

Abstract
The emergence of robust single-cell ‘omics techniques enables studies of uncultivable species, allowing for the (re)discovery of diverse genomic features. In this study, we combine single-cell genomics and transcriptomics to explore genome evolution in ciliates (a > 1 Gy old clade). Analysis of the data resulting from these single-cell ‘omics approaches show: 1) the description of the ciliates in the class Karyorelictea as “primitive” is inaccurate because their somatic macronuclei contain loci of varying copy number (i.e., they have been processed by genome rearrangements from the zygotic nucleus); 2) gene-sized somatic chromosomes exist in the class Litostomatea, consistent with Balbiani’s (1890) observation of giant chromosomes in this lineage; and 3) gene scrambling exists in the underexplored Postciliodesmatophora (the classes Heterotrichea and Karyorelictea, abbreviated here as the Po-clade), one of two major clades of ciliates. Together these data highlight the complex evolutionary patterns underlying germline genome architectures in ciliates and provide a basis for further exploration of principles of genome evolution in diverse microbial lineages.
Keywords: genome evolution, single-cell genomics, single-cell transcriptomics, epigenetics, Ciliophora
Introduction
Although genomes are often described as being conserved within species, a plethora of data demonstrate their inherently dynamic nature (Parfrey et al. 2008; Oliverio and Katz 2014; Maurer-Alcalá and Katz 2015). In eukaryotes, examples of dynamic genomes include the separation of germline and somatic genetic material, and variation throughout life cycles (such as changes in ploidy or DNA content during development; Parfrey et al. 2008; Maurer-Alcalá and Katz 2015). These changes are often regulated by epigenetic mechanisms that are involved in analogous (and perhaps homologous) processes among anciently diverged lineages of eukaryotes (Matzke and Mosher 2014; Rogato et al. 2014; Maurer-Alcalá and Katz 2015), which has led to the hypothesis that such mechanisms existed in the last common ancestor of extant eukaryotes (Zufall et al. 2005; Parfrey 2008; Oliverio and Katz 2014; Maurer-Alcalá and Katz 2015).
Dynamic genomes, including the separation of germline and somatic DNA into distinct nuclei, are present in ciliates, an ancient clade of predominantly single-celled eukaryotic microorganisms. Unlike multicellular organisms, where germline (i.e., gametic) and somatic (i.e., leaves, hyphae, muscle) genomes are in distinct cell types, the germline micronucleus (MIC) and somatic macronucleus (MAC) share a common cytoplasm in ciliates (Raikov 1982; Prescott 1994). As in other eukaryotes, both the germline and somatic genomes differentiate from a zygotic nucleus after sex (conjugation in ciliates). However, the development of a new somatic genome includes complex epigenetically guided processes (i.e., large-scale genome rearrangements, DNA elimination, chromosome fragmentation, de novo telomere addition, and chromosome amplification; Wang and Blackburn 1997; Heyse et al. 2010; Chalker and Yao 2011; Bellec and Katz 2012; Xu et al. 2012; Chen et al. 2014; Huang and Katz 2014; Cheng et al. 2016; Fuhrmann et al. 2016; Hamilton et al. 2016; Guérin et al. 2017; Maurer-Alcalá et al. 2018).
Molecular studies of germline-soma differentiation in ciliates are largely limited to a few cultivable “model” ciliates—Oxytricha trifallax (Swart et al. 2013; Chen et al. 2014), Paramecium tetraurelia (Aury et al. 2006; Arnaiz et al. 2012; Guérin et al. 2017), and Tetrahymena thermophila (Eisen et al. 2006; Hamilton et al. 2016). All of these models fall within the “Intramacronucleata” (referred to as the Im-clade for this study), which is one of the two major clades of ciliates (fig. 1). The other major clade, the “Postciliodesmatophora” (referred to as the Po-clade), shares a common ancestor with the model ciliates over 1.0 Ga (see Fig. 2 in Parfrey et al. 2011; Eme et al. 2014; Vďačný 2015). Yet, the Po-clade remain largely undersampled for genome features.
Fig. 1.
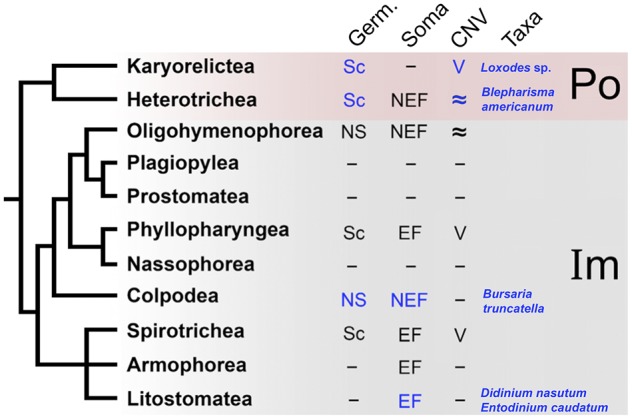
—Summary of general ciliate features demonstrates large gaps in knowledge for many ciliate classes and indicates data generated in this manuscript in blue. Absence of available data is denoted as “–.” Germline (Germ) genomes are denoted as either scrambled (Sc) or non-scrambled (NS). Somatic genomes (Soma) are marked as either extensively fragmented (EF) or non-extensively fragmented (NEF). Similarly, copy number variation (CNV) of chromosomes containing protein coding genes are indicated as variable (V) or approximately equal (≈). The lineages in the Po-clade (Po) are highlighted by red. The remaining ciliate classes are found in the Im-clade (Im).
Fig. 2.

—Relative chromosome copy numbers for members of the Po-clade show contrasting patterns of high copy number but stochasticity in Blepharisma and variable but repeatable copy number in Loxodes. Expected plots of chromosome copy number for Blepharisma americanum (A) and Loxodes spp. (C) are based on previous studies. The observed variable copy number for B. americanum (B) is consistent with the expected results for both the population sample (pop-DNA) and the three individuals (WGA). However, for all four Loxodes spp. individuals (WGA-1/2 and WGA-3/4 representing two distinct morphospecies), the observed chromosome copy number (D) deviates substantially from the expected copy numbers (C). “*” indicate relative chromosome copy number values less than three.
Arguably, one of the most notable differences among the model ciliates is the dramatic variation in somatic genomic architecture: ciliates can be grouped into those with “long” versus “nano” sized somatic chromosomes. In the models T. thermophila and P. tetraurelia (Class Oligohymenophorea), the somatic chromosomes are “large” (by ciliate standards) being on an average 100’s of kilobases to ∼1–2 megabases in length, lack centromeres (Aury et al. 2006; Eisen et al. 2006) and are substantially gene-rich (∼60–80% of their length composed of open reading frames). By contrast, the somatic genome of O. trifallax (Cl: Spirotrichea) is predominantly composed of ∼16,000 unique “nano-chromosomes,” most of which contain a single ORF (ranging from <1 kb to ∼66 kb; Swart et al. 2013). Prior to our work, evidence for the phylogenetic distribution of somatic nano-chromosomes was limited to only three ciliate classes: Spirotrichea, Armophorea, and Phyllopharyngea (e.g. Riley and Katz 2001; fig. 1).
In addition to variable chromosome size among ciliate somatic genomes, there are differences in patterns of chromosome copy number. For example, in Tetrahymena thermophila, each of its 225 unique somatic chromosomes is maintained at ∼45 copies in the somatic nucleus (Doerder et al. 1992; Eisen et al. 2006). In the ciliates Chilodonella uncinata and Oxytricha trifallax, both with nano-sized somatic chromosomes, the macronuclei contain millions of chromosomes maintained at variable but heritable copy numbers (Heyse et al. 2010; Bellec and Katz 2012; Xu et al. 2012; Huang and Katz 2014). The range of copy numbers of these chromosomes can span multiple orders of magnitude from several hundred to >50,000 (Bellec and Katz 2012; Xu et al. 2012; Huang and Katz 2014). Current data suggest that differential chromosome amplification is limited to those ciliates with macronuclear nano-chromosomes (fig. 1; Heyse et al. 2010; Bellec and Katz 2012; Xu et al. 2012; Huang and Katz 2014).
Ciliates in the Po-clade represent two presumed extremes in genome architecture. Ciliates in the Heterotrichea are often very large (some species are > 1 mm in length) with correspondingly large somatic nuclei that contain from ∼1,000 to >13,000 times more DNA than their germline nuclei (Ovchinnikova et al. 1965; Wancura et al. 2017). The other class, Karyorelictea, can be of similar sizes yet often have numerous clusters of somatic nuclei with relatively low DNA content (∼1.1–12 times more DNA in their somatic nuclei; reviewed in Yan et al. 2017). Based on this observation, Karyorelictea are the only group of ciliates to be described as paradiploid (i.e., nearly diploid) and their name (karyo = nucleus; relictea—relict) suggests a primitive state (Kovaleva and Raikov 1978; Bobyleva et al. 1980; Raikov 1982, 1985; Raikov and Karadzhan 1985; Yan et al. 2017).
Karyorelictean ciliates have been described as primitive based on three features: 1) relatively simple ciliature, 2) unusually low DNA content (“paradiploid,” or nearly diploid; Kovaleva and Raikov 1978; Raikov and Karadzhan 1985; Yan et al. 2017), and 3) the inability to divide their macronuclei during asexual divisions, which has only been described in members of this class (Raikov 1985, 1994; Yan et al. 2017). Molecular phylogenies place the Karyorelictea sister to the Heterotrichea (i.e., the Po-clade), inconsistent with the idea that this lineage represents the ancestral state for ciliates (e.g. Gao and Katz 2014; Gao et al. 2016). We use qPCR to estimate chromosome copy number to address the putative paradiploidy of Karyorelictea.
The complex processing underlying development of somatic nuclei from zygotic nucleus in ciliates relies on the elimination of germline-limited DNA (i.e., internally eliminated sequences; IESs) and the accurate “assembly” of functional somatic regions (i.e., macronuclear-destined sequences; MDS). The removal of IESs during the development of the somatic genome is analogous to intron-splicing during mRNA maturation, though IES excision occurs within the DNA (Jönsson et al. 2009; Wahl et al. 2009; Allen and Nowacki 2017). The organization of MDS/IES in germline genomes falls into two major categories: scrambled and nonscrambled (Prescott 1994; Ardell et al. 2003; Möllenbeck et al. 2006; Wong and Landweber 2006; Chen et al. 2014; Maurer-Alcalá et al. 2018). We define non-scrambled germline loci as those with MDSs that are on the same DNA strand and joined in “order” during DNA elimination in ciliates (fig. 5A). By contrast, scrambled germline loci are characterized by MDSs being found on opposing DNA strands and/or in nonconsecutive order (fig. 5B). Germline scrambling has only been documented in the Phyllopharyngea and Spirotrichea clades (fig. 1; Ardell et al. 2003; Wong and Landweber 2006; Katz and Kovner 2010; Chen et al. 2014; Gao et al. 2015; Maurer-Alcalá et al. 2018).
Fig. 5.

—Exemplar cases of ciliate germline genome architecture generated in this study from Bursaria and Loxodes. Left, representative images of Bursaria truncatella (A) and Loxodes sp. (B) with their germline (solid blue circles) and somatic nuclei (blue-bordered). Right, germline loci are represented as a single line harboring MDSs (blue-bordered rectangles). All identifiable germline loci from Bursaria truncatella (A) were nonscrambled, whereas for Loxodes sp. (B) there is a mixture of scrambled and non-scrambled loci (only scrambled shown here). MDSs are numbered according to the order in which they are found in the soma and the corresponding arrows indicate their directionality in the germline genome. Bottom right scale bar (black) is 300 bp. Scale bar (bottom left of each ciliate) is 25 µm.
The details on germline genome architecture and the transformations that underlie the development of the somatic genome have largely been studied in only three classes of ciliates (Oligohymenophorea, Phyllopharyngea, and Spirotrichea; fig. 1). Taking advantage of single-cell genomics and transcriptomics technologies, we explore the genomes of Blepharisma americanum (Heterotrichea, Po-clade), several Loxodes species (Karyorelictea; Po-clade), the large Bursaria truncatalla (Colpodea, Im-clade) and voracious predatory ciliate Didinium nasutum (Litostomatea, Im-clade), capturing a deep (>1.0 Gy) divide between the Im and Po clades (fig. 1; see Fig. 2 in Parfrey et al. 2011). We present insights into genome evolution from these nontraditional models, which demonstrate a greater diversity of genomic architectures than we expected from the literature.
Materials and Methods
Ciliate Culturing and Isolation
Blepharisma americanum, Bursaria truncatella, and Didinium nasutum cultures were ordered from Carolina Biological Supply whereas Loxodes spp. were collected from a small pond in Hawley Bog (Hawley, MA; 42°35′N, 72°53′W) by collecting water at the sediment-water column interface. From these wild-caught Loxodes spp., we observed two dominant morphospecies which we used for our analyses in this study. Cultures of B. americanum were maintained in filtered pond water with a sterilized rice grain to support bacterial growth. For isolation, individual cells were picked from cultures and then washed through a series of dilutions with filtered pond or bog water to dilute any contaminating bacteria and microeukaryotes that may have been carried over with the cell.
Total DNA Extraction
For Blepharisma americanum, ∼1,300 cells were collected on a 10-µm filter and rinsed thoroughly with filtered pond water. DNA extraction from the filter was done using the ZR Soil Microbe DNA MiniPrep kit (Zymo Research, catalog number D6001) following the manufacturer’s instructions. The eluted gDNA was stored at −20°C prior to the qPCR analyses performed, described below.
Single-Cell Whole Genome Amplification
For whole genome amplification (WGA), each washed cell was placed into a minimal volume of media in an individual sterile 0.2 ml tube containing 1 µl of molecular grade water. For each morphospecies this was done in triplicate. Cell lysis and genome amplification were then carried out following the manufacturer’s instructions (Qiagen; Repli-g Single Cell Kit, catalog number 150343). Of the resulting WGA products, we selected the most robust products (e.g., with the best amplification plots over time) for high-throughput sequencing and subsequent use in our analyses. In the end, we used a single WGA product for B. americanum, B. truncatella, and D. nasutum. For the two distinct Loxodes spp. morphospecies, several WGAs were produced, although only two WGA products for each of the morphospecies were used in our study. Of Loxodes WGAs, only a portion of a single WGA product for each morphospecies was used for high-throughput sequencing, but all four products were used for the qPCR analyses in this study (detailed below).
Single-Cell Whole Transcriptome Amplification
For the morphospecies with successful whole genome amplifications, freshly isolated (and washed) individual cells of the same morphospecies were placed in a minimal volume of their media in individual sterile 0.2 ml centrifuge tubes containing 1 µl of molecular grade water. The whole transcriptome amplification (WTA) reactions for each of the cells followed the manufacturer’s protocols (Clontech; SMART-Seq v4 Ultra Low Input RNA Kit, catalog number 634888) adjusting all volumes to ¼ reaction volumes. For B. americanum, five WTA products were prepared, three of which were from “typical” individuals from a log-phase culture and the remaining two from “giant” individuals with obvious signs of predation on other B. americanum (e.g., bright red vacuoles). For B. truncatella, D. nasutum, and each of the two morhospecies of Loxodes, two WTA products from “vegetative” individuals (e.g., no apparent signs of conjugation, division, or gigantism) were used for downstream analyses. Overall 13 WTA products were sequenced and used in this study.
Library Preparation, Genome and Transcriptome Sequencing
Libraries of the amplified WGAs and WTAs were constructed using the Nextera XT DNA Library Preparation kit, following the manufacturer’s instructions (Illumina). The prepared libraries were sequenced at the IGM Genome Center at University of California at San Diego on a portion of a single channel of a HiSeq4000. For Loxodes spp., WGA and WTAs were also later sequenced at the IGS Genome Resource Center at the University of Maryland on a portion of a single channel of a HiSeq4000.
Genome and Transcriptome Assembly
The raw reads from all data sources were processed using BBDuK (sourceforge.net/projects/bbmap/ (last accessed January 2018); Bushnell 2015) with a minimum quality score of 24 and minimum length of 120 bp. Single-cell genomes were assembled with SPAdes (v3.10.0; Bankevich et al. 2012) using the single-cell and careful parameters. For Loxodes spp. WGAs, we pooled the raw reads by morphospecies prior to assembly as they had been resequenced at a later date. All single-cell transcriptomes were assembled individually using rnaSPAdes, which is part of the SPAdes package, using default parameters. Prior to the single-cell genome assembly of Bursaria truncatella, trimmed read pairs were mapped against the P. tetraurelia genome (CAAL00000000), the dominant food source in the culture, using BBDuK. Those pairs that remained unalignable to the P. tetraurelia genome were then used for the B. truncatella genome assembly.
Post-Assembly Preparation of Transcriptome Data
A suite of custom python scripts was used to process the transcriptomic data generated from our single-cell WTAs (github.com/maurerax/KatzLab/tree/HTS-Processing-PhyloGenPipeline). In brief the processing includes: 1) the removal of contaminating rRNAs and bacterial transcripts; 2) the identification of putative ORFs from the transcripts; 3) the removal of transcripts of near identity (>98% nucleotide identity) across ≥70% of their length to larger transcripts. For all of our taxa, the pooling of “redundant” transcripts was performed after we concatenated the assemblies by taxon, resulting in a single “core” transcriptome for each.
Identification of Telomeric Repeats
Prior to the identification of potential telomeric repeats from the taxa whose genomes, we partially sequenced, we also downloaded the genomes of Entodinium caudatum, Stentor coeruleus, and Condylostoma magnus (NBJL00000000, MPUH00000000, and CVLX00000000, respectively) from GenBank. These additional taxa were downloaded as they represent the only currently available large-scale genomic data from the same classes of ciliates to those in our studies (with the exception of B. truncatella and Loxodes spp. as no genomic data for members of the Colpodea and Karyorelictea, respectively, was publicly released). For all of the genome assemblies, we isolated the first and last 30 bp of every scaffold. These scaffold ends were run through MEME (v4.11.4; Bailey et al. 2009) twice to evaluate the presence (or absence) of repetitive motifs, once without shuffling the sequences of the scaffolds’ ends and the second time with random shuffling of the sequences. Putative telomeric ends (e.g., significant motifs that were not found in the “shuffled” run of MEME) were only found for Stentor coeruleus, Didinium nasutum, and Entodinium caudatum. Afterwards, we used custom python scripts and these potential telomeric repeats to identify and extract scaffolds that were capped on both ends with telomeric repeats (allowing for a single mismatch; github.com/maurerax/KatzLab/tree/SingleCellGermSoma).
Evaluation of Putative Germline Genome Scaffolds
Genomic scaffolds of the taxa we sequenced in this study that were not capped by telomeric repeats were used to identify putative germline loci that may have been amplified by the WGA reaction (given its previously demonstrated ability to amplify portions of the germline genome in ciliates; Maurer-Alcalá et al. 2018). For the identification of putative germline genome scaffolds and identification of germline-soma architecture, we used previously outlined protocols (Maurer-Alcalá et al. 2018). Briefly, this includes identification of ORF-poor genomic scaffolds, alignment of transcripts to those scaffolds and evaluation of common signatures of germline-soma architectures found in other ciliates.
Evaluation of Germline Genome Architecture
After identifying a set of putative germline (micronuclear) scaffolds from Blepharisma amercianum, Bursaria truncatella, and a single Loxodes sp. (due to poor assembly of the second morphospecies; fragmented and strong signatures of contamination), we used BLAST (v2.4.0; Camacho et al. 2009), with parameters of “-ungapped -perc_identity 97 -outfmt 6 -word_size 25,” to map each taxon’s transcriptomic data to its germline scaffolds. Custom python scripts (github.com/maurerax/KatzLab/tree/SingleCellGermSoma) analyzed the output from BLAST and categorized the loci and transcriptome data into three broad categories: nonscrambled, scrambled, and unmapped. Based on data from a previous study exploiting single-cell genomics and transcriptomics for analyses of germline architecture, we also only used germline loci where ≥60% of the length of a transcript was successfully mapped for subsequent analyses.
As a precaution to ensure that these loci were more likely germline than soma (which often comprised a substantial proportion of the overall initial genome assembly), we explored the portions of the mapped transcripts that represented alignment boundaries with the genome assembly (e.g., genome assembly limited DNA). To be considered a true putative germline sequence these boundaries must not be nearly identical to the canonical GT-YAG intron–exon boundaries. Similarly, to characterize the genomic-loci as being germline (e.g., harboring an IES), the genome-limited DNA must be flanked by identical pointer sequences that are present at these mapped–unmapped boundaries.
Quantitative PCR
Quantitiave real-time PCR (qPCR) was used to estimate patterns of gene copy number in Loxodes spp. and Blepharisma americanum. Ten-fold serially diluted plasmids (1 to 10−7 ng/μl) containing gene fragments of interest were prepared and used to generate the standard curve for each gene. Primers were designed using sequences obtained from both the WGA and WTA products (supplementary table S3, Supplementary Material online) of B. americanum and Loxodes spp. The DyNAmo Flash SYBR Green qPCR kit (Fisher Scientific) was used for all quantitative PCR experiments in 96-well plates on an ABI StepOnePlus thermal-cycler. Reactions were conducted in a final volume of 20 μl, containing 10 μl 2×master mix, 150 nM of each primer, 1 μl of template DNA (at 1 ng/μl), and 8 μl of water. qPCR of each targeted gene fragment and WGA sample was performed in triplicate for each experiment. Each experiment was replicated 2 times. To estimate the copy number of chromosomes (copies/ng per cell), we divided the copy numbers of each cell (copies/µl per cell) by the concentration of the WGA (X ng/µl). To mitigate the potential impact of genome amplification on absolute copy number (and allow comparisons between species), we estimated the relative copy number for each gene of interest by setting the nSSU-rDNA copy number to 1×106 while maintaining the genes’ copy number ratio to the nSSU-rDNA locus.
Statistical Analyses
All statistical analyses were performed using R (Team 2017). For qPCR data, we used a mixed effects ANOVA evaluating patterns of copy number abundance between and within cells for both B. americanum and Loxodes spp.
Code Availability
All custom python scripts used in this study are available from: github.com/maurerax/KatzLab/tree/SingleCellGermSoma and github.com/maurerax/KatzLab/tree/HTS-Processing-PhyloGen Pipeline.
Results and Discussion
Differential Chromosome Amplification in the Po-Clade
We explore patterns of somatic chromosome copy number in the Po-Clade, focusing on the genera Blepharisma (Heterotrichea) and Loxodes (Karyorelictea), to test whether either of these ciliates differentially amplifies somatic chromosomes. In fact, many eukaryotes extensively amplify extrachromosomal copies of their ribosomal RNA genes (e.g. Sinclair and Guarente 1997; Zufall et al. 2005; Cohen et al. 2008), so we compare the nuclear small subunit ribosomal RNA gene (nSSU-rRNA) to several protein coding genes. We analyze chromosome copy number using DNA isolated from a population of ∼1,300 Blepharisma americanum individuals (pop-DNA) and compare this to copy number estimates from three individual B. americanum following whole genome amplification (WGA). Given that we do not find any significant bias produced by the WGA reactions, we then use this single-cell ‘omics approach on the uncultivable genus Loxodes (Karyorelictea).
In the analyses of both total genomic DNA (pop-DNA) and single-cell WGA (sc-WGA) of B. americanum, the nSSU-rRNA gene is characteristically high, with an estimated 2.55 × 107 ± 8.42×106 copies/ng per cell and 7.90 × 107 ±1.02 × 107 copies/ng per cell, respectively. Copy numbers were estimated from DNA contents of quantitative PCR (qPCR) results and were adjusted to per ng per cell. Estimates of copy numbers for protein coding genes between the different preparations of Blepharisma (pop-DNA and sc-WGA) are similarly consistent, ranging from 1.18×106 ± 4.38×104 copies/ng per cell and 8.45×105 ± 1.14×105 copies/ng per cell (for one α-tubulin paralog). The least abundant of the protein coding genes from the total gDNA and single-cells are 9.77×104 ± 2.41×104 copies/ng per cell and 6.06×102 ± 2.87×102 copies/ng per cell for EF-1α and an α-tubulin paralog, respectively (supplementary table S1, Supplementary Material online).
To compare relative abundances across taxa, we set the nSSU-rRNA copy number to 106 (a value based on evidence from diverse ciliates; Heyse et al. 2010; Gong et al. 2013; Huang and Katz 2014). We find that the relative copy numbers for chromosomes containing protein coding genes (two paralogs of Actin and α-Tubulin, and EF-1α) in B. americanum span ∼2 orders of magnitude (fig. 2B) with the exception of actin paralog 2, which is consistently at low copy number across all samples (P = 0.093 from an ANOVA). Despite greater variability in absolute copy numbers from the population of cells (pop-DNA) compared with the individual cells, we observe no significant biases between methods (total pop-DNA vs. single-cell WGA; P = 0.474 from a Kruskal–Wallis test; fig. 2B). In other words, the sc-WGA method can be used to assess patterns of individual chromosome copy numbers because this method yields the same results as studies of a population of cells.
We then deployed the same methods (qPCR after single-cell WGA) to study the uncultivable genus Loxodes in the “paradiploid” class Karyorelictea, which is predicted to have ∼2 copies of every protein coding gene (Raikov 1982; Yan et al. 2017). We performed a similar qPCR experiment using five genes (nSSU-rRNA, EF-1α, Actin, Rs11, and α-Tubulin) from sc-WGAs of wild-caught individuals of Loxodes, representing two distinct species based on rRNA gene diversity (supplementary table S2, Supplementary Material online). As we only have relative numbers here, we again set the nSSU-rRNA gene to 106 copies to allow comparison of patterns of chromosome copy numbers. By contrast to the stochastic patterns of chromosome copy number in B. americanum, the differences in copy number among protein coding genes in Loxodes spp. consistently spanned a far greater range with the highest being Actin at ∼2.87×104 copies relative to the rRNA gene and the lowest being EF-1α at ∼4.39×10−1 copies/ng per cell (∼4 orders of magnitude; supplementary table S2, Supplementary Material online and fig. 2D). We observe significant differences in gene copy number within each cell of Loxodes spp. (P ≪ 0.05), implicating the differential amplification of chromosomes. For both of the Loxodes species, gene copy numbers are maintained in a mostly conserved order: nSSU-rRNA ≫ Actin > Rs11 > α-Tubulin > EF-1α (fig. 2D), which contrasts with the stochastic pattern in Heterotrichea.
The contrasting pattern of stochasticity in chromosome copy number in B. americanum and the predictability in chromosome number in Loxodes spp. likely reflects differences in genome architecture of their somatic nuclei. The macronuclei of Blepharisma house large quantities of DNA and possess the ability to divide by amitosis, while Loxodes spp.’ macronuclei are DNA poor and do not divide with cell division (Raikov 1982; Katz 2001; Yan et al. 2017). The stochasticity in chromosome copy number for Blepharisma may be a byproduct of the massive genome amplification that occurs during development (Santangelo and Barone 1987), as the somatic nucleus is estimated to have >1,000× more DNA than the germline nucleus (Ovchinnikova et al. 1965; Wancura et al. 2017). Variable chromosome copy number among individuals is likely an inherent feature of Blepharisma and its relatives (in the class Heterotrichea; fig. 1), exemplified by Stentor coeruleus, whose chromosome copy numbers of the nSSU-rDNA are clearly correlated to cell size (Slabodnick et al. 2017) and likely nuclear volume (Cavalier-Smith 1978). This suggests that the observed stochasticity from our measurements is likely the result of biological differences (e.g., cell volume or life-cycle stages; fig. 2A and B).
Although Loxodes spp. are found in the sister class to B. americanum (both in the Po-clade), Loxodes and its relatives have long been considered as “primitive” ciliates (Raikov 1985, 1994; Raikov and Karadzhan 1985; Orias 1991). This presumption arose from early studies that found that the somatic macronucleus is unable to divide (instead needing to be differentiated from a germline nucleus with each cell division; reviewed in Yan et al. 2017) as well as from estimates of DNA content based on autoradiographic measurements from the somatic and germline nuclei of Loxodes and its relatives (Kovaleva and Raikov 1978; Bobyleva et al. 1980). From these early measurements, where the somatic nuclei typically harbor only ∼1.1 to ∼12 times the amount of DNA compared with the germline nuclei, karyorelictid lineages were labeled as paradiploid (“nearly-diploid”). This has led to the expectation that the relative copy number among protein-coding genes would be approximately equal in this class of ciliates (fig. 2C). Such low ploidy is unusual among ciliates. For example, ploidy is species-dependent and ranges from ∼45 N in Tetrahymena thermophila (Woodard et al. 1972) to ∼800 N in Paramecium tetraurelia (Duret et al. 2008) and an average of ∼2,000 N in the differentially amplified nano-chromosomes in Oxytricha trifallax (Swart et al. 2013).
Surprisingly, our data demonstrate that Loxodes spp. is neither paradiploid nor are all chromosomes equally amplified. Our estimates of relative chromosome copy number show that instead of being present in roughly equal abundance, chromosomes containing our target genes differ by several orders of magnitude (fig. 2C and D). Though nondividing macronuclei in Loxodes spp. (and other members of the class Karyorelictea) age over time (at most seven generations; Raikov 1982, 1985, 1994; Yan et al. 2017), we do not believe aging alone is sufficient to explain our data as the replicability of estimates across cells suggests heritable differences in copy number (from 2 to >1,000 copies). These copy number data suggest that the long-held description of Loxodes spp. as “primitive,” based upon DNA content estimates and the inability to divide their macronuclei, is inaccurate.
Unexpected Extensive Fragmentation of Somatic Genomes from the Im-Clade
Extensive fragmentation of chromosomes into gene-sized “nano-chromosomes” during the development of somatic macronuclei is well documented in only three ciliate classes (e.g., in Chilodonella uncinata [cl: Phyllopharyngea; McGrath et al. 2007], Oxytricha trifallax [cl: Spirotrichea; Swart et al. 2013], and Nycotherus ovalis [cl: Armophorea; McGrath et al. 2007; Ricard et al. 2008]; fig. 1). We searched for evidence of extensive fragmentation in the class Litosomatea (Im-clade; fig. 1), analyzing a single-cell WGA assembly for Didinium nasutum and the recently released genome assembly of Entodinium caudatum (a distantly related member of the same class). We evaluated the ends of scaffolds for both D. nasutum and E. caudatum to look for telomeres as no record of telomeres has been reported for members in this class. This approach resulted in a common repetitive motif in both taxa, C4A2T. As telomeric sequences seem well conserved over broad phylogenetic scales in ciliates (Aury et al. 2006; Eisen et al. 2006; McGrath et al. 2007; Swart et al. 2013; Aeschlimann et al. 2014), this simple repeat may be specific to Litostomatea.
To assess the size distributions of somatic chromosomes, we used the telomeric motif to identify scaffolds bounded by repeats at both ends (e.g., complete assembled chromosomes) for both D. nasutum and E. caudatum. To our surprise, we identified 328 complete nano-chromosomes in D. nasutum’s telomere-bound scaffolds and 7,560 complete chromosomes from the released E. caudatum genome assembly (figs. 3 and 4; supplementary fig. S1 and supplementary data 1 and 2, Supplementary Material online). Although the nano-chromosome estimates are very disparate among D. nasutum and E. caudatum, previous work has demonstrated the bias in the genome amplification reaction against ciliate nano-chromosomes, which may be present in genome assemblies as “by-catch” (Maurer-Alcalá et al. 2018) and which account for the order of magnitude difference between Didinium and Entodinium. To further check that these were not simply assembly artefacts, we mapped transcripts from single D. nasutum individuals to the pool of 328 putatively complete nano-chromosomes (i.e., those with telomeres on each end). Of these 328 chromosomes, 254 (77.4%) harbor a single ORF and overall 316 (96.3%) of the chromosomes are actively transcribed as evidenced in the single-cell transcriptomes. As no transcriptome data are publicly available for E. caudatum, we used more relaxed conditions (e.g., chromosomes with ≥50% identity and align to ≥50% of the translated ORF) to map 5,692 translated ORFs from our D. nasutum transcriptome to 5,293 (70.0%) of E. caudatum’s complete chromosomes.
Fig. 3.

—Distribution of chromosome lengths among diverse lineages reveals unexpected presence of nano-chromosomes in Didinium nasutum and Entodinium caudatum. Representative images of each taxon are next to their names and are not drawn to scale. Tetrahymena thermophila’s germline chromosomes are noted, whereas the ciliate’s drawing is next to its somatic chromosomes.
Fig. 4.

—Exemplar chromosomes harboring α-tubulin from diverse ciliates highlights the unusual ciliate genome architecture. Telomere sequences bound complete ciliate chromosomes, with nonprotein coding regions found in gray, and α-tubulin in blue (orientation of the coding sequence is noted by arrows). For Entodinium caudatum, we found a single nanochromosome with two divergent α-tubulin genes which are noted by differences in the shade of blue. Black scale bar represents 1 kb.
Having demonstrated the presence of nano-chromosomes in the D. nasutum and E. caudatum genome assemblies, we determined that the size range of these complete chromosomes are nearly identical for both, ranging from ∼0.4 kb to ∼26 kb, despite differences in the methods used to obtain the genomic data (e.g., use of sc-WGA techniques for D. nasutum and more traditional DNA isolation approaches used for E. caudatum; supplementary fig. S1, Supplementary Material online). However, previous work using pulsed-field gel electrophoresis of total gDNA from D. nasutum did not report chromosomes <50 kb (Popenko et al. 2015), which suggests that the nano-chromsomes may be present at relatively low copy numbers and/or that the retention of these chromosomes is strongly dependent on the DNA isolation approaches. Comparisons of the size distribution of these complete chromosomes for D. nasutum and E. caudatum to genomic data from other ciliates, demonstrate that these chromosomes’ sizes are consistent with the “gene-sized” chromosomes found in divergent ciliate taxa (e.g., Chilodonella uncinata and Oxytricha trifallax; McGrath et al. 2007; Swart et al. 2013; figs. 3 and 4; supplementary fig. S1, Supplementary Material online).
The data on nano-chromosomes in the class Litostomatea are consistent with the 1890 description of giant germline chromosomes, which are presumably generated through endoreplication during development of a new macronucleus (Balbiani 1890). The correspondence between the appearance of giant chromosomes during development and the presence of nano-sized chromosomes in somatic genomes has been extensively documented (most notably in Chilodonella and Stylonychia, classes Phyllopharygnea and Spirotrichea, respectively; Pyne 1978; Ammermann 1986; Katz 2001; Riley and Katz 2001; Juranek et al. 2005; Postberg et al. 2008; Katz and Kovner 2010; figs. 1 and 4). In these ciliate classes, polytenization occurs just prior to the extensive genome remodeling that ultimately leads to the formation of the thousands of unique nano-chromosomes through epigenetically guided DNA elimination, large-scale genome rearrangements, and de novo telomere addition (Spear and Lauth 1976; Pyne 1978; Ammermann 1986; Postberg et al. 2008; Chen et al. 2014; Fuhrmann et al. 2016). The absence of polytenization of germline chromosomes from the model ciliates Paramecium tetraurelia and Tetrahymena thermophila, which possess “large” macronuclear chromosomes (ranging from ∼0.2 Mb to several Mb in size; Aury et al. 2006; Eisen et al. 2006; fig. 3 and supplementary fig. S1, Supplementary Material online), further implicates generation of giant chromosomes as being limited to nano-chromosome formation.
Well over 100 years ago, Éduoard-Gérard Balbiani, who provided the original description of polytene chromosomes in the dipteran Chironomus (Balbiani 1881), described the presence of polytene chromosomes in the ciliate Loxophyllum meleagris (also in the class Litostomatea; Balbiani 1890). Unfortunately, there had been little work able to corroborate the observations of Balbiani (1890). However, given the sister relationships between the classes Litostomatea, Spirotrichea, and Armophorea (“SAL” clade; Gentekaki et al. 2014; Gao et al. 2016), all of which possess both nano-chromosomes (Riley and Katz 2001; Ricard et al. 2008; Swart et al. 2013) and giant chromosomes (Wichterman 1937; Golikova 1965), these unusual genome architectural features could be a synapomorphy that further unites these classes within the Im-clade (fig. 1).
Germline Genome Architecture from Diverse Ciliates
Previous studies of germline genome architecture in ciliates are from even more sparsely sampled lineages than studies of somatic genomes, with data available from only a few model species in the classes Oligohymenophorea, Phyllopharyngea, and Spirotrichea (e.g. Landweber et al. 2000; Nowacki et al. 2008; Arnaiz et al. 2012; Chen et al. 2014; Gao et al. 2015; Hamilton et al. 2016; Guérin et al. 2017; Maurer-Alcalá et al. 2018). This has largely been a result of the lack of robust methods for the efficient extraction of high-quality germline DNA from uncultivable lineages. To overcome these limitations, we use a combination of single-cell genomics transcriptomics and bioinformatics to gain insights into the germline genome organization of three ciliate taxa, representing members of both the Im (Bursaria truncatella; cl: Colpodea) and Po clades (B. americanum; cl: Heterotrichea and Loxodes sp.; cl: Karyorelictea; fig. 1) and building on our previous work in C. uncinata (Maurer-Alcalá et al. 2018).
To explore the germline genome architecture of three ciliates, Blepharisma, Loxodes, and Bursaria, we mapped transcripts from single-cell transcriptome assemblies to the respective germline scaffolds generated by WGA. By following established methods for characterizing germline scaffolds (Maurer-Alcalá et al. 2018), we identified numerous putative germline scaffolds for all three taxa. We also find several scrambled germline loci in both B. americanum and Loxodes sp. (24 and 23, respectively; fig. 5B andsupplementary table S4, Supplementary Material online) as well as non-scrambled germline loci (15 and 11, respectively; supplementary table S4, Supplementary Material online). By contrast, we find no evidence of scrambling among the 162 transcripts mapped in Bursaria (supplementary table S4, Supplementary Material online). We define non-scrambled loci as those where macronuclear destined sequences are maintained in consecutive order (e.g., “MDS 1—MDS 2—MDS 3”; fig. 5A) while scrambled loci meet at least one of two criteria: 1) MDSs are present in a non-consecutive order (e.g., “MDS 2—MDS 3—MDS 1”) and/or 2) MDSs can be found on both strands of the germline scaffolds (i.e., some are inverted; fig. 5B).
Our pilot study also reveals that scrambled germline loci in members of the Po-clade vary from patterns in C. uncinata and O. trifallax, both of which are members of the Intramacronucleata. For example, the data on gene scrambling in the classes Spirotrichea and Phyllopharyngea (Im-clade) reveal small MDSs separated by relatively large distances in the germline genome (Chen et al. 2014; Maurer-Alcalá et al. 2018). This is not the case in the germline scaffolds of B. americanum and Loxodes sp. (Po-clade), where differences in the distances between MDSs for both scrambled and nonscrambled germline loci were insignificant (P = 0.301). Similarly, in both C. uncinata and O. trifallax, scrambled germline loci are composed of a greater number of MDSs than nonscrambled loci (Chen et al. 2014; Maurer-Alcalá et al. 2018), yet for both B. americanum and Loxodes sp. nearly all germline loci (i.e., scrambled and non-scrambled) are composed of only two large MDSs and are most often found on opposing DNA strands (i.e., inverted; fig. 5B andsupplementary table S4, Supplementary Material online).
The observations from the members of the Po-clade contrast with those from Bursaria truncatella (Im clade), whose last common ancestor with the model ciliates Paramecium and Tetrahymena was more recent (∼800–1,000 Ma; Fig. 2 of Parfrey et al. 2011). We did not find any evidence of scrambled germline loci from the mapping of transcriptomic data back to the putative germline scaffolds for B. truncatella, with all 162 identifiable germline loci being non-scrambled (fig. 5A). This suggests that B. truncatella’s germline genome lacks substantial amounts of gene-scrambling and that the single-cell genomic methods used here do not introduce false evidence of scrambling.
Given the absence of germline gene-scrambling in B. truncatella, we evaluated the similarity its nonscrambled germline genome architecture might have by comparison to the model ciliates Paramecium and Tetrahymena. The germline-limited IESs present in the B. truncatella germline scaffolds do interrupt the protein-coding domains (fig. 5A), as is the case in P. tetraurelia (Arnaiz et al. 2012; Guérin et al. 2017) but not its close relative, T. thermophila where the majority of IESs occur in the intergenic regions (Hamilton et al. 2016). The pointer sequences for Paramecium tetraurelia and Tetrahymena thermophila, which are involved in aiding the guided genome rearrangements during development, are redundant and are often delineated by a terminal “TA” di-nucleotide in Paramecium and Tetrahymena (Arnaiz et al. 2012; Hamilton et al. 2016; Guérin et al. 2017). Unlike these model ciliates, the identified pointer sequences from the germlines of B. americanum, B. truncatella, and Loxodes sp. were unique at each germline locus. This suggests that the observable “simple” consensus sequences found in Paramecium and Tetrahymena are likely to be unique to the Oligohymenophorea and not a general feature of across the ciliate phylogeny.
Synthesis
In this study, we use single-cell ‘omics to explore the somatic and germline genome architectures of diverse ciliates. Our analyses demonstrate the presence of differential chromosome amplification and some scrambled germline loci in what has long been considered as a “primitive” class of ciliates, the Karyorelictea (Loxodes). These data, and those from their sister class (Heterotrichea, Blepharisma), suggest that the last common ancestor of ciliates was fairly complex, with a polyploid somatic nucleus and a complex developmental life-cycle that may have included un-scrambling of germline loci. Our analyses support the >100-year-old observations of Balbiani, who used light microscopy to identify unusual genome features in ciliates in the class Litostomatea. The interrelated insights presented here highlight how the power of single-cell genomics techniques can be harnessed to critically evaluate long-standing questions in genome biology, especially uncultivable lineages.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
The work in this study was supported by an NIH award (1R15GM113177) and NSF Go-LIFE (DEB-1541511) to L.A.K. and a Blakeslee award to Smith College. We thank Kelsie Maurer-Alcalá for contributing artistic renderings of the organisms used in the figures of this manuscript. We also thank members of the Katz Lab, particularly Monica Wilson, for frequent and insightful discussion and James Gaffney of the Knight Lab for his invaluable technical guidance.
Data deposition: The raw reads generated in this study are available in GenBank’s Short Read Archive (SRA) under BioProject number PRJNA427655.
Literature Cited
- Aeschlimann SH, et al. 2014. The draft assembly of the radically organized Stylonychia lemnae macronuclear genome. Genome Biol Evol. 6(7):1707–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen SE, Nowacki M.. 2017. Necessity is the mother of invention: ciliates, transposons, and transgenerational inheritance. Trends Genet. 33(3):197–207. [DOI] [PubMed] [Google Scholar]
- Ammermann D. 1986. Giant chromosomes in ciliates. Biol Chem Hoppe-Seyler 367(1):80–1102. [Google Scholar]
- Ardell DH, Lozupone CA, Landweber LF.. 2003. Polymorphism, recombination and alternative unscrambling in the DNA polymerase alpha gene of the ciliate Stylonychia lemnae (Alveolata; class Spirotrichea). Genetics 165(4):1761–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaiz O, et al. 2012. The Paramecium germline genome provides a niche for intragenic parasitic DNA: evolutionary dynamics of internal eliminated sequences. PLoS Genet. 8(10):e1002984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aury JM, et al. 2006. Global trends of whole-genome duplications revealed by the ciliate Paramecium tetraurelia. Nature 444(7116):171–178. [DOI] [PubMed] [Google Scholar]
- Bailey TL, et al. 2009. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37(Web Server issue):W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird SE, Fino GM, Tausta SL, Klobutcher LA.. 1989. Micronuclear genome organization in Euplotes crassus – a transposon-like element is removed during macronuclear development. Mol Cell Biol. 9(9):3793–3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbiani E. 1881. Sur la structure du noyau des cellules salivaires chez les larves de Chironomus. Zool Anz. 4:637–641. [Google Scholar]
- Balbiani EG. 1890. Sur la structure intime du noyau du Loxophyllum meleagris. Zool Anz. 13(110–115):132–136. [Google Scholar]
- Bankevich A, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 19(5):455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellec L, Katz LA.. 2012. Analyses of chromosome copy number and expression level of four genes in the ciliate Chilodonella uncinata reveal a complex pattern that suggests epigenetic regulation. Gene 504(2):303–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobyleva NN, Kudrjavtsev BN, Raikov IB.. 1980. Changes of the DNA content of differentiating and adult macronuclei of the ciliate Loxodes-Magnus (Karyorelictida). J Cell Sci. 44:375–394. [DOI] [PubMed] [Google Scholar]
- Bushnell B. 2015. BBMap Short-read aligner, and other bioinformatics tools.
- Camacho C, et al. 2009. BLAST plus: architecture and applications. BMC Bioinformatics 10:421.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalier-Smith T. 1978. Nuclear volume control by nucleoskeletal DNA, selection for cell-volume and cell-growth rate, and solution of DNA C-value paradox. J Cell Sci. 34:247–278. [DOI] [PubMed] [Google Scholar]
- Chalker DL, Yao MC.. 2011. DNA elimination in ciliates: transposon domestication and genome surveillance. Annu Rev Genet. 45(1):227–246. [DOI] [PubMed] [Google Scholar]
- Chen X, et al. 2014. The architecture of a scrambled genome reveals massive levels of genomic rearrangement during development. Cell 158(5):1187–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CY, et al. 2016. The PiggyBac transposon-derived genes TPB1 and TPB6 mediate essential transposon-like excision during the developmental rearrangement of key genes in Tetrahymena thermophila. Genes Dev. 30(24):2724–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Houben A, Segal D.. 2008. Extrachromosomal circular DNA derived from tandemly repeated genomic sequences in plants. Plant J. 53(6):1027–1034. [DOI] [PubMed] [Google Scholar]
- Doerder FP, Deak JC, Lief JH.. 1992. Rate of phenotypic assortment in Tetrahymena thermophila. Dev Genet. 13(2):126–132. [DOI] [PubMed] [Google Scholar]
- Duret L, et al. 2008. Analysis of sequence variability in the macronuclear DNA of Paramecium tetraurelia: a somatic view of the germline. Genome Res. 18(4):585–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen JA, et al. 2006. Macronuclear genome sequence of the ciliate Tetrahymena thermophila, a model eukaryote. PLoS Biol. 4(9):e286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eme L, Sharpe SC, Brown MW, Roger AJ.. 2014. On the age of eukaryotes: evaluating evidence from fossils and molecular clocks. Cold Spring Harb Perspect Biol. 6(8):a016139.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann G, Jönsson F, Weil PP, Postberg J, Lipps HJ.. 2016. RNA-template dependent de novo telomere addition. RNA Biol. 13(8):733–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F, et al. 2016. The all-data-based evolutionary hypothesis of ciliated protists with a revised classification of the phylum Ciliophora (Eukaryota, Alveolata). Sci Rep. 6(1):24874.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F, Katz LA.. 2014. Phylogenomic analyses support the bifurcation of ciliates into two major clades that differ in properties of nuclear division. Mol Phylogenet Evol. 70:240–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F, Roy SW, Katz LA.. 2015. Analyses of alternatively processed genes in ciliates provide insights into the origins of scrambled genomes and may provide a mechanism for speciation. Mbio 6(1):e01998-14.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentekaki E, et al. 2014. Large-scale phylogenomic analysis reveals the phylogenetic position of the problematic taxon Protocruzia and unravels the deep phylogenetic affinities of the ciliate lineages. 78:36–42 [DOI] [PubMed]
- Golikova M. 1965. Der Aufbau des Kernapparates und die Verteilung der Nukleinsäuren und Proteine bei Nyctotherus cordiformis stein. Archiv für Protistenkunde 108:191–216. [Google Scholar]
- Gong J, Dong J, Liu XH, Massana R.. 2013. Extremely high copy numbers and polymorphisms of the rDNA operon estimated from single cell analysis of oligotrich and peritrich ciliates. Protist 164(3):369–379. [DOI] [PubMed] [Google Scholar]
- Guérin F, et al. 2017. Flow cytometry sorting of nuclei enables the first global characterization of Paramecium germline DNA and transposable elements. BMC Genomics 18(1):327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale CA, Jacobs ME, Estes HG, Ghosh S, Klobutcher LA.. 1996. Micronuclear and macronuclear sequences of a Euplotes crassus gene encoding a putative nuclear protein kinase. J Eukaryot Microbiol. 43(5):389–392. [DOI] [PubMed] [Google Scholar]
- Hamilton EP, et al. 2016. Structure of the germline genome of Tetrahymena thermophila and relationship to the massively rearranged somatic genome. Elife 5:e19090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyse G, Jönsson F, Chang WJ, Lipps HJ.. 2010. RNA-dependent control of gene amplification. Proc Natl Acad Sci USA. 107(51):22134–22139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Katz LA.. 2014. Nanochromosome copy number does not correlate with RNA levels though patterns are conserved between strains of the ciliate morphospecies Chilodonella uncinata. Protist 165(4):445–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jönsson F, Postberg J, Lipps HJ.. 2009. The unusual way to make a genetically active nucleus. DNA Cell Biol. 28(2):71–78. [DOI] [PubMed] [Google Scholar]
- Juranek SA, Rupprecht S, Postberg J, Lipps HJ.. 2005. snRNA and heterochromatin formation are involved in DNA excision during macronuclear development in stichotrichous ciliates. Eukaryot Cell 4(11):1934–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz LA. 2001. Evolution of nuclear dualism in ciliates: a reanalysis in light of recent molecular data. Int J Syst Evol Microbiol. 51(Pt 4):1587–1592. [DOI] [PubMed] [Google Scholar]
- Katz LA, Kovner AM.. 2010. Alternative processing of scrambled genes generates protein diversity in the ciliate Chilodonella uncinata. J Exp Zoolog B 314B(6):480–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klobutcher LA, et al. 1998. Conserved DNA sequences adjacent to chromosome fragmentation and telomere addition sites in Euplotes crassus. Nucleic Acids Res. 26:4230–4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovaleva VG, Raikov IB.. 1978. Diminution and re-synthesis of DNA during development and senescence of diploid macronuclei of ciliate Trachelonema sulcata (Gymnostomata-Karyorelictida). Chromosoma 67(2):177–192. [Google Scholar]
- Landweber LF, Kuo TC, Curtis EA.. 2000. Evolution and assembly of an extremely scrambled gene. Proc Natl Acad Sci USA. 97(7):3298–3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzke MA, Mosher RA.. 2014. RNA-directed DNA methylation: an epigenetic pathway of increasing complexity. Nat Rev Genet. 15(6):394–408. [DOI] [PubMed] [Google Scholar]
- Maurer-Alcalá XX, Katz LA.. 2015. An epigenetic toolkit allows for diverse genome architectures in eukaryotes. Curr Opin Genet Dev. 35:93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer-Alcalá XX, Knight R, Katz LA.. 2018. Exploring the germline genome of the ciliate Chilodonella uncinata through single-cell omics (transcriptomics and genomics). mBio 9(1):e01836-17.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath CL, Zufall RA, Katz LA.. 2007. Variation in macronuclear genome content of three ciliates with extensive chromosomal fragmentation: a preliminary analysis. J Eukaryot Microbiol. 54(3):242–246. [DOI] [PubMed] [Google Scholar]
- Möllenbeck M, Cavalcanti ARO, Jönsson F, Lipps HJ, Landweber LF.. 2006. Interconversion of germline-limited and somatic DNA in a scrambled gene. J Mol Evol. 63(1):69–73. [DOI] [PubMed] [Google Scholar]
- Nowacki M, et al. 2008. RNA-mediated epigenetic programming of a genome-rearrangement pathway. Nature 451(7175):153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliverio AM, Katz LA.. 2014. The dynamic nature of genomes across the tree of life. Genome Biol Evol. 6(3):482–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orias E. 1991. Evolution of amitosis of the ciliate macronucleus: gain of the capacity to divide. J Protozool. 38(3):217–221. [DOI] [PubMed] [Google Scholar]
- Ovchinnikova L, Cheissin E, Selivanova G.. 1965. Photometric study of the DNA content in the nuclei of Spirostomum ambiguum (Ciliata, Heterotricha) 3(7):69–78. [Google Scholar]
- Parfrey LW, Lahr DJG, Katz LA.. 2008. The dynamic nature of eukaryotic genomes. Mol Biol Evol. 25(4):787–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parfrey LW, Lahr DJG, Knoll AH, Katz LA.. 2011. Estimating the timing of early eukaryotic diversification with multigene molecular clocks. Proc Natl Acad Sci USA. 108(33):13624–13629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popenko VI, Potekhin AA, Karajan BP, Skarlato SO, Leonova OG.. 2015. The size of DNA molecules and chromatin organization in the macronucleus of the ciliate Didinium nasutum (Ciliophora). J Eukaryot Microbiol. 62(2):260–264. [DOI] [PubMed] [Google Scholar]
- Postberg J, Heyse K, Cremer M, Cremer T, Lipps HJ.. 2008. Spatial and temporal plasticity of chromatin during programmed DNA-reorganization in Stylonychia macronuclear development. Epigenet Chromatin 1(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott DM. 1994. The DNA of ciliated protozoa. Microbiol Rev. 58(2):233–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyne CK. 1978. Electron-microscopic studies on macronuclear development in ciliate Chilodonella uncinata. Cytobiologie 18(1):145–160. [PubMed] [Google Scholar]
- R Core Team 2017. R: a language and environment for statistical computing. Vienna (Austria: ): R Foundation for Statistical Computing. [Google Scholar]
- Raikov IB. 1982. The protozoan nucleus: morphology and evolution. Wien: Springer-Verlag. [Google Scholar]
- Raikov IB. 1985. Primitive never-dividing macronuclei of some lower ciliates. Int Rev Cytol. 95:267–325. [DOI] [PubMed] [Google Scholar]
- Raikov IB. 1994. The nuclear-apparatus of some primitive ciliates, the karyorelictids – structure and divisional reorganization. Boll Zool. 61(1):19–28. [Google Scholar]
- Raikov IB, Karadzhan BP.. 1985. Fine-structure and cyto-chemistry of the nuclei of the primitive ciliate Tracheloraphis crassus (Karyorelictida). Protoplasma 126(1–2):114–129. [Google Scholar]
- Ricard G, et al. 2008. Macronuclear genome structure of the ciliate Nyctotherus ovalis: single-gene chromosomes and tiny introns. BMC Genomics 9:587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley JL, Katz LA.. 2001. Widespread distribution of extensive chromosomal fragmentation in ciliates. Mol Biol Evol. 18(7):1372–1377. [DOI] [PubMed] [Google Scholar]
- Rogato A, et al. 2014. The diversity of small non-coding RNAs in the diatom Phaeodactylum tricornutum. BMC Genomics 15(1):698.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santangelo G, Barone E.. 1987. Experimental results on cell-volume, growth-rate, and macronuclear DNA variation in a ciliated protozoan. J Exp Zool. 243(3):401–407. [Google Scholar]
- Sinclair DA, Guarente L.. 1997. Extrachromosomal rDNA circles – a cause of aging in yeast. Cell 91(7):1033–1042. [DOI] [PubMed] [Google Scholar]
- Slabodnick MM, et al. 2017. The macronuclear genome of Stentor coeruleus reveals tiny introns in a giant cell. Curr Biol. 27(4):569–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear BB, Lauth MR.. 1976. Polytene chromosomes of Oxytricha – biochemical and morphological changes during macronuclear development in a ciliated protozoan. Chromosoma 54(1):1–13. [DOI] [PubMed] [Google Scholar]
- Swart EC, et al. 2013. The Oxytricha trifallax macronuclear genome: a complex eukaryotic genome with 16,000 tiny chromosomes. PLoS Biol. 11(1):e1001473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vďačný P. 2015. Estimation of divergence times in litostomatean ciliates (Ciliophora: intramacronucleata), with Bayesian relaxed clock and 18S rRNA gene. Eur J Protistol. 51(4):321–334. [DOI] [PubMed] [Google Scholar]
- Wahl MC, Will CL, Luhrmann R.. 2009. The spliceosome: design principles of a dynamic RNP machine. Cell 136(4):701–718. [DOI] [PubMed] [Google Scholar]
- Wancura MW, Yan Y, Katz LA, Maurer-Alcalá XX.. 2017. Nuclear features of the heterotrich ciliate Blepharisma americanum: genome amplification, life cycle, and nuclear inclusion. J Eukaryot Microbiol. 65(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Blackburn EH.. 1997. De novo telomere addition by Tetrahymena telomerase in vitro. EMBO J. 16(4):866–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichterman R. 1937. Division and conjugation in Nyctotherus cordiformis (Ehr.) Stein (Protozoa, Ciliata) with special reference to the nuclear phenomena. J Morphol. 60(2):563–611. [Google Scholar]
- Wong LC, Landweber LF.. 2006. Evolution of programmed DNA rearrangements in a scrambled gene. Mol Biol Evol. 23(4):756–763. [DOI] [PubMed] [Google Scholar]
- Woodard J, Gorovsky MA, Kaneshiro E.. 1972. Cytochemical studies on problem of macronuclear subnuclei in Tetrahymena. Genetics 70(2):251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, et al. 2012. Copy number variations of 11 macronuclear chromosomes and their gene expression in Oxytricha trifallax. Gene 505(1):75–80. [DOI] [PubMed] [Google Scholar]
- Yan Y, Rogers AJ, Gao F, Katz LA.. 2017. Unusual features of non-dividing somatic macronuclei in the ciliate class Karyorelictea. Eur J Protistol. 61:399.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zufall RA, Robinson T, Katz LA.. 2005. Evolution of developmentally regulated genome rearrangements in eukaryotes. J Exp Zool. B Mol Dev Evol. 304B(5):448–455. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.