
Fig. 1.
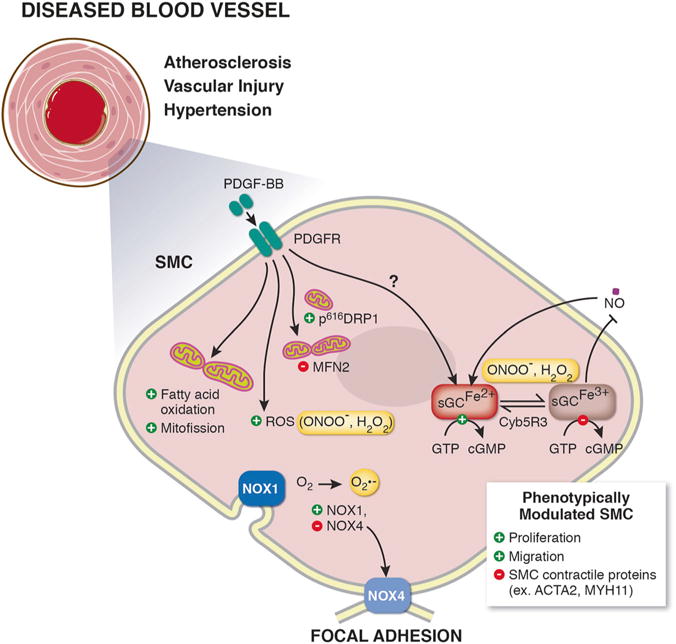
Schematic illustration showing mechanisms of smooth muscle cell (SMC) phenotypic switching. In response to vascular disease, SMC phenotypically switch from contractile cells to proliferative and migratory SMC characterized by downregulation of SMC contractile proteins (ex. smooth muscle α actin (ACTA2), smooth muscle myosin heavy chain 11 (MYH11)). SMC mitogen platelet derived growth factor beta (PDGF-BB) is known to induce SMC phenotypic switching. PDGF-BB can change cell bioenergetics, including shifting SMC metabolism to fatty-acid oxidation, increasing reactive oxygen species (ROS) production, and inducing mitochondrial fission, via increasing phosphorylation of dynamin-related protein 1 (DRP1) and decreasing mitofusin 2 (MFN2) levels. Vascular nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOX) NOX1 and NOX4 generate superoxide (O2−) and are known to drive SMC switching. The soluble guanylate cyclase (sGC)-cyclic guanosine monophosphate (cGMP)-protein kinase G (PKG) pathway signaling maintains SMC quiescence and mediates physiological vessel dilation. Excessive ROS generation results in oxidation of the sGC heme iron (Fe2+ to Fe3+) inhibiting binding of nitric oxide (NO) to sGC. Restoration of the sGC heme iron back to its NO-sensitive Fe2+ state is done endogenously by NADH cytochrome B5 reductase 3 (Cyb5R3)