DG. Delivery technologies for genome editing. Nat Rev Drug Discov. 2017;16:387–399. doi: 10.1038/nrd.2016.280. [DOI] [PubMed] [Google Scholar]
- Kim S, Kim D, Cho SW, Kim J, Kim JS. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014;24:1012–1019. doi: 10.1101/gr.171322.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeussler M, et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016;17:148. doi: 10.1186/s13059-016-1012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paix A, Folkmann A, Seydoux G. Precision genome editing using CRISPR-Cas9 and linear repair templates in C. elegans. Methods. 2017. [DOI] [PMC free article] [PubMed]
- Prior H, Jawad AK, MacConnachie L, Beg AA. Highly Efficient, Rapid and Co-CRISPR-Independent Genome Editing in Caenorhabditis elegans. G3 (Bethesda) 2017;7:3693–3698. doi: 10.1534/g3.117.300216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JoVE Science Education Database. Basic Methods in Cellular and Molecular Biology. PCR: The Polymerase Chain Reaction. J Vis Exp. 2017.
- Berkowitz LA, Knight AL, Caldwell GA, Caldwell KA. Generation of stable transgenic C. elegans using microinjection. J Vis Exp. 2008. [DOI] [PMC free article] [PubMed]
- JoVE Science Education Database. Basic Methods in Cellular and Molecular Biology. Restriction Enzyme Digests. J Vis Exp. 2017.
- JoVE Science Education Database. Basic Methods in Cellular and Molecular Biology. DNA Gel Electrophoresis. J Vis Exp. 2017.
- JoVE Science Education Database. Basic Methods in Cellular and Molecular Biology. Gel Purification. J Vis Exp. 2017.
- Estrada-Rivadeneyra D. Sanger sequencing. FEBS J. 2017. [DOI] [PubMed]
- Ludolph AC, Brettschneider J, Weishaupt JH. Amyotrophic lateral sclerosis. Curr Opin Neurol. 2012;25:530–535. doi: 10.1097/WCO.0b013e328356d328. [DOI] [PubMed] [Google Scholar]
- Evans TC. WormBook. The C. elegans Research Community, WormBook; 2006. [Google Scholar]
- Mello CC, Kramer JM, Stinchcomb D, Ambros V. Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd SD, et al. A Balanced Look at the Implications of Genomic (and Other "Omics") Testing for Disease Diagnosis and Clinical Care. Genes (Basel) 2014;5:748–766. doi: 10.3390/genes5030748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccon RA, Bunton-Stasyshyn RK, Fisher EM, Fratta P. Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain. 2013;136:2342–2358. doi: 10.1093/brain/awt097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acevedo-Arozena A, et al. A comprehensive assessment of the SOD1G93A low-copy transgenic mouse, which models human amyotrophic lateral sclerosis. Dis Model Mech. 2011;4:686–700. doi: 10.1242/dmm.007237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney ME, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]