

Abstract
Bacteria, one of the most important causative agents of various plant diseases, secrete a set of effector proteins into the host plant cell to subvert the plant immune system. During infection cytoplasmic effectors are delivered to the host cytosol via a type III secretion system (T3SS). After delivery into the plant cell, the effector(s) targets the specific compartment(s) to modulate host cell processes for survival and replication of the pathogen. Although there has been some research on the subcellular localization of effector proteins in the host cells to understand their function in pathogenicity by using fluorescent proteins, investigation of the dynamics of effectors directly injected from bacteria has been challenging due to the incompatibility between the T3SS and fluorescent proteins.
Here, we describe our recent method of an optimized split superfolder green fluorescent protein system (sfGFPOPT) to visualize the localization of effectors delivered via the bacterial T3SS in the host cell. The sfGFP11 (11th β-strand of sfGFP)-tagged effector secreted through the T3SS can be assembled with a specific organelle targeted sfGFP1-10OPT (1-10th β-strand of sfGFP) leading to fluorescence emission at the site. This protocol provides a procedure to visualize the reconstituted sfGFP fluorescence signal with an effector protein from Pseudomonas syringae in a particular organelle in the Arabidopsis and Nicotiana benthamiana plants.
Keywords: Biology, Issue 135, Type III secretion system, effector dynamics, Pseudomonas, superfolder green fluorescent protein system, subcellular localization
Introduction
Plants are sessile organisms that encounter numerous invading pathogens including bacteria, fungi, viruses, insects, and nematodes throughout their life cycle. Among the phytopathogens, the gram-negative bacterial pathogens such as Pseudomonas spp. and Ralstonia spp., infect their host plants by entering through wounds or natural openings, such as the stomata and hydathode1. To successfully colonize host plants, bacterial pathogens have evolved to develop a variety of virulence factors2. When bacteria invade a host plant, they inject a series of virulence proteins — known as effectors — directly into the plant cells to promote their pathogenicity. These effectors suppress or modulate the plant innate immunity, and manipulate the host cellular processes to result in bacterial survival3.
Pathogenic bacteria mainly use a T3SS to deliver effector proteins directly into host cells4. The T3SS resembles a molecular syringe with a needle-like channel connecting from a scaffold protein structure across the inner and the outer bacterial membranes to the injection site of the host cell5. This T3SS-mediated effector (T3E) secretion mechanism is well-conserved in various gram-negative bacterial pathogens of the plant as well as human. One of the representative plant pathogens, the P. syringae pv. Tomato DC3000 hrcC mutant which typically has a defective T3SS, has restricted growth in plants likely due to the inability of this mutant to fully suppress the plant immunity (by injecting effector proteins)6. Upon translocation into the host cells, effectors target various host proteins that are important for the host cell system, including plant defense responses, gene transcription, cell death, proteasome, vesicle trafficking, and hormone pathways7,8,9,10. Therefore, tracking of the cellular localization of the effector proteins in the host cells is an attractive target to understand their functions with respect to modulation of the plant immunity.
Most of the localization studies of the T3Es have employed Agrobacterium-mediated overexpression with a large fluorescence protein in the host plant9. However, the heterologous expression method for genes that are introduced in other species has been shown to be mis-localized or occasionally non-functional11,12,13. In addition, several studies revealed that bacterial effectors undergo modification for proper targeting in the host cells14,15,16,17. Therefore, transiently expressed effectors in the cytosol of the plant cells may not be functionally or quantitatively identical to the effectors which are delivered by the T3SS upon pathogen infection18. Moreover, the fusion of large fluorescent tags to effector proteins may disrupt the proper effector delivery and visualization18,19. Therefore, these approaches to assay the T3E function may not fully reflect the native localization of the T3SS-secreted effectors.
A green fluorescent protein (GFP) is composed of an 11-stranded β-barrel enclosing a central strand that includes a chromophore20. Waldo et al. reported a novel split-GFP system that consists of a small component (GFP β strand 11; GFP11) and a large complementary fragment (GFP β strand 1-10; GFP1-10)21. The fragments do not fluoresce by themselves but fluoresce upon their self-association when both fragments are in close proximity with each other. For the optimization of the protein folding efficiency, robust folding variants of the GFP, i.e., sfGFP and sfGFPOPT, were subsequently developed for the split GFP system20,21,22. Recently, single amino acid mutated variants of sfGFP1-10OPT- sfYFP1-10OPT and sfCFP1-10OPT- that can reconstitute with a sfGFP11 fragment, and show yellow and cyan fluorescence respectively, were generated23. Moreover, sfCherry, a derivative of mCherry, can be split into sfCherry1-10 and sfCherry11 fragments in the same manner as sfGFP23.
This system has been adapted to label and track the T3SS effectors in HeLa cells during infection using the effectors from Salmonella24. However, it was previously not optimized for the host plant-bacterial pathogen system. Recently, we optimized the split GFP system based on the improved sfGFP1-10OPT to monitor the subcellular localization of the T3Es delivered from P. syringae into plant cells25. To facilitate the localization studies of the T3Es to different subcellular compartments in the plant cells, a set of transgenic Arabidopsis thaliana plants were generated to express sfGFP1-10OPT in the various subcellular compartments25. Moreover, the plasmids carrying a variety of organelle-targeted sfGFP1-10OPT for the Agrobacterium-mediated transient overexpression and the sfGFP11-tagged vectors for the T3SS-based effector delivery were also generated. The seeds of various transgenic Arabidopsis lines and the plasmids to express the T3Es of interest can be obtained from sources mentioned in the Table of Materials26,27.
In the following protocol, we describe an optimized system to monitor the dynamics of effectors delivered by bacteria in the host cells using the split sfGFP system. Infection of plants expressing sfGFP1-10OPT with transgenic Pseudomonas carrying recombinant sfGFP11 plasmid results in a delivery of the sfGFP11-tagged effector from Pseudomonas into the host cell. Consequently, these proteins are reconstituted and translocate to the specific effector target compartment(s). The Pseudomonas syringae pv. tomato CUCPB5500 strain in which 18 effectors are deleted, was used because this strain showed low or no cell death in both A. thaliana and N. benthamianas28. However, all of the materials and steps described here can be replaced or modified to adapt the split sfGFP system for investigation of other biological questions or optimization in the given laboratory conditions.
Protocol
Note: All steps are performed at room temperature, unless stated otherwise.
1. Preparation of Plant Materials (4 Weeks)
- Preparation for the N. benthamiana plants
- Sow 2 seeds of N. benthamiana on the soil surface of each pot, cover the tray with a plastic dome, and allow seeds to germinate in a 25 °C, 60% humidity growth chamber with a 16/8-h light/dark photoperiod cycle.
- After two weeks, pick out and discard the smallest seedling in each pot and continue to grow plants under the same growth conditions as applied for the germination in step 1.1.1. Add 1 L of water per tray every two days. Note: The growth conditions for plants may vary across labs. Therefore, follow the regular watering protocol to grow the plant in a healthy condition.
- In a week, transfer the plants to a new tray and arrange them with adequate space for further growth. Keep growing the plants under the conditions described in step 1.1.1 until they are ready to be infiltrated at 4 weeks of age. Note: Plant growth may differ depending on the growth condition across labs. Usually, we find that the 4-week-old N. benthamiana plants bear about six leaves.
- Preparation for A. thaliana transgenic plants
- Refer to the Table of Materials and order the transgenic Arabidopsis seeds.
- Soak ~50 - 100 transgenic Arabidopsis seeds in 1 mL of distilled water and store them at 4 °C for 3 days in the dark to synchronize the onset of germination.
- Sow ~2 - 3 seeds on the soil surface of a plug plant tray and cover the tray with a plastic dome. Allow seeds to germinate at 23 °C, 60% humidity with a 10/14-h light/dark photoperiod cycle. Note: The seeds should be homozygotes. However, we recommend reconfirming the presence of the transgene in the batch. In this case, sterilize the seeds by washing with 70% ethanol for 2 min, 50% bleach (about 2% hypochlorite) containing 0.05% triton X-100 for 5 min. Follow by washing 5 - 6 times with sterile double distilled water (ddH2O). After sterilization, stratify at 4 °C for 3 days and plate them on plant germination media containing 25 µg/L of hygromycin B to select the transgenic plants.
- After a week, leave only one plant per plug and continue growing plants under the same growth conditions used for step 1.2.3. Note: Four-week-old plants were used for the P. syringae infection. Water plants every other day to keep plants healthy.
2. Preparation of Pseudomonas Culture (~1 Week)
- Construction of the plasmid for Pseudomonas transformation
- Refer to the Table of Materials and order the desired vector(s) of the T3SS-based effector delivery system vector25.
- Insert the effector gene of interest into the effector delivery vector using site-specific recombination cloning25. Note: When monitoring subcellular localization of full-length effector protein, put the full-length gene into pBK-GW-1-2 or pBG-GW-1-2. It is also possible to choose pBK-GW-1-4 or pBG-GW-1-4 containing 2x sfGFP11 for increased fluorescence signal. In the case of a partial effector lacking signal peptide, use pBK-GW-2-2 or pBK-GW-2-4. Refer to Park et al. for detailed information about effector delivery vectors25.
Transform the plasmid carrying an effector fused to the sfGFP11 tag to P. syringae pv. Tomato (Pst) CUCPB5500 using standard electroporation29. Note: Other Pseudomonas strains can be used if necessary. The sfGFP11 tag system is constructed for a broad range of vectors30 and the gene expression of the effector is regulated by the AvrRpm1 promoter, which is comparable with, e.g., Pseudomonas fluorescens (EthAn)31.
Spread the transformed bacterial cells gently over the surface of the King’s B agar plates containing 100 µg/mL rifampicin and 25 µg/mL kanamycin or 25 µg/mL gentamycin. Incubate at 28 °C for 2 days.
Inoculate one colony into King’s B liquid media with antibiotics appropriate for the vector used, and grow the cells overnight at 28 °C with shaking at 200 rpm.
Make a glycerol stock. Add autoclaved glycerol to a final concentration of 50% and store at -80 °C.
3. Transient Expression of Organelle-targeted sfGFP1-10OPT in N. benthamiana (4 Days)
- Preparation of Agrobacterium culture
- Order the desired vector(s) of organelle-targeted sfGFP1-10OPT plasmid(s) (refer to the Table of Materials).
- Transform the plasmid(s) into Agrobacterium tumefaciens strain GV3101 cells32. Grow the cells on Luria-Bertani (LB) agar medium supplemented with 50 µg/mL kanamycin and 50 µg/mL rifampicin at 28 °C for 2 days.
- From a single colony on the LB agar medium, inoculate the cells into 5 mL of liquid LB media supplemented with 50 µg/mL kanamycin and 50 µg/mL rifampicin. Grow the cells overnight at 28 °C with shaking at 200 rpm.
- Harvest the cells by centrifugation at 3,000 x g for 10 min. Pour off the supernatant media and resuspend the pellet in 1 mL of freshly made infiltration buffer.
- Measure the quantity of Agrobacterium by obtaining the optical density (OD) value at an absorbance of 600 nm (Abs 600 nm). Adjust the OD600 of the bacteria to 0.5 with infiltration buffer. Note: 1 mL of suspension is enough to infiltrate on two spots.
- Leave the culture at room temperature on a gentle rocker for 1 - 5 h before infiltration.
- Poke a hole into the center of the leaves to be infiltrated with a 10 µL tip. Use a 1-mL needleless syringe to infiltrate the Agrobacterium suspensions. Carefully and slowly inject about 500 µL of the suspensions prepared from step 3.1.5 into the leaf adaxial side via the syringe. Repeat the infiltration on at least three different plants for experimental replicates. Note: For health and safety reasons, eye protection should be worn during infiltration.
- Wipe off the remaining bacterial suspension on the leaves and mark the boundary of the infiltrated region.
- Keep the infiltrated plants under the same growth conditions used for step 1.1.1 for 2 days.
4. Inoculation of Pseudomonas (4 Days)
Streak the transformed Pseudomonas strain from the glycerol stock in step 2.5 on King’s B agar media with the appropriate antibiotics at 28 °C for 2 days. Note: The health of Pseudomonas is very critical. If colonies do not form well, streak the cells again or propagate the cells in the King’s liquid media prior to proceeding.
Inoculate a loopful of Pseudomonas cells in Mannitol-Glutamate (MG) liquid media at 28 °C with shaking at 200 rpm for overnight.
Harvest the cells by centrifugation at 3,000 x g for 10 min. Pour off the supernatant media, resuspend the pellet in 10 mM MgCl2, and adjust the OD600 to 0.02 (1 x 107 cfu/mL) for N. benthamiana leaves and to 0.002 (1 x 106 cfu/mL) for Arabidopsis leaves.
For the N. benthamiana, infiltrate the Pseudomonas suspension into the area of the leaves where the Agrobacterium carrying sfGFP1-10OPT construct was infiltrated 2 days previously (as in step 3.1.7). For the sfGFP1-10OPT transgenic Arabidopsis, infiltrate the Pseudomonas suspension into two 4-week-old short day-grown leaves. Note: At least three plants are needed for experimental replicates.
5. Observation of sfGFP Signal via Confocal Microscopy (1 Day)
Cut out the leaf disc from the Pseudomonas-inoculated leaves. At specific time points after infiltration of Pseudomonas, image two 2-cm2 leaf discs from the single plant using a laser scanning confocal system with 40X/1.2 NA C-Apochromat water immersion objective or 63X/0.8 NA C-Apochromat oil immersion objective. To avoid dead cells killed by wounding, observe the cells away from the infiltration hole.
- Start the observation at a low power setting of the 488-nm argon laser. Increase the laser power to detect sfGFP. Note: We usually use 2 - 15% of the laser intensity to detect the fluorescence signal. However, the laser power and detection setting should be adjusted based upon the user’s microscopy system. Here, the emission filters were set to 520 - 550 nm. The dead cells often emit auto-fluorescence under the 488-nm laser excitation. Therefore, as a negative control, the same effector without the sfGFP11 tag should be infiltrated and observed under the same observation conditions. In addition, high laser excitation can induce chlorophyll autofluorescence. Therefore, adjust the laser intensity using the control plant cells so as not to induce chlorophyll autofluorescence.
- For a counterstaining of the cell wall, infiltrate 20 mM propidium iodide (PI) into the leaf disc at 5 - 10 min prior to observation.
- For the nucleus staining, submerge the leaf discs into 0.1% paraformaldehyde for 5 min followed by washing with water. Then, infiltrate the 10 mM PI into the leaf disc at 5 - 10 min before microscopic observation. Repeat the experiments at least three times. Note: This step is not critical but helpful to define the effector localization at the plasma membrane or the nucleus. If the effectors of interest showed their localization in a specific organelle, use a marker for the given organelle to confirm the localization of the effector.
Representative Results
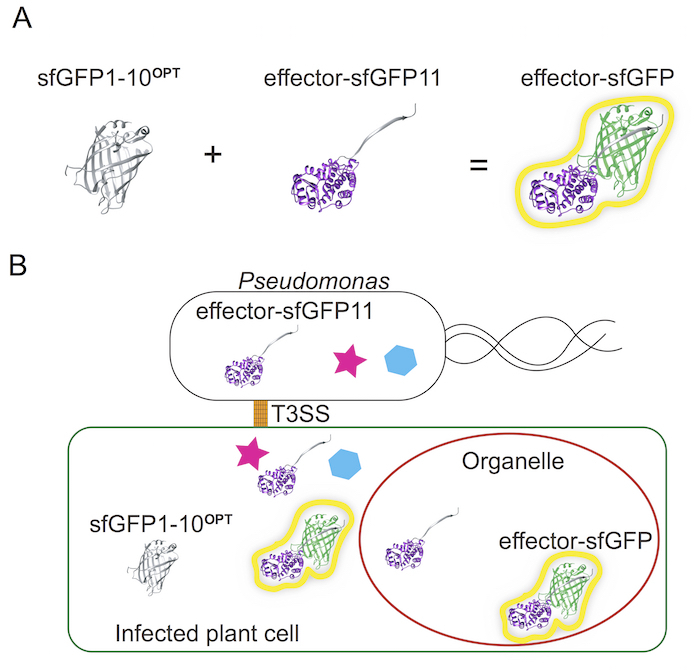
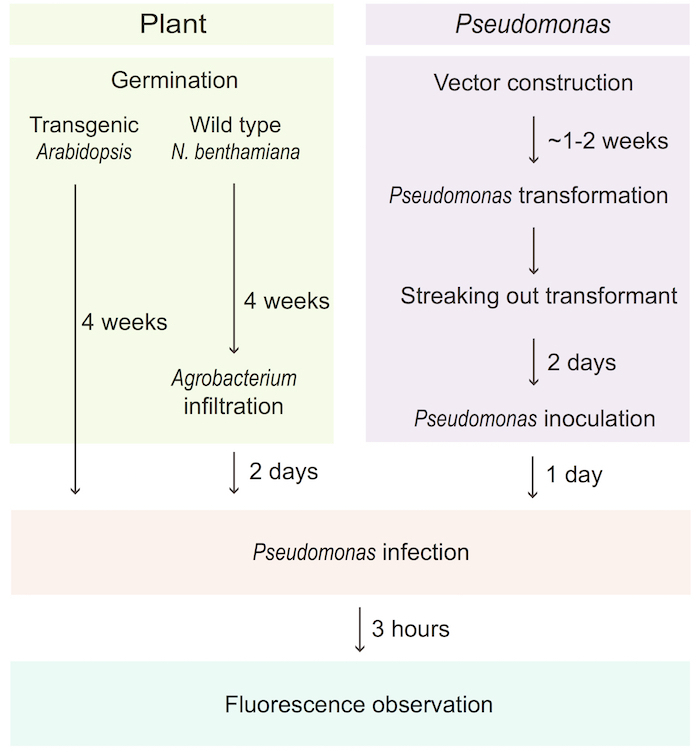
The β-barrel structure of GFP is composed of eleven β strands and can be divided into two fragments, the 1 - 10th strand (GFP1-10OPT) and the 11th (GFP11) strand. Although neither of two fragments fluorescent by themselves, self-assembled sfGFP can emit the fluorescence when the two fragments exist in close proximity (Figure 1A). In this system, sfGFP1-10OPT-expressing Arabidopsis or N. benthamiana plants are inoculated with Pseudomonas carrying a sfGFP11 tagged effector. The sfGFP11 tagged effector delivered by Pseudomonas into the cytosol of the host cell is reconstituted to the sfGFP1-10OPT expressed in the cytosol and then together translocate into the target compartment (Figure 1B). Figure 2 represents the overall procedure, from the preparation of the plant materials and Pst to the detection of the fluorescence signal in the plant cell.
As an example of our method, we used the P. syringae effector protein, AvrB, which is delivered into the host plant cells thorough the T3SS during infection. In Arabidopsis, AvrB is recognized by a corresponding resistance protein, RPM1, and triggers immune responses including hypersensitive cell death33. In the previous study, the GFP reporter assay and the biochemical assay revealed that AvrB and RPM1 are localized in the plasma membrane of the plant cell33,34,35. To examine the localization of AvrB using our method, Agrobacterium harboring the CYTO sfGFP1-10OPT gene was infiltrated in N. benthamiana. In two days, AvrB-sfGFP11-transformed Pseudomonas cells were inoculated into the Agrobacterium-infiltrated region. The complemented sfGFP fluorescence signals were observed in the Pst CUCPB5500 containing AvrB-sfGFP11 at the plasma membrane (Figure 3B). In contrast, no signals were found in the infected cell by Pst carrying native AvrB (Figure 3A).
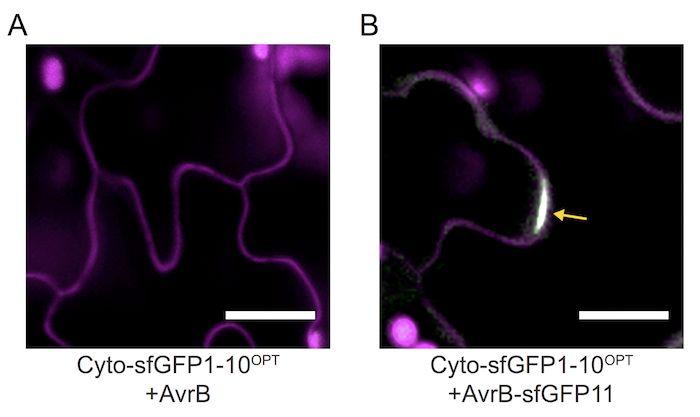
Figure 1. The optimized split GFP system. (A) The β-barrel structure of GFP is made of eleven β-strands and can be split into the 1-10th β-strand (GFP1-10) and the 11th β-strand (GFP11). (B) In this method, the sfGFP1-10OPT fragments were expressed in plant cells, while the sfGFP11 strand was fused to AvrB and transformed into Pseudomonas. Only plants cell infected by Pseudomonas containing sfGFP11 will show the sfGFP fluorescence (where the effector localizes). Please click here to view a larger version of this figure.
Figure 2. Overview of the procedure. The transgenic Arabidopsis or the sfGFP1-10OPT-infiltrated N. benthamiana plant are infected with Pst CUCPB5500 carrying a sfGFP11-tagged effector through syringe infiltration. The fluorescence of the assembled sfGFP can be detected via a confocal microscopy system at the site of the effector localization. Please click here to view a larger version of this figure.
Figure 3. AvrB-sfGFP detection on N. benthamiana leaves 3 h after infection. The Pst CUCPB5500 harboring a sfGFP11-tagged AvrB gene was infiltrated on the 5th or 6th leaf of N. benthamiana, which was infiltrated by Agrobacterium carrying the sfGFP1-10OPT 2 days previously. The cell wall was stained by propidium iodide. While the cells expressing sfGFP1-10 OPT with AvrB-only do not show any fluorescence signal (A). The GFP signals (yellow arrow) were observed at the infected cells by the Pst containing AvrB-sfGFP11 gene at 3 h post-infiltration (B). This result represents that only AvrB-sfGFP11, not AvrB, is reconstituted with sfGFP1-10OPT at the cytosol and then the assembled sfGFP is translocated to the plasma membrane. Magenta pseudo color represents the cell wall and chlorophyll autofluorescence. Green represents the assembled sfGFP fluorescence. Scale bars = 40 µm. Please click here to view a larger version of this figure.
Discussion
The protocol described here is used for monitoring the accurate localization of the effector proteins injected by the bacterial T3SS into the host plant cell upon infection. Previously, the split GFP system was used as a tool to study the subcellular localization of mammalian proteins23,36, Salmonella T3E localization, and Agrobacterium VirE2 delivery through the T4SS into the plant cells37. To apply this system in the plant cells, previous studies used a transgenic maize plant that constitutively expresses the cytoplasmic GFP1-10 and transgenic fungi, Ustilago maydis, that expresses the GFP11-tagged effector protein. However, the maize-U. maydis system has not been successful because the expression levels of the translocated effector proteins were weak and high level of background autofluorescence impeded the detection of the reconstituted GFP signal38. To remedy this problem, we used a sfGFP1-10 variant, sfGFP1-10OPT, that significantly improves the solubility and fluorescence intensity21,22.
Despite the advantages of the split sfGFP system, there are some limitations to the study of the effector localization and dynamics. First, the observed reconstituted sfGFP fluorescent signal is relatively weak. This could be due to a small amount of effector molecules delivered directly from P. syringae. This weak signal could be improved by using the multimerizing sfGFP11 tag. The vectors carrying 2x sfGFP11 tag are also available from the sources listed in the Table of Materials. Second, the cytosolic sfGFP1-10 OPT failed to be reconstituted with sfGFP11 targeted to Golgi, ER, and plastid25. Co-expression of mitochondria-targeted sfGFP11 and cytosolic sfGFP1-10OPT resulted in mislocalization of the re-constituted sfGFP to the cytosol and nucleus25. Therefore, localization of the effector of interest should be re-validated by applying the appropriate organelle-targeted sfGFP1-10OPT. When any GFP signal is not detected in the infected cells expressing cytosolic sfGFP1-10OPT, we recommend using N. benth expressing Golgi, ER, and plastid targeted sfGFP1-10OPT or using the corresponding organelle targeted sfGFP1-10OPT transgenic Arabidopsis25.
This method demonstrated the use of the split fluorescent protein system as a useful tool to examine the temporal and spatial dynamics of effectors in host plants25. Future research will focus on the advances in single-molecule imaging, which may enable detailed studies of the dynamics of plasma membrane-localized effectors. Furthermore, a secretion assay using a bacterial system may be useful alternative to study localization of fungal effectors; it prevents the aggregation of effectors and high autofluorescence at the fungal penetration site.
It should be noted that there are some key points in this method. Firstly, both plant materials and bacteria should be healthy and fresh. To ensure visualization of the effector signal, both the sfGFP1-10OPT from plants and the sfGFP11 from Pseudomonas should be strongly expressed. Therefore, it is important to grow plants under the optimal growth conditions and protect them from environmental stressors, such as pests. We also recommend that all bacteria used for infiltration be taken from the 2 day-grown cells upon the agar plates and not from glycerol stocks. Secondly, in the case of transient expression in N. benthamiana, the length of time required for the maturation of the organelle-targeted sfGFP1-10OPT might vary for each organelle marker protein. For example, maturation of PM-sfGFP1-10OPT requires more than 48 h after infiltration. Lastly, the time point and the expression levels of the reconstituted sfGFP signals depend upon the type of effector proteins that are required to optimize some experimental conditions.
Disclosures
The authors have no conflicts of interest to disclose.
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and future Planning (NRF-2018R1A2A1A05019892) to DC and by a grant from Plant Molecular Breeding Center (PMBC) of the Next Generation Biogreen 21 program of the Rural Development Administration (PJ013201) to EP. We thank the imaging center of the National Instrumentation Center for Environmental Management to provide a confocal microscope for filming.
References
- Melotto M, Underwood W, He SY. Role of stomata in plant innate immunity and foliar bacterial diseases. Annu Rev Phytopathol. 2008;46:101–122. doi: 10.1146/annurev.phyto.121107.104959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melotto M, Underwood W, Koczan J, Nomura K, He SY. Plant stomata function in innate immunity against bacterial invasion. Cell. 2006;126(5):969–980. doi: 10.1016/j.cell.2006.06.054. [DOI] [PubMed] [Google Scholar]
- Toruno TY, Stergiopoulos I, Coaker G. Plant-Pathogen Effectors: Cellular Probes Interfering with Plant Defenses in Spatial and Temporal Manners. Annu Rev Phytopathol. 2016;54:419–441. doi: 10.1146/annurev-phyto-080615-100204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttner D. Behind the lines-actions of bacterial type III effector proteins in plant cells. FEMS Microbiol Rev. 2016;40(6):894–937. doi: 10.1093/femsre/fuw026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewoody RS, Merritt PM, Marketon MM. Regulation of the Yersinia type III secretion system: traffic control. Front Cell Infect Microbiol. 2013;3:4. doi: 10.3389/fcimb.2013.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng WL, Preston G, Collmer A, Chang CJ, Huang HC. Characterization of the hrpC and hrpRS operons of Pseudomonas syringae pathovars syringae, tomato, and glycinea and analysis of the ability of hrpF, hrpG, hrcC, hrpT, and hrpV mutants to elicit the hypersensitive response and disease in plants. J Bacteriol. 1998;180(17):4523–4531. doi: 10.1128/jb.180.17.4523-4531.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JD, Guttman DS, Desveaux D. The targeting of plant cellular systems by injected type III effector proteins. Semin Cell Dev Biol. 2009;20(9):1055–1063. doi: 10.1016/j.semcdb.2009.06.003. [DOI] [PubMed] [Google Scholar]
- Alfano JR, Collmer A. Type III secretion system effector proteins: double agents in bacterial disease and plant defense. Annu Rev Phytopathol. 2004;42:385–414. doi: 10.1146/annurev.phyto.42.040103.110731. [DOI] [PubMed] [Google Scholar]
- Aung K, Xin X, Mecey C, He SY. Subcellular Localization of Pseudomonas syringae pv. tomato Effector Proteins in Plants. Methods Mol Biol. 2017;1531:141–153. doi: 10.1007/978-1-4939-6649-3_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay S, Bonas U. How Xanthomonas type III effectors manipulate the host plant. Curr Opin Microbiol. 2009;12(1):37–43. doi: 10.1016/j.mib.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Bassham DC, Raikhel NV. Plant cells are not just green yeast. Plant Physiol. 2000;122(4):999–1001. doi: 10.1104/pp.122.4.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courbot M, et al. A major quantitative trait locus for cadmium tolerance in Arabidopsis halleri colocalizes with HMA4, a gene encoding a heavy metal ATPase. Plant Physiol. 2007;144(2):1052–1065. doi: 10.1104/pp.106.095133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler M, Murphy AS. The ABC of auxin transport: the role of p-glycoproteins in plant development. FEBS Lett. 2006;580(4):1094–1102. doi: 10.1016/j.febslet.2005.11.054. [DOI] [PubMed] [Google Scholar]
- Boucrot E, Beuzon CR, Holden DW, Gorvel JP, Meresse S. Salmonella typhimurium SifA effector protein requires its membrane-anchoring C-terminal hexapeptide for its biological function. J Biol Chem. 2003;278(16):14196–14202. doi: 10.1074/jbc.M207901200. [DOI] [PubMed] [Google Scholar]
- Reinicke AT, et al. A Salmonella typhimurium effector protein SifA is modified by host cell prenylation and S-acylation machinery. J Biol Chem. 2005;280(15):14620–14627. doi: 10.1074/jbc.M500076200. [DOI] [PubMed] [Google Scholar]
- Patel JC, Hueffer K, Lam TT, Galan JE. Diversification of a Salmonella virulence protein function by ubiquitin-dependent differential localization. Cell. 2009;137(2):283–294. doi: 10.1016/j.cell.2009.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Alvarez A, et al. Identification of O-mannosylated virulence factors in Ustilago maydis. PLoS Pathog. 2012;8(3):e1002563. doi: 10.1371/journal.ppat.1002563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan JE. Common themes in the design and function of bacterial effectors. Cell Host Microbe. 2009;5(6):571–579. doi: 10.1016/j.chom.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigó G, Ayaydin F, Szabados L, Koncz C, Cséplô Á. Suspension protoplasts as useful experimental tool to study localization of GFP-tagged proteins in Arabidopsis thaliana. Proceedings of the 9th Hungarian Congress on Plant Biology. 2008;52(1):59. [Google Scholar]
- Pedelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS. Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol. 2006;24(1):79–88. doi: 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- Cabantous S, Terwilliger TC, Waldo GS. Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat Biotechnol. 2005;23(1):102–107. doi: 10.1038/nbt1044. [DOI] [PubMed] [Google Scholar]
- Cabantous S, et al. A new protein-protein interaction sensor based on tripartite split-GFP association. Sci Rep. 2013;3:2854. doi: 10.1038/srep02854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiyama D, et al. Versatile protein tagging in cells with split fluorescent protein. Nat Commun. 2016;7:11046. doi: 10.1038/ncomms11046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Engelenburg SB, Palmer AE. Imaging type-III secretion reveals dynamics and spatial segregation of Salmonella effectors. Nat Methods. 2010;7(4):325–330. doi: 10.1038/nmeth.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E, Lee HY, Woo J, Choi D, Dinesh-Kumar SP. Spatiotemporal Monitoring of Pseudomonas syringae Effectors via Type III Secretion Using Split Fluorescent Protein Fragments. Plant Cell. 2017;29(7):1571–1584. doi: 10.1105/tpc.17.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addgene. 2018. Available from: https://www.addgene.org.
- Arabidopsis Biological Resource Center. 2018. Available from: https://abrc.osu.edu.
- Kvitko BH, et al. Deletions in the repertoire of Pseudomonas syringae pv. tomato DC3000 type III secretion effector genes reveal functional overlap among effectors. PLoS Pathog. 2009;5(4):e1000388. doi: 10.1371/journal.ppat.1000388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue H, Nojima H, Okayama H. High efficiency transformation of Escherichia coli with plasmids. Gene. 1990;96(1):23–28. doi: 10.1016/0378-1119(90)90336-p. [DOI] [PubMed] [Google Scholar]
- Kovach ME, et al. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene. 1995;166(1):175–176. doi: 10.1016/0378-1119(95)00584-1. [DOI] [PubMed] [Google Scholar]
- Upadhyaya NM, et al. A bacterial type III secretion assay for delivery of fungal effector proteins into wheat. Mol Plant Microbe Interact. 2014;27(3):255–264. doi: 10.1094/MPMI-07-13-0187-FI. [DOI] [PubMed] [Google Scholar]
- Weigel D, Glazebrook J. Transformation of agrobacterium using the freeze-thaw method. CSH Protoc. 2006;2006(7) doi: 10.1101/pdb.prot4666. [DOI] [PubMed] [Google Scholar]
- Boyes DC, Nam J, Dangl JL. The Arabidopsis thaliana RPM1 disease resistance gene product is a peripheral plasma membrane protein that is degraded coincident with the hypersensitive response. Proc Natl Acad Sci U S A. 1998;95(26):15849–15854. doi: 10.1073/pnas.95.26.15849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimchuk Z, et al. Eukaryotic fatty acylation drives plasma membrane targeting and enhances function of several type III effector proteins from Pseudomonas syringae. Cell. 2000;101(4):353–363. doi: 10.1016/s0092-8674(00)80846-6. [DOI] [PubMed] [Google Scholar]
- Gao Z, Chung EH, Eitas TK, Dangl JL. Plant intracellular innate immune receptor Resistance to Pseudomonas syringae pv. maculicola 1 (RPM1) is activated at, and functions on, the plasma membrane. Proc Natl Acad Sci U S A. 2011;108(18):7619–7624. doi: 10.1073/pnas.1104410108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonetti MD, Sekine S, Kamiyama D, Weissman JS, Huang B. A scalable strategy for high-throughput GFP tagging of endogenous human proteins. Proc Natl Acad Sci U S A. 2016;113(25):E3501–E3508. doi: 10.1073/pnas.1606731113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Yang Q, Tu H, Lim Z, Pan SQ. Direct visualization of Agrobacterium-delivered VirE2 in recipient cells. Plant J. 2014;77(3):487–495. doi: 10.1111/tpj.12397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, et al. Experimental approaches to investigate effector translocation into host cells in the Ustilago maydis/maize pathosystem. Eur J Cell Biol. 2015;94(7-9):349–358. doi: 10.1016/j.ejcb.2015.06.007. [DOI] [PubMed] [Google Scholar]