

ABSTRACT
The bioactive sphingolipid metabolite sphingosine-1-phosphate (S1P) and the enzyme that produces it, SPHK1 (sphingosine kinase 1), regulate many processes important for the etiology of cancer. It has been suggested that SPHK1 levels are regulated by the tumor suppressor protein TP53, a key regulator of cell cycle arrest, apoptosis, and macroautophagy/autophagy. However, little is still known of the relationship between TP53 and SPHK1 activity in the regulation of these processes. To explore this link, we examined the effects of inhibiting SPHK1 in wild-type and TP53 null cancer cell lines. SK1-I, an analog of sphingosine and isozyme-specific SPHK1 inhibitor, suppressed cancer cell growth and clonogenic survival in a TP53-dependent manner. It also more strongly enhanced intrinsic apoptosis in wild-type TP53 cells than in isogenic TP53 null cells. Intriguingly, SK1-I induced phosphorylation of TP53 on Ser15, which increases its transcriptional activity. Consequently, levels of TP53 downstream targets such as pro-apoptotic members of the BCL2 family, including BAX, BAK1, and BID were increased in wild-type but not in TP53 null cells. Inhibition of SPHK1 also increased the formation of autophagic and multivesicular bodies, and increased processing of LC3 and its localization within acidic compartments in a TP53-dependent manner. SK1-I also induced massive accumulation of vacuoles, enhanced autophagy, and increased cell death in an SPHK1-dependent manner that also required TP53 expression. Importantly, downregulation of the key regulators of autophagic flux, BECN1 and ATG5, dramatically decreased the cytotoxicity of SK1-I only in cells with TP53 expression. Hence, our results reveal that TP53 plays an important role in vacuole-associated cell death induced by SPHK1 inhibition in cancer cells.
KEYWORDS: Apoptosis, autophagy, autophagic cell death, p53, TP53, S1P, SPHK1, SK1-I
Abbreviations
- ER
endoplasmic reticulum
- GFP
green fluorescent protein
- LC3
microtubule associated protein 1 light chain 3
- PBS
phosphate-buffered saline
- RFP
red fluorescent protein
- SPHK1
sphingosine kinase 1
- S1P
sphingosine-1-phosphate
- AMPK
AMP-activated protein kinase
Introduction
Sphingosine-1-phosphate (S1P) is a bioactive sphingolipid metabolite produced from sphingosine by 2 sphingosine kinase isoforms, SPHK1 and SPHK2, that regulate many processes important for the etiology of cancer, such as cell growth, resistance to apoptosis, angiogenesis, invasion, and inflammation, to name a few.1-3 SPHK1 levels are elevated in many types of cancers including lung,4,5 kidney,6 colon,7,8 breast,9,10 prostate,11 stomach,12 non-Hodgkin lymphoma,13 chronic myeloid leukemia,14 astrocytoma,15 glioblastoma,16 and hepatocellular carcinoma,17 and are associated with higher tumor grade and worse prognosis.18 Therefore, targeting SPHK1 to reduce S1P levels has been suggested to be an attractive chemotherapeutic approach, and several SPHK1 inhibitors show significant potential in preclinical cancer models.18-20
The tumor suppressor TP53, often referred to as the “guardian of the genome,” is a critical transcription factor that can induce cell cycle arrest, promote cell death, regulate autophagy,21 and contribute to the ability of cells to adapt to and survive metabolic stresses.22 The importance of TP53 to tumorigenesis is clear as mutations of this gene occur in well over 50% of all human cancers.23 Previous studies suggest an association between SPHK1 and TP53, as genotoxic stresses that activate TP53 reduce SPHK1 protein levels in cultured cells.24 Accumulation of TP53 in response to genotoxic insults leads to activation of the lysosomal protease CTSB (cathepsin B), which in turn degrades SPHK1.24 An important role for SPHK1 in TP53 actions has been suggested based on the observation that deletion of Sphk1 in trp53 null mice reduces thymic lymphomas and prolongs life span.25 TP53 tumor suppression by loss of SPHK1 is due to increased levels of sphingosine that are accompanied by increased expression of cell cycle inhibitors and tumor cell senescence.25 However, whether SPHK1 activity affects TP53 or its targets has not been investigated.
TP53 has a complex role in regulation of autophagy.21,26 On the one hand, TP53 stabilization and activation stimulate autophagy through transcription-independent mechanisms involving AMPK activation, MTOR inhibition, or transcription-dependent mechanisms by upregulation of PTEN27,28 or DRAM.29 On the other hand, cytosolic TP53 inhibits autophagy.26 Yet, much less is known of the functions of SPHK1 and S1P in autophagy. Previous studies suggested that SPHK1 and S1P induce autophagy to protect cells from apoptosis during nutrient starvation.30 Similarly, in primary neurons cytosolic S1P was reported to regulate neuronal autophagy. SPHK1-S1P enhances flux through autophagy and, conversely, the S1P-metabolizing enzyme SGPL1 (sphingosine-1-phosphate lyase 1), decreases this flux.31 Depletion of SGPP1 (sphingosine-1-phosphate phosphatase 1), which specifically dephosphorylates S1P and increases intracellular pools of S1P, also induces autophagy and ER stress.32 However, more recently it was shown that knocking out SPHKs in macrophages increases sphingosine and results in the accumulation of autophagosomes,33 yet inhibition of SPHK1 dynamically upregulates autophagic flux in primary mouse embryonic fibroblasts.34
Autophagy plays an important physiological role in maintaining cell homeostasis in the face of various types of stress. However, high levels of autophagy can lead to a special type of cells death known as autophagic cell death.35-38 Little is still known about the relationship between TP53 and SPHK1 activity in the regulation of autophagy, and particularly of the mechanisms that govern the switch between survival and autophagic cell death. Therefore, in this study we used a SPHK1 isotype-specific inhibitor to explore this link. Our results revealed that inhibiting SPHK1 in human colon cancer cells leads to activation of TP53 and subsequent regulation of pro-apoptotic mediators of the BCL2 family. In addition, we observed that inhibition of SPHK1 greatly increased autophagic flux and cell death that was dependent on the autophagic regulators BECN1 and ATG5 in a TP53-dependent manner. Therefore, our results suggest that TP53 is an important determinant of autophagic cell death induced by SPHK1 inhibition in cancer cells.
Results
The SPHK1 inhibitor SK1-I suppresses cancer cell growth and survival in a TP53-dependent manner
TP53 is a key tumor suppressor that is mutated in a majority of human cancers.23 Previous studies suggest a link between TP53 and the bioactive sphingolipid metabolites ceramide and S1P.39-41 Because TP53 is required for degradation of SPHK1 in response to chemotherapy,24 and deletion of Sphk1 attenuates cancer in trp53 null mice,25 it was of interest to examine the involvement of TP53 in cancer cell killing induced by SK1-I, a SPHK1-specific inhibitor.42 In agreement with numerous studies showing that inhibition of SPHK1 reduces growth and survival,18 SK1-I treatment resulted in death of HCT116 colon carcinoma cells as demonstrated by live-dead staining with calcein-AM to visualize live cells, and with red-fluorescent ethidium homodimer-1 for dead cells (Figure 1A-C). SK1-I also greatly inhibited growth of HCT116 cells in time- (Figure 1D) and dose-dependent manners (Figure 1E). Surprisingly, however, isogenic HCT116 cells in which TP53 was inactivated by targeted homologous recombination were significantly more resistant to SK1-I than wild-type TP53 HCT116 cells (Figure 1A-E). Next, we used a clonogenic assay to determine cell reproductive death induced by SK1-I. Wild-type TP53 HCT116 cells were more effectively killed by low concentrations of SK1-I than their TP53-deleted counterparts (Figure 1F). However, HCT116 cells deleted for the TP53-regulated protein P21 were as sensitive to SK1-I as wild-type HCT116 cells (Figure 1F). Likewise, colon cancer cells with known TP53 mutations, such as DLD1 and HT29,43 had increased survival over a broad range of SK1-I concentrations (Figure 1F). Similar results were observed with breast cancer cell lines: MCF7 cells that express wild-type TP53 were also more sensitive to SK1-I killing than BT474 breast ductal carcinoma cells with mutant TP53 (Figure 1F).
Figure 1.
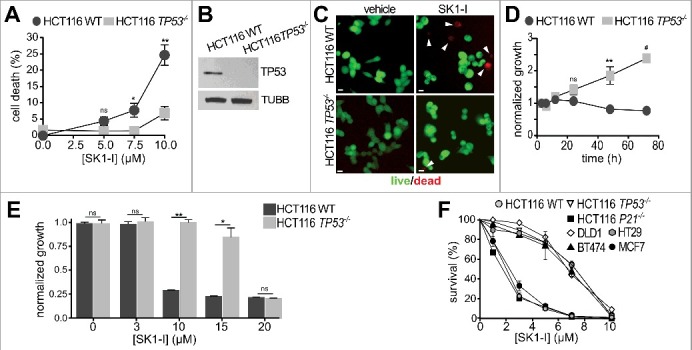
SK1-I decreases cancer cell growth and survival in a TP53-dependent manner. (A-C) Wild-type and TP53−/− HCT116 cells were treated with vehicle or with the indicated SK1-I concentrations for 24 h and the percentage of dead cells determined by live-dead assays. n = 4. (B) Immunoblots with the indicated antibodies for cells in panel (A). (C) Representative images of cells treated with 10 µM SK1-I for 24 h: live, green; dead, red. White arrowheads point to dead cells. (D-F) Wild-type and TP53−/− HCT116 cells were treated without or with 10 µM SK1-I for the indicated times (D) or with the indicated concentrations of SK1-I for 72 h (E) and cell proliferation determined. n = 3. (F) Wild-type, TP53−/− and P21−/− HCT116, DLD (mutant TP53), HT29 (mutant TP53), MCF7 (wild-type TP53), and BT474 (mutant TP53) cells were plated as single cell suspensions in 60-mm dishes and cultured 10 d for clonogenic assays. Colonies were fixed, stained, and colonies of >50 cells were counted. Survival data are expressed as percentage of colonies formed for each cell type treated with vehicle. n = 3. In (A,D,E,F) data are mean ± SEM; ns, not significant; *p ≤ 0.05; **p ≤ 0.005; #p≤ 0.0005. Scale bars: 10 µm.
Inhibition of SPHK1 with SK1-I activates TP53 and intrinsic apoptotic pathways
In agreement with previous studies,42 SK1-I induced the cleavage of CASP3 (Figure 2A), a hallmark of apoptosis.44 However, cells lacking TP53 had significantly less CASP3 cleavage compared to their wild-type TP53 isogenic counterparts following SK1-I treatments (Figure 2A,B). SK1-I also induced cleavage of the initiator CASP9 to its catalytically active form in both colon (Figure 2C) and H460 lung cancer cells (Figure 2D) expressing wild-type TP53. There was significantly less CASP9 cleavage in HCT116 TP53−/− cells (Figure 2C) and H1299 lung cancer cells that lack TP53 expression45 (Figure 2D,E). H1299 TP53-null cells were also significantly less sensitive to SK1-I in clonogenic assays (Figure 2F).
Figure 2.
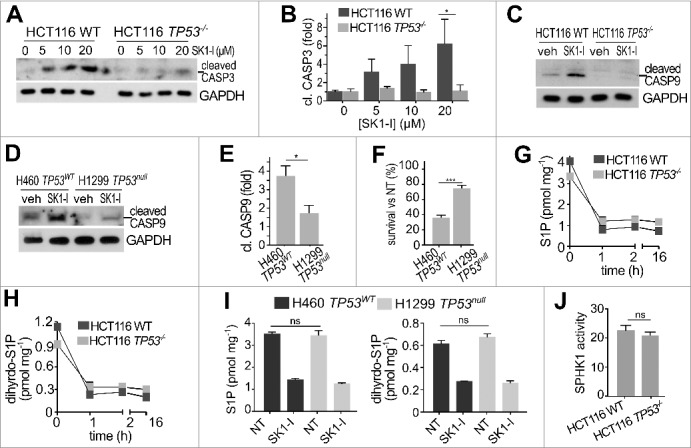
Inhibition of SPHK1 with SK1-I induces cleavage of CASP3 and CASP9 in a TP53-dependent manner. (A-C) Wild-type and TP53−/− HCT116 cells were treated with vehicle or with increasing concentrations of SK1-I for 12 h (A,C) and analyzed by immunoblotting with the indicated antibodies, and cleaved CASP3 quantified by densitometry (B). n = 3. (D,E) H460 lung cancer cells with wild-type TP53, and H1299 TP53 null lung cancer cells were treated with vehicle or with SK1-I for 24 h, analyzed by immunoblotting with the indicated antibodies (D), and cleaved CASP9 quantified (E). n = 3. (F) Survival of H460 and H1299 cells was evaluated in clonogenic assays with 10 µM SK1-I. Cells were plated as single cell suspensions in 6-well dishes and cultured 10 d. Colonies were fixed, stained, and colonies of >50 cells were counted. Survival data are expressed as percentage of colonies formed for each cell type treated with vehicle (NT, not treated). n = 3. (G-I) LC-ESI-MS/MS analysis of S1P and dihydro-S1P in the indicated cell lines treated with 10 µM SK1-I for the indicated times (G,H) or for 2 h (I). Units are lipid per mg protein. (J) SPHK1 activity in wild-type and TP53−/− HCT116 cell extracts was measured with NBD-sphingosine as substrate.60 Data are mean ± SEM. n = 3. *p ≤ 0.05; ns, not significant.
Because targeting the SPHK1-S1P axis reduces cancer cell survival in vitro and in pre-clinical cancer models,18 we examined whether different reductions in S1P levels might account for the observed resistance to SK1-I-induced cell death in cells lacking TP53. However, mass spectrometry analysis revealed no differences in basal levels of S1P and dihydro-S1P between wild-type and TP53 null cells (Figure 2G-I), and SK1-I reduced their levels to a similar extent (Figure 2G-I). In vitro activity assays also revealed no differences in basal SPHK1 activity between cells with or without TP53 (Figure 2J). Therefore, we focused on investigating the effects of SK1-I on TP53-dependent pathways of programmed cell death. Although activation of TP53 by genotoxic stress induces proteolysis of SPHK1,24,25 and some SPHK1 inhibitors also induce SPHK1 degradation,46 no reduction in SPHK1 levels after SK1-I treatment was detected (Figure 3A). However, SK1-I treatment induced rapid phosphorylation of TP53 on Ser15 (Figure 3A), which stabilizes and promotes its activity.47 Phosphorylation of TP53 on Ser15 was increased nearly 6-fold by SK1-I and remained elevated for at least 6 h (Figure 3B). SK1-I did not affect the total levels of TP53 (Figure 3A). Consistent with the notion that activation of AMPK in response to energetic stress induces phosphorylation of TP53 at Ser15,48 SK1-I also enhanced phosphorylation of AMPK (Figure 3C), as did sphingosine, and sphingosine in combination with PF543, a potent SPHK1 inhibitor (Figure 3C). Moreover, similar to other water-soluble synthetic sphingolipids,49,50 SK1-I triggered internalization of the glucose transporter SLC2A1/GLUT1 (Figure 3E).
Figure 3.
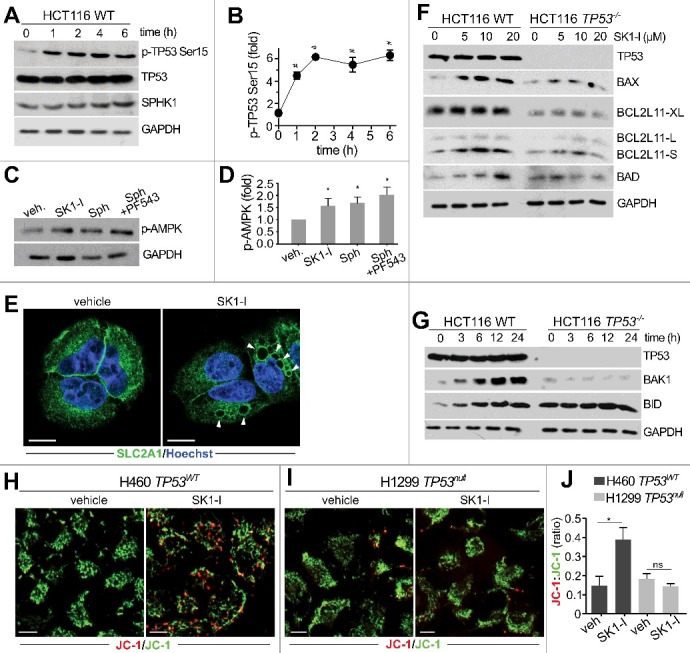
SK1-I increases TP53 phosphorylation and activation of BCL2-related pro-apoptotic proteins. Wild-type TP53 HCT116 cells (A,C,F,G) and TP53−/− HCT116 cells (F,G) were treated with 10 µM SK1-I for the indicated times (A,G), or with the indicated concentrations of SK1-I for 12 h (F), or for 5 h with 10 µM SK1-I (C), 10 µM sphingosine (C), or pre-treated with 1 µM PF543 for 30 min followed by10 µM SK1-I (C). Cell lysates were analyzed by immunoblotting with the indicated antibodies. (B,D) Densitometric quantification of phospho-TP53 (Ser15) shown in panel A (n = 4), or phospho-AMPK in panel C (n = 3). (E) Confocal images of H460 cells treated with vehicle or with 10 µM SK1-I for 4 h and immunostained with anti-SLC2A1/GLUT1. Nuclei were labeled with Hoechst. (H-I) Confocal live cell images (H,I) and accompanying red:green fluorescence quantification (J) of TP53 wild-type H460 (H) and TP53 null H1299 (I) cells labeled with JC-1 dye following treatment for 8 h without or with 7.5 µM SK1-I. n = 6. Data are mean ± SEM. ns, not significant. *p ≤ 0.05; #p ≤ 0.0005.
In addition to increased phosphorylation of TP53 on Ser15, which regulates its transcriptional activity,47 SK1-I treatments also increased the levels of TP53 apoptotic target genes, including BAX (BCL2 associated X, apoptosis regulator),51 BAK1 (BCL2 antagonist/killer 1),52 and BID (BH3 interacting domain death agonist),53 in time- and concentration-dependent manners (Figure 3F,G). In contrast, expression of these pro-apoptotic proteins was not increased by SK1-I in TP53 null cells (Figure 3F,G). Treatment with SK1-I also increased the alternatively spliced highly apoptotic short form of BCL2L11/BIM (BCL2 like 11) in colon cancer cells expressing wild-type TP53, but not in TP53−/− cells (Figure 3F), in agreement with studies suggesting that BCL2L11/BIM can be indirectly upregulated by TP53.54 SK1-I treatment did not increase levels of the extra-long form of BCL2L11/BIM (Figure 3F).
Activation of pro-apoptotic members of the BCL2 family and BAX-BAK1 oligomerization in the mitochondrial outer membrane results in loss of mitochondrial transmembrane potential and cell death.55 The cationic fluorochrome tracer JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide) incorporates into the mitochondrial membrane and emits green light (indicates normal potential) in the monomeric state or red (indicates loss of membrane potential) in the aggregated state, and can reliably provide a measure of the inner mitochondrial transmembrane potential.55 In cells with wild-type TP53, SK1-I treatment resulted in a significant loss of mitochondrial membrane potential as shown by a red shift in JC-1 dye staining (Figure 3H-J). However, the JC-1 red:green ratio in cells lacking TP53 expression was unchanged after SK1-I treatment (Figure 3 H-J). Thus, inhibition of SPHK1 activates TP53 and its targets, leading to loss of mitochondrial membrane potential, and suggests that it can induce intrinsic apoptotic cell death.
Involvement of TP53 in autophagy induced by SK1-I
TP53 also plays a critical, albeit complex role in the regulation of autophagy, and can act as a positive or negative regulator, depending on its cellular location26 and the nature of the stimulus.56 Because SPHK1 has also been implicated in autophagy,31,33,34,57 it was of interest to investigate the effects of inhibiting SPHK1 on autophagic processes in cells with or without wild-type TP53. In cells with TP53, and in agreement with previous results,34 SK1-I increased processing of the cytosolic soluble MAP1LC3/LC3 (microtubule associated protein 1 light chain 3) form (LC3-I) to its lipidated membrane-bound form (LC3-II)58 (Figure 4A). In addition, SK1-I treated cells developed dilated vesicles that were positively stained with the cationic amphiphilic tracer Cyto-ID (Figure 4B) that selectively labels autophagic bodies.59 Redistribution of GFP-LC3 fusion protein from a diffuse cytoplasmic pattern to a punctate pattern, indicative of the formation of autophagic vesicles, was also observed (Figure 4C,D). Electron micrograph images of cells 4 h after SK1-I treatment confirmed formation of dilated amphisomes, and 24-h post-treated cells had membrane-bound electron dense autophagic and multivesicular body structures (Figure 4E, zoom box). Intriguingly, based on the redistribution of GFP-LC3, almost 50% of the TP53 wild-type HCT116 cells treated with 10 µM SK1-I for 6 h showed an accumulation of autophagosomes compared with <15% of the untreated controls (Figure 4D), whereas in TP53 null HCT116 cells SK1-I-induced autophagy was greatly reduced, with only 25% of the cells showing increased autophagosomes (Figure 4D). Similar differences in GFP-LC3 redistribution between HCT116 with wild-type TP53 and HCT116 TP53 null cells were observed at lower SK1-I concentrations (Figure 4D).
Figure 4.
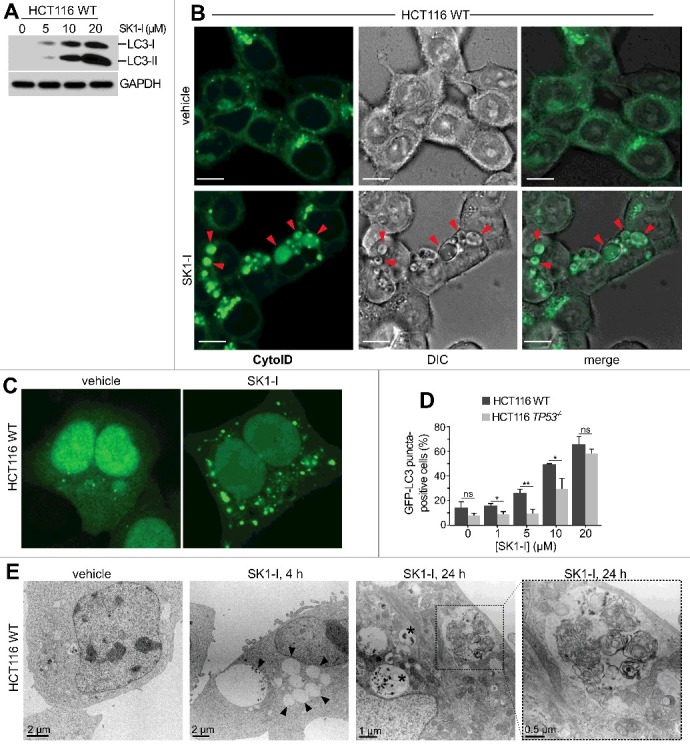
SK1-I induces autophagy in HCT116 cells. (A) Wild-type HCT116 cells were treated for 12 h with the indicated SK1-I concentrations and extracts immunoblotted for LC3 and GAPDH. (B) Live cell confocal images of HCT116 cells with wild-type TP53 treated with vehicle or 10 µM SK1-I for 7 h and labeled with Cyto-ID. Red arrowheads indicate autophagic vacuoles. (C, D) Wild-type HCT116 (C, D) or TP53−/− HCT116 cells (D) transfected with GFP-LC3 were treated with vehicle or 10 µM SK1-I for 3 h (C), or with the indicated concentrations of SK1-I for 3 h (D). Cells were examined by confocal fluorescence microscopy, and representative images are shown (C). Percentage of cells showing 5 or more intense GFP-LC3 fluorescent puncta was quantified (D). At least 100 cells were analyzed for each. Data are means ± S.D. n = 3. ns, not significant. *p ≤ 0.05; **p ≤ 0.005. (E) Transmission electron micrographs of HCT116 cells treated with 10 µM SK1-I for the indicated times. Representative micrographs are shown. Black arrowheads point to dilated vesicles; asterisks (*) indicate multivesicular bodies; and zoom box demarcates a morphologically multilamellar vesicle. Scale bars: (B) 10 μm; (E) as indicated.
SK1-I-induced autophagic flux and accumulation of enlarged vacuoles requires TP53
We further examined differences in autophagic activity using immunoblotting to determine processing of LC3-I to LC3-II in cells with and without wild-type TP53. Consistent with the massive formation of autophagic vesicles observed with SK1-I, LC3-II was increased in these cells in concentration- (Figure 5A) and time-dependent manners (Figure 5B). However, in TP53 null HCT116 cells, LC3-II levels were significantly lower compared to their isogenic counterparts following SK1-I treatment (Figure 5A-C). In HCT116 cells, SK1-I increased LC3-II in the presence of the lysosomal protease inhibitors E64d, leupeptin, and pepstatin A (E/L/P) at 5 h and more significantly at 20 h (Figure 5D,E), compared to E/L/P treatment alone (Figure 5D,E), suggesting that the enhanced LC3 lipidation was due to induced autophagosome biogenesis and increased autophagic flux at later time points. However, this enhancement of LC3-II accumulation in the presence of lysosomal inhibitors was greatly reduced in TP53 null HCT116 cells (Figure 5D,E). As an additional approach to monitor autophagic flux, cells were transfected with tandem-fluorescent LC3 (RFP-GFP-LC3) and the RFP:GFP fluorescence ratio was used to monitor its association with autophagosomes (less acidic) or amphisomes and autolysosomes (more acidic), which attenuates GFP but not RFP fluorescence. In cells with wild-type TP53, intermediate (7 h) and long exposures (18 h) to SK1-I led to a red-shift (Figure 5F,I), and significant increases in RFP:GFP ratios (Figure 5H,K). However, consistent with reduced levels of LC3-II over a similar period, cells lacking TP53 had much less intensely red vesicles (Figure 5G,J), and had significantly lower RFP:GFP ratios (Figure 5H,K). These results suggest that SK1-I-induced accumulation of RFP-GFP-LC3 within the acidic environment of autolysosomes is lower in cells lacking TP53 expression.
Figure 5.
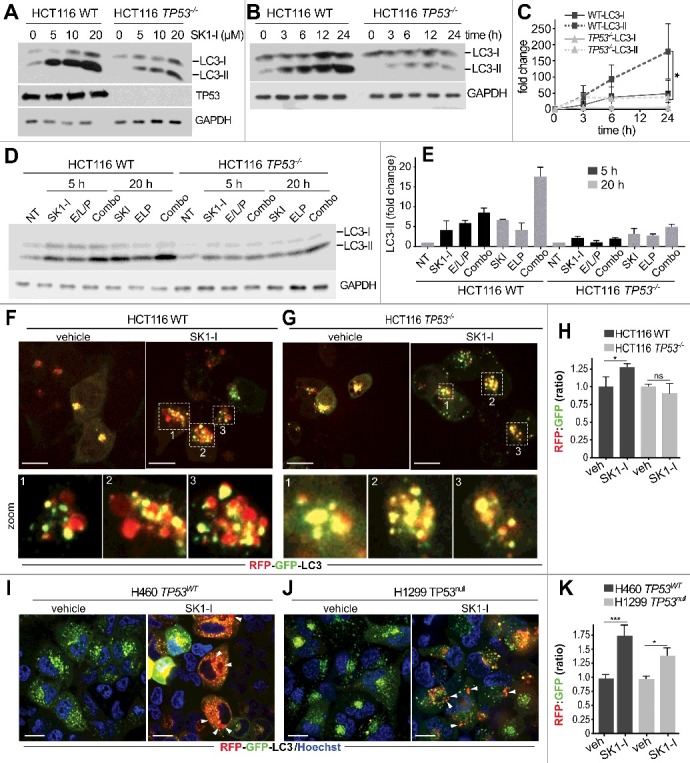
SK1-I increases LC3 processing in a TP53-dependent manner. (A-E) Wild-type HCT116 cells and TP53−/− HCT116 cells were treated with the indicated SK1-I concentrations for 12 h (A), with 10 µM SK1-I for the indicated times (B), or with the indicated treatments and times (D); E/L/P was 10 µM E64d, 1 µg/ml leupeptin, 10 µg/ml pepstatin A, and 10 µM SK1-I. For combo, cells were pretreated with E/L/P for 30 min followed by SK1-I. Cell lysates were immunoblotted with the indicated antibodies. (C,E) Densitometric analysis of LC3-I and LC3-II shown in panels (B) and (D), respectively. n = 3. Note: no time-dependent changes in LC3-II were observed after treatment with vehicle in (D). (F-K) Confocal images of wild-type HCT116 (F), TP53−/- HCT116 (G), H460 cells with wild-type TP53 (I), or H1299 TP53 null cells (J) expressing RFP-GFP-LC3 treated with a vehicle or 7.5 µM SK1-I for 7 h (F,G) or 18 h (I, J), and RFP:GFP fluorescence ratio determined (H, K). n = 4. Data are means ± S.E.M. ns, not significant. *p ≤ 0.05; ***P≤0.001. Scale bars: 10 µm. In panels (D, E), zoom boxes outline LC3 puncta that were distinctly red, yellow, and/or green.
We have previously shown that the SPHK1-selective inhibitor and sphingosine mimetic SK1-I induces the formation and accumulation of intracellular dilated vacuoles that require the sphingosine backbone and that further vacuole enlargement depends on the inhibition of SPHK1 and accumulation of sphingosine in these vesicles.34,60 In agreement, SK1-I induced the formation of dilated vacuoles in HCT116 and H460 cells expressing wild-type TP53. However, significantly fewer vacuoles were present in HCT116 TP53−/− cells or in H1299 cells lacking TP53 (Figure 6A,B). We also generated CRISPR-Cas9-induced TP53 gene deletion in H460 cells (H460crTP53; Figure 6C), in which SK1-I induced fewer vacuoles (Figure 6D,E). Re-expression of wild-type TP53 in H460crTP53 restored vacuole formation (Figure 6D,E). Furthermore, transfection of wild-type TP53 into HCT116 TP53−/− cells also significantly enhanced LC3 lipidation induced by SK1-I, whereas transfection with S15A mutant TP53 did not (Figure 6F,G).
Figure 6.
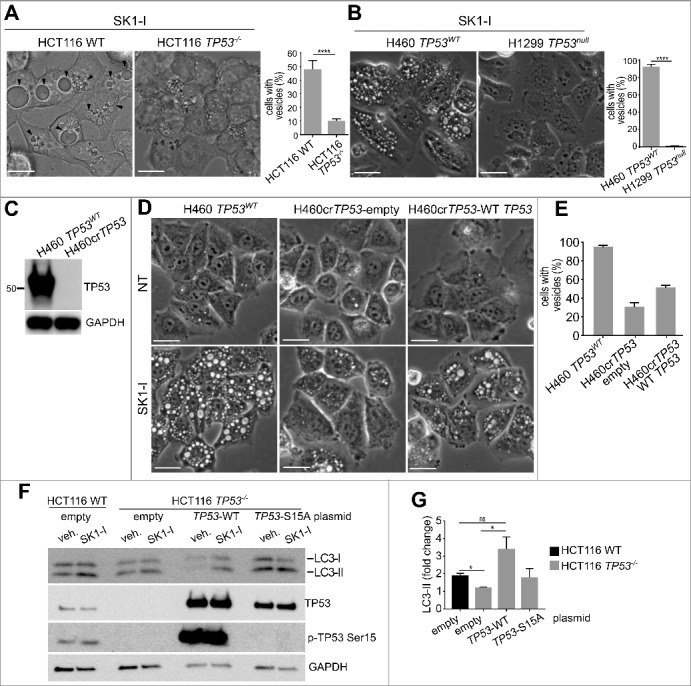
TP53 is required for SK1-I-induced accumulation of enlarged vacuoles and autophagy. (A, B) Phase contrast images and quantification of TP53WT and TP53−/− HCT116 cells (A), or H460 cells with wild-type TP53 and H1299 TP53-null cells (B), treated with 10 µM SK1-I for 1 h. (C) Immunoblot of H460 and CRISPR-Cas9-deleted TP53 H460 cells (H460crTP53). (D) Phase contrast images of H460, H460crTP53 cells transfected with an empty plasmid, or H460crTP53 cells transected with a plasmid expressing wild-type TP53, treated with vehicle or 10 µM SK1-I for 2 h. (E) Quantification of vesicle-positive cells in panel (D). (F,G) HCT116 wild-type cells transfected with empty vector, and HCT116 TP53−/− cells transfected with empty vector, or plasmids encoding wild-type TP53, or the TP53S15A mutant treated with 10 µM SK1-I for 12 h. Cell lysates were immunoblotted with anti-LC3 or the indicated antibodies. (K) Densitometric analysis of LC3-I and LC3-II shown in panel (J) n = 3. Scale bars (A, B, D): 10 µm.
SK1-I enhanced autophagic flux and increased vacuolization-associated cell death in an SPHK1-dependent manner
Consistent with our observations that sphingosine-like inhibitors generate small vacuoles in sphk1−/− MEFs that do not fuse upon prolonged treatment,34,60 SK1-I induced fewer and much smaller vacuoles in H460 cells in which SPHK1 was deleted using CRISPR-Cas9 (H460crSPHK1) as compared to the parental cell line (Figure 7A-D), suggesting that vacuole enlargement is SPHK1 dependent. These SPHK1 null cells were also resistant to SK1-I killing, as demonstrated by clonogenic assays (36.4% vs 99.2% cell survival of H460 cells versus H460crSPHK1 cells following treatment with 10 µM SK1-I). Moreover, as revealed by examining LC3 lipidation by immunoblotting, SK1-I-induced autophagosome formation and enhanced autophagic flux at 20 h were significantly decreased in Sphk1−/− cells, and LC3-II did not further accumulate in the presence of lysosomal inhibitors in contrast to their effects in Sphk1+/+ cells (Figure 7E, F). These results further confirm the specificity of SK1-I as a SPHK1 inhibitor42 and suggest that accumulation of vacuoles is a determinant in SK1-I-induced cell death.
Figure 7.
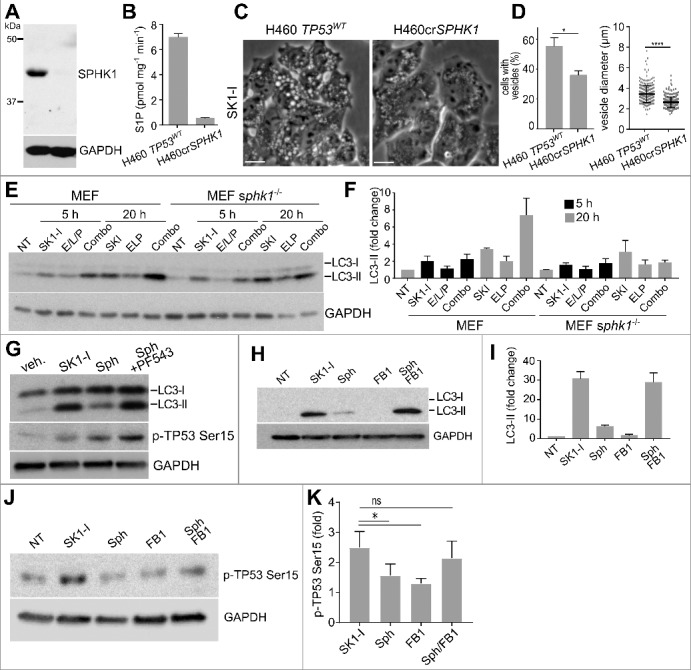
SK1-I induces accumulation of vacuoles, autophagy, and cell death in an SPHK1-dependent manner. (A, B) Immunoblots with the indicated antibodies (A) and SPHK1 isotype-specific activity assay (B) of H460 and CRISPR-Cas9-deleted SPHK1 H460 (H460crSPHK1) cells. (C, D) Phase contrast images (C) of H460 and H460crSPHK1 treated with 10 µM SK1-I for 1 h, and quantification of vacuole-positive cells and vacuole size (D) in panel C. n = 6. (E) Wild-type and sphk1−/− MEFs treated with vehicle, 10 µM SK1-I alone, or E64d (10 µM), leupeptin (1 µg/ml), and pepstatin A (10 µg/ml) (E/L/P), or a combination of SK1-I and E/L/P for the indicated times. Cell lysates were immunoblotted with the indicated antibodies. n = 3. (F) Densitometric analysis of LC3-II shown in panel (D). Note: no time-dependent changes in LC3-II were observed after treatment with vehicle. (G) HCT116 cells were treated with SK1-I (10 μM), or sphingosine (10 μM) in the absence or presence of PF543 (1 μM) for 5 h. Cell lysates were immunoblotted with the indicated antibodies. (H,I) H460 cells were treated with SK1-I (10 μM), or sphingosine (10 μM) in the absence or presence of fumonisin B1 (FB1, 50 μM) for 12 h. Cell lysates were immunoblotted with the indicated antibodies and blots quantified by densitometric analysis (I). n = 3. (J, K) HCT116 cells were treated with SK1-I (10 μM), or sphingosine (10 μM) in the absence or presence of FB1 (1 μM). NT, not treated. Cell lysates were analyzed by immunoblotting with anti-phospho-TP53 (Ser15) and blots quantified by densitometric analysis (K). n = 3. Data are means ± S.E.M. ns, not significant. *p ≤ 0.05.
Because vacuoles do not accumulate in SPHK1 null cells under basal conditions, it was suggested that SPHK2 compensates for the loss of SPHK1 in specific cell types to prevent the accumulation of sphingosine in vacuoles.34,60 Indeed, only sphk1 and sphk2 double-knockout mice and not single knockout mice are embryonic lethal with defective vascularization.61 We have previously shown that vacuoles are not observed upon SPHK1 inhibition by the more potent inhibitor PF543. However, inhibition of SPHK1 by PF543 significantly prolongs sphingosine-induced vacuole accumulation.34,60 Therefore, to more closely mimic the effects of SK1-I, we treated cells with PF543 together with sphingosine. Consistent with its effect on vacuoles, PF543 also increased LC3 lipidation induced by sphingosine as well as phosphorylation of TP53 on Ser15 (Figure 7G). Because both sphingosine and ceramide have been implicated in autophagy,34 it was also important to examine whether conversion of sphingosine to ceramide was involved. To this end, cells were treated with fumonisin B1 (FB1), which inhibits all ceramide synthases.62,63 Consistent with a previous report,34 FB1 significantly increased LC3-II in response to sphingosine (Figure 7H,I), as well as phosphorylation of TP53 (Figure 7J,K).
SK1-I mediates ATG5- and BECN1-dependent cell death in a TP53-dependent manner
As SK1-I triggers both autophagic and apoptotic pathways in a TP53-dependent manner, it was important to examine in more detail the predominant cytotoxic mechanism of SK1-I action. To this end, following SK1-I treatments, flow cytometry was used to analyze cell populations stained with ANXA5, a marker of early apoptotic cell death, and 7-AAD, a fluorescent DNA intercalator that is excluded from live cells. SK1-I caused an increase in the population of cells stained by ANXA5 and 7-AAD (Figure 8A-C). However, because necroptotic cells can also be stained with ANXA5 prior to the loss of plasma membrane integrity, suggesting that phospholipid scrambling occurs during several mechanisms of cell death,64 it was important to determine whether apoptosis was the primary mechanism of cell death in SK1-I-treated cells. To this end, cells not stained with 7-AAD but positively stained with ANXA5 were also assessed for CASP3-CASP7 activation using CellEvent TM CASP3-CASP7 Green, a cell-permeable fluorogenic probe that is intrinsically nonfluorescent; CASP3-CASP7-dependent cleavage results in unquenching that makes the probe detectable by fluorescence. In contrast to the apoptosis-inducing agent TNFSF10/TRAIL in which nearly all of the ANXA5+ 7-AAD− cells contained activated CASP3-CASP7 (indicating early apoptosis), CASP3-CASP7 was only activated in a small population of the SK1-I-treated ANXA5+ 7-AAD− population (Figure 8C, D), indicating that apoptosis is not a major component of cell death induced by SK1-I.
Figure 8.
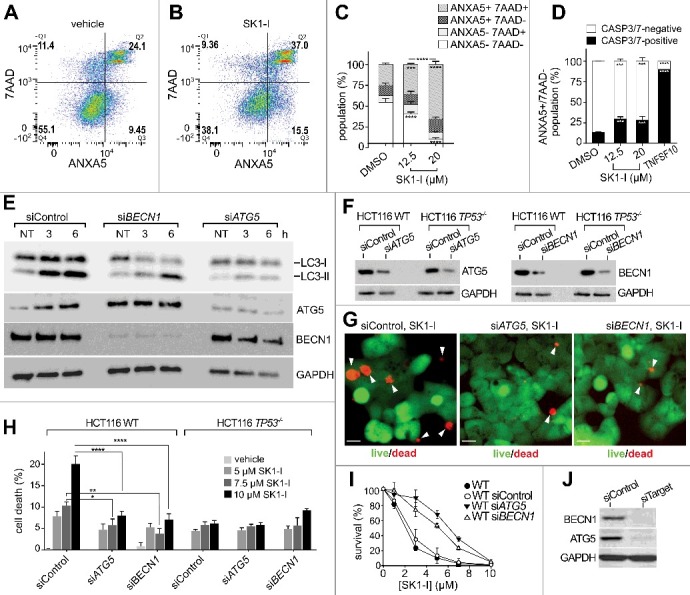
Depleting BECN1 or ATG5 reduces SK1-I cytotoxicity. (A, B) Representative flow cytometry analysis of wild-type HCT116 cells treated with the vehicle (A) or 12.5 µM SK1-I (B) for 24 h followed by ANXA5 and 7-AAD staining. (C) Flow cytometric analysis of cells treated with the indicated concentrations of SK1-I for 24 h and labeled with CellEvent TM CASP3/7 Green for the final 6 h of SK1-I treatment followed by ANXA5 and 7-AAD staining. (D) Percentage of ANXA5+ 7-AAD- cells containing activated CASP3 and CASP7 in panel (C). TNFSF10/TRAIL-treated (100 ng/mL for 6 h) HCT116 wild-type cells were included as a positive control. (E) Wild-type HCT116 cells transfected with scrambled siRNA (siControl), siRNA targeting ATG5 (siATG5), or siRNA targeting BECN1 (siBECN1) were treated with 10 µM SK1-I for 20 h and cell lysates immunoblotted with the indicated antibodies. (F-H) HCT116 wild-type (F-H) and TP53-/- null (F, H) cells transfected with the indicated siRNAs were treated with 10 µM SK1-I (F, G) or with the indicated SK1-I concentrations for 12 h (H). n = 3. (G,H) Representative live or dead images (G) and live or dead quantification (H). White arrowheads in (G) point to dead cells. (I) Clonogenic assay of wild-type HCT116 cells transfected with siControl, siATG5, or siBECN1 treated with vehicle or the indicated SK1-I concentrations, cultured for 10 d, and colonies of >50 cells counted. n = 3. (J) Cell lysates of duplicate cultures in panel (I) were immunoblotted with the indicated antibodies (F-I). n = 3, mean ± S.D. ns, not significant; *p≤0.05; **p≤0.005; ***P≤0.001;****P≤0.0005.
Autophagy is a critical process that recycles cellular components to provide energy and metabolites during nutrient stress conditions, and plays housekeeping roles that remove damaged organelles or misfolded proteins from the cell. However, excessive autophagy can also lead to a type of cell death distinct from apoptosis,35 and is defined as being dependent on autophagic regulator proteins and increased autophagic flux.21 BECN1 and ATG5 are autophagy-related proteins that play integral roles in the regulation of autophagy.65 Because SK1-I induced formation of autophagic vesicles and increased processing of LC3 and its localization within acidic compartments, suggesting increased autophagic flux, we next examined the role of autophagy in the cytotoxic effects of SK1-I. To this end, expression of ATG5 or BECN1 was downregulated (Figure 8E,F), which severely impaired SK1-I-induced LC3 lipidation levels (Figure 8E). Importantly, in cells transfected with a nontargeting control siRNA (siControl), SK1-I increased cell death (Figure 8G,H) in a concentration-dependent manner (Figure 8H), and reduced survival in clonogenic assays (Figure 8I). In stark contrast, cells transfected with siRNAs targeting BECN1 or ATG5 were significantly protected from the cytotoxic effects of SK1-I (Figure 8G,H), and had an increased number of surviving colonies (Figure 8I). In cells lacking TP53 expression, depletion of either BECN1 or ATG5 (Figure 8F) had no significant effects on SK1-I killing efficacy (Figure 8H). Therefore, these results indicate that SK1-I can trigger autophagic cell death in a TP53-dependent manner.
Discussion
The autophagic and apoptotic pathways intersect under stress conditions that significantly deprive cells of nutrients or upon exposure to chemotoxic agents. Autophagy is activated as a cellular survival and adaptation program. However, in severe cellular stress conditions, it can proceed as an alternative cell death pathway.21,66 The bioactive sphingolipid metabolites ceramide, sphingosine, and S1P have been implicated in regulation of both autophagy and apoptosis.18,67,68 Cells tightly regulate levels of these interconvertible sphingolipid metabolites as their opposing signaling pathways are determinants of cell fate, often referred to as the “sphingolipid rheostat”.69 SPHK1 is a key regulator of the sphingolipid rheostat and the balance between pro-death sphingosine and ceramide and prosurvival S1P. Indeed, and in agreement with previous studies,42 inhibition of SPHK1 with SK1-I induced apoptosis, autophagic flux, and accumulation of enlarged vacuoles, and decreased reproductive viability. Remarkably, however, these were dependent on the presence of TP53. Interestingly, we observed that inhibition of SPHK1 led to increased phosphorylation of TP53 on Ser15 and subsequent upregulation of pro-apoptotic BCL2 family members including BAD, BAK1, and BID, This is consistent with previous observations that overexpression of SGPL1, which reduces S1P levels, promotes apoptosis in response to DNA damage through a pathway involving TP53.41
Similar to other sphingosine analogs that reduce the cell surface availability of nutrient transporters,49,50 SK1-I also induced translocation of the glucose transporter SLC2A1/GLUT1 from the cell surface to dilated intracellular vacuoles. The vacuolization triggered by these compounds has been suggested to limit nutrient acquisition pathways and lead to nutrient stress, thus limiting tumor growth.49,50 Accompanying the accumulation of dilated intracellular vacuoles, SK1-I, or sphingosine alone and in combination with the SPHK1 inhibitor PF543, induced phosphorylation of AMPK. AMPK is one of the central regulators of cellular metabolism70 and autophagy,71 which under low glucose conditions induces phosphorylation of TP53 on Ser15.48 Hence, consistent with previous work,72 our data suggest that following SK1-I treatment, reduction of cell surface bioavailability of SLC2A1/GLUT1, and the resulting metabolic stress, can activate AMPK to phosphorylate TP53 leading to subsequent activation of the intrinsic mitochondrial apoptotic machinery. In the intrinsic apoptotic pathway, induction of BH3-only proteins, such as the membrane permeabilizing proteins BAX and BAK1, is usually accompanied by mitochondrial outer membrane permeabilization.21 SK1-I treatment resulted in the induction of these TP53-dependent targets and significant depolarization of the membrane potential only in cells expressing wild-type TP53. Consequently, the active form of the apoptotic cell death regulators CASP3 and CASP9 were markedly decreased in cells lacking TP53, consistent with a previous report demonstrating that the cytotoxic effects of SK1-I are mediated in part by a caspase-dependent mechanism.73
Although the role of SPHK1 and S1P in cell growth and suppression of apoptosis is well accepted, their role in the regulation of autophagy is still a matter of debate. Initially, it was shown that SPHK1 activity increases during nutrient starvation, and its downregulation suppresses autophagy and exacerbates cell death.30 Similarly, overexpressed SPHK1 increases autophagic flux in primary cortical neurons and expression of a dominant-negative form of SPHK1 inhibits autophagosome formation.31 Moreover, pharmacological stimulation of autophagy in primary neurons leads to formation of SPHK1-GFP-positive puncta that colocalize with autophagosomal markers, supporting the notion that SPHK1 plays a role in the biogenesis of autophagosomes.34,57 In contrast, however, in macrophages with deletion of both Sphk1 and Sphk2 there is an increase in sphingosine and enhanced autophagy.33 Furthermore, inhibition of SPHK1 or treatment with sphingosine leads to accumulation of enlarged, dysfunctional late endosomes and amphisomes in multiple cell lines.34,60 In agreement, we found that SK1-I increased autophagic flux and enhanced accumulation of dilated vacuoles and autophagosomes. Importantly, however, accumulation of dilated vacuoles and the accompanying increase in LC3 lipidation were attenuated in the absence of SPHK1 or TP53.
TP53 functions at the junction of autophagy and apoptosis and can activate or repress autophagy.74,75 In its cytosolic form, TP53 suppresses autophagy76 by interacting with RB1CC1/FIP200, the putative human ortholog of yeast Atg17, and therefore blocking autophagosome formation.77 However, upon phosphorylation, TP53 translocates to the nucleus, where it activates transcription of multiple pro-autophagic and pro-apoptotic genes.22 In addition to their involvement in apoptosis, the BH3-only proteins BAD and BID can also promote autophagy by disrupting the inhibitory interactions between BECN1, a central scaffold protein that assembles components important for autophagy, and anti-apoptotic BCL2 family proteins.75 Consistent with these regulatory mechanisms, in our study, treating cells with the SPHK1 inhibitor SK1-I led to the phosphorylation of TP53, induction of BID and BAD, and concomitant activation of the intrinsic mitochondrial apoptotic pathway. However, our results indicate that in SK1-I-treated cells caspase-dependent apoptosis is only a minor contributor. Furthermore, neither the pan-caspase inhibitor Q-VD-Oph, the RIPK1 inhibitor necrostatin-1, nor antagonists of Na+/K+-ATPase (cardiac glycosides) that inhibit autosis, rescue SK1-I-induced cell death (data not shown). In parallel, SK1-I also caused massive accumulation of autophagic bodies, dilated intracellular vacuoles, and a dramatic increase in autophagic flux in a TP53-dependent manner. Importantly, however, in TP53-positive cells, depleting BECN1 and ATG5 markedly suppressed SK1-I-induced lethality, whereas in TP53-null cells downregulation of these proteins had no effect. Taken together, our data indicate that cell death induced by SK1-I requires the functional basic machinery of autophagy and active autophagic flux, classified as autophagic cell death.78 Therefore, our work highlights a critical role for SPHK1 and its substrate sphingosine in the crosstalk between TP53 and autophagic cell death. Although the concept that cells can die by autophagy was proposed several decades ago,79 the detailed molecular mechanisms are still not clear. Recently, it has been suggested that this type of cell death results from excessive degradation of cytosolic components.36 While further work is needed to understand what converts autophagy from a protective to a lethal mechanism, activating such an alternative cell death pathway, with SK1-I for example, may be a promising avenue to overcome apoptosis resistance found in many cancers.
Materials and methods
Cell culture
HCT116 TP53+/+, HCT116 TP53−/− and HCT116 CDKN1A/p21−/− cells were generous gifts from B. Vogelstein, John Hopkins University. All other established cell lines, including DLD1 (CCL-221), HT29 (HTB-38), MCF7 (HTB-22), H460 (HTB-177) and BT474 (HTB-20) cells were from ATCC. All cells were cultured in Dulbecco's modified Eagle medium (DMEM) containing 4,500 mg/l D-glucose, 4 mM L-glutamine, and 110 mg/l sodium pyruvate (Life Technologies, 11995065), supplemented with 10% fetal bovine serum (FBS; Serum Source International, FB22-500). SK1-I was from Enzo Life Sciences (BML-EI411) and was dissolved in water. H1299 control and H1299 cells in which TP53 was inducible by doxycycline were obtained from S. Torti (University of Connecticut Health Sciences) and cultured in DMEM supplemented with 10% FBS and doxycycline (10 µg/ml; Fisher Scientific, AAJ67043AD) as previously described.80
Cell death and proliferation assays
Live-dead assays were performed in 96-well plates with a Hermes Wiscan instrument (IDEA Bio-medical, Israel) as previously described.81 Briefly, after treatment, floating cells were collected by centrifuging plates at 500 × g or 5 min. Cells were simultaneously stained with green-fluorescent calcein-AM as a measure of intracellular esterase activity of live cells and red-fluorescent ethidium homodimer-1 to determine dead cells by loss of plasma membrane integrity (LIVE/DEAD Viability/Cytotoxicity Kit; Life Technologies, L3224). Cells were visualized at 10X magnification and viable and dead cells in 5 fields per well were counted manually to determine percent dead cells as described previously.81
Cell-proliferation assays were conducted in 96-well plates with 5000 cells seeded per well in full serum medium. Cells were allowed to attach for 18 h and following SK1-I treatments, cell viability was determined with WST-8 [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt] using the Cell Counting Kit-8 (Dojindo Molecular Technologies, CK04).
2D-colony formation assay
Single cells were plated in 6-well dishes at a density of 1,500 cells per well; 24 h later, cells were treated with SK1-I as indicated in the figure legends. Cells were washed after 24 h and cultured in DMEM containing 10% FBS for 10 d in drug-free medium. Cells were then fixed with methanol, stained with crystal violet (5%, w:v; Fisher Scientific, C581-25), and colonies with >50 cells per colony counted as described previously.82
Assessment of autophagy
Cells were transfected with a plasmid encoding GFP-LC3 and treated with vehicle or SK1-I, washed with PBS (Life Technologies, 10010023) and fixed with 4% paraformaldehyde. Coverslips were examined with a Zeiss LSM 510 laser confocal microscope (Zeiss, Germany). Cells with 5 or more intense GFP-LC3 puncta were considered autophagic, whereas those with diffuse cytoplasmic GFP-LC3 staining were considered non-autophagic. Autophagy was quantified in at least 100 cells in 3 independent experiments and percent autophagy was determined in a double-blind manner.32 In other experiments, cells were infected with 20 µl of viral particles (RFP-GFP-LC3; Life Technologies, P36239) per 50,000 cells for 48 h to express LC3 fused to green fluorescent and red fluorescent proteins. GFP fluorescence is decreased in acidic pH environments whereas RFP fluorescence is not. For analysis of cells expressing the RFP-GFP-LC3 construct, GFP- and RFP-positive cells were visualized at 63X in a Zeiss Cell Observer Spinning Disc (Zeiss, Germany) confocal microscope equipped with a growth chamber that was maintained at 37°C and 5% CO2 saturation. RFP and GFP fluorescence were collected with identical settings across all wells and RFP:GFP ratios were quantified using ImageJ.83
siRNA
Cells were transfected with control and specific Smartpool siRNAs (Thermo Scientific Dharmacon) using Lipofectamine 2000 (Thermo Fisher Scientific, 11668027) following the supplier's guidelines. Twenty-four h later, cells were cultured in DMEM containing 10% FBS and treated with SK1-I as indicated in the figure legends.
Imaging
Fluorescence confocal microscopy with live cells plated on 24-well glass bottom plates (Cellvis, CELLVIS P241.5HN) was performed with a Zeiss Cell Observer Spinning Disc confocal microscope with a 63X objective and equipped with an Axiocam MRm camera and 2 Photometrics Evolve 512 cooled emCCD cameras, and a growth chamber that was maintained at 37°C with 5% CO2 saturation.
JC-1 and Cyto-ID labeling
Cells cultured in 24-well glass bottom plates were incubated with JC-1 dye (10 µg/ml; Life Technologies, M34152) in medium containing 10% serum for 10 min followed by a wash, and then were immediately imaged by fluorescence microscopy as described above using 488/509 nm excitation/emission for JC-1 monomer form (green), and 545/572 nm excitation/emission for JC-1-aggregate form (red). Confocal fluorescence images were collected as described above using identical settings across all wells. JC-1 red:green ratios were quantified using ImageJ.
For Cyto-ID (Enzo Life Sciences, ENZ-51031-0050) labeling, cells were seeded as described above and were stained in medium containing 10% serum with Cyto-ID (1 µl per ml medium), and were imaged on a confocal microscope with 463/534nm excitation/emission.
Electron microscopy
Cells were seeded on Thermanox coverslips (Thermo Fisher Scientific, 12–565), treated, and fixed with a solution containing 2% paraformaldehyde and 2% glutaraldehyde in 0.1 M phosphate buffer (pH 7.3) for 30 min at room temperature. After 1 h at 4°C, cells were washed with PBS and post-fixed with 1% osmium tetroxide in 0.1 M phosphate buffer for 30 min at 4°C. Fixed cells were rinsed with water and incubated with 1% uranyl acetate at room temperature for 1.5 h, followed by dehydration in graded solutions of ethanol and propylene oxide (70–100%). Coverslips were embedded in Embed 812 resin (Electron Microscopy Sciences, 14120) and sections prepared. Images were collected on a Jeol JEM-1230 transmission electron microscope equipped with a Gatan UltraScan 4000SP 4K × 4K CCD camera and a Gatan Orius SC1000 side mount CCD camera.
Immunoblotting
Following treatments, cells were harvested by washing twice with ice-cold PBS, and then scraped in 200 µl buffer containing 20 mM HEPES, pH 7.4, 250 mM NaCl, 1% Triton X-100 (Sigma-Aldrich, T8787), 1 mM DTT, 1 mM EDTA, 20% glycerol, and Halt protease plus phosphatase inhibitors (Thermo Fisher Scientific, 78440). Cell suspensions were sonicated, centrifuged (15,000 × g, 10 min), and equal amounts of supernatant proteins analyzed by SDS-PAGE. Protein concentrations were estimated using the Bio-Rad Protein Assay kit (Bio-Rad, 5000006). Immunoblotting was performed using the SNAP i.d. 2.0 Protein Detection System (Millipore) with the following antibodies diluted 1:500 in TBS (50 mM Tris-Cl, pH 7.5, 150 mM NaCl) containing 0.1% Tween-20 (Sigma-Aldrich, P9416) and 0.05% blotting grade milk (Bio-Rad, 1706404): LC3 (12741), GAPDH (2118), p-TP53 Ser15 (9284), BIM (2933), BAX1 (5023), BAD (9239), BIM (2933), BAK1 (12105), cleaved CASP9 (9505), cleaved CASP3 (9664), BECN1 (3495), TUBB (2146), p-AMPK (2535), and ATG5 (12994) all obtained from Cell Signaling Technology; SPHK1 antibody (1:250) was from Sigma-Aldrich (HPA022829); TP53 (1:500) was from Millipore (OP43). Immunopositive bands were visualized by enhanced chemiluminescence using secondary antibodies conjugated with horseradish peroxidase (anti-rabbit: Jackson Immuno Research Labs, 111035045; anti-mouse: Jackson Immuno Research Labs, 115035166) and Super-Signal West Pico (Pierce, PI-34078) or Dura (Pierce, PI-34076) chemiluminescent substrates. For imaging, exposure times were adjusted to avoid saturation and produce images that were within the linear range of the ChemiDoc MP Imaging System (Bio-Rad).
Mass spectroscopy
Cells were seeded in 6-well plates at 350,000 cells per well and allowed to attach for 18 h. Prior to harvesting, cells were washed 3 times with ice-cold PBS, and then scraped in ice-cold PBS plus Halt protease and phosphatase inhibitors. An equal aliquot from each well was mixed with 1 ml of ice-cold methanol, internal standards were added (Avanti, d17:1 S1P, 860641P; d17:0 dihydro-S1P, 860655P), and sphingolipids extracted for mass spectrometry analysis.84 Sphingolipids were quantified by liquid chromatography, electrospray ionization-tandem mass spectrometry (LC-ESI-MS/MS, 5500 QTRAP, ABI) as described previously.84
SPHK1 activity
Enzymatic activity was determined with NBD-sphingosine (Avanti Polar Lipids, 810205P-250 μg) as previously described.60
CRISPR-Cas9 deletion of TP53 and SPHK1
H460 cells were from ATCC (NCI-H460). TP53 and SPHK1 knockout H460 cells were generated by co-transfection (3 × 106 cells in a 10-cm dish) with 1 μg CRISPR-Cas9 plasmid targeting the TP53 loci (Santa Cruz Biotechnologies, sc-416469) and 1 μg of a homology-directed repair plasmid for TP53 (Santa Cruz Biotechnologies, sc-416469-HDR), or a plasmid encoding a double nickase targeting the SPHK1 loci and a puromycin resistance cassette (Santa Cruz Biotechnologies, sc-401274). Cells were transfected using PolyJet reagent (Signagen, SL100688) following the manufacturers guidelines. After 72 h, cells were exposed to 2.5 μg/ml puromycin (Corning, 62111–170) with daily media exchanges to replenish selection agent. After all cells transfected with 1 μg of a control CRISPR/Cas9 plasmid (Santa Cruz Biotechnologies, sc-418922) were killed (∼96 h), puromycin was removed and the cells allowed to recover and grow as individual colonies, which were then selected and examined for expression of TP53 or SPHK1 using western blotting.
Cell death and CASP3-CASP7 flow cytometric assay
Wild-type HCT116 cells (1.5 × 105) were seeded in a 12-well plate. The following day, cells were treated in triplicate with DMSO vehicle control or SK1-I (12.5 or 20 μM) for 24 h. Six h prior to harvesting, cells were labeled with CellEvent TM CASP3/7 Green ReadyProbes TM Reagent (Invitrogen, R37111) according to the manufacturer's instructions. Cells were detached and harvested using non-enzymatic CellStripper solution (Corning, 25-056-CI), washed once in ice-cold PBS and stained with APC-Annexin V (1:80) (BioLegend, 640919) and 7-AAD (1:40) (BD Biosciences, 559925) for 15 min prior to flow cytometric analyses using a BD FACSCanto (10-Color) (BD Biosciences, USA) instrument in the Penn State College of Medicine Flow Cytometry Core Facility. Data were analyzed using FlowJo V10 software (FlowJo).
Statistical analyses
Results are expressed as means ± standard error of the mean, and statistical analyses performed using unpaired 2-tailed Student t test for comparison of 2 groups (GraphPad Prism) or ANOVA with post hoc analyses for multiple groups. All experiments were repeated independently at least times and representative data are shown. For all analyses, p ≤ 0.05 was considered statistically significant.
Disclosure of potential conflicts of interest
No potential conflicts of interest were reported by the authors.
Acknowledgments
We thank Dr. Sandrine Lepine who made some of the initial observations and Dr. Jeremy Allegood for skillful sphingolipid analyses. We acknowledge the VCU Lipidomics and Microscopy Cores, which are supported in part by funding from the NIH-NCI Cancer Center Support Grant P30 CA016059.
References
- 1.Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9(2):139–150. https://doi.org/ 10.1038/nrm2329. PMID:18216770 [DOI] [PubMed] [Google Scholar]
- 2.Maceyka M, Spiegel S. Sphingolipid metabolites in inflammatory disease. Nature. 2014;510(7503):58–67. https://doi.org/ 10.1038/nature13475. PMID:24899305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pyne NJ, Pyne S. Sphingosine 1-phosphate and cancer. Nat Rev Cancer. 2010;10(7):489–503. https://doi.org/ 10.1038/nrc2875. PMID:20555359 [DOI] [PubMed] [Google Scholar]
- 4.Johnson KR, Johnson KY, Crellin HG, Ogretmen B, Boylan AM, Harley RA, Obeid LM. Immunohistochemical distribution of sphingosine kinase 1 in normal and tumor lung tissue. J Histochem Cytochem. 2005;53:1159–1166. https://doi.org/ 10.1369/jhc.4A6606.2005. PMID:15923363 [DOI] [PubMed] [Google Scholar]
- 5.Song L, Xiong H, Li J, Liao W, Wang L, Wu J, Li M. Sphingosine kinase-1 enhances resistance to apoptosis through activation of PI3K/Akt/NF-kappaB pathway in human non-small cell lung cancer. Clin Cancer Res. 2011;17(7):1839–1849. https://doi.org/ 10.1158/1078-0432.CCR-10-0720. PMID:21325072 [DOI] [PubMed] [Google Scholar]
- 6.Koch A, Pfeilschifter J, Huwiler A. Sphingosine 1-phosphate in renal diseases. Cell Physiol Biochem. 2013;31(6):745–760. https://doi.org/ 10.1159/000350093. PMID:23736205 [DOI] [PubMed] [Google Scholar]
- 7.Kawamori T, Osta W, Johnson KR, Pettus BJ,Bielawski J, Tanaka T, Wargovich MJ, Reddy BS, Hannun YA, Obeid LM, et al.. Sphingosine kinase 1 is up-regulated in colon carcinogenesis. FASEB J. 2006;20(2):386–388. https://doi.org/ 10.1096/fj.05-4331fje. PMID:16319132 [DOI] [PubMed] [Google Scholar]
- 8.Kawamori T, Kaneshiro T, Okumura M, Maalouf S, Uflacker A, Bielawski J, Hannun YA, Obeid LM. Role for sphingosine kinase 1 in colon carcinogenesis. FASEB J. 2009;23(2):405–414. https://doi.org/ 10.1096/fj.08-117572. PMID:18824518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruckhaberle E, Rody A, Engels K, Gaetje R, von Minckwitz G, Schiffmann S, Grösch S, Geisslinger G, Holtrich U, Karn T, et al.. Microarray analysis of altered sphingolipid metabolism reveals prognostic significance of sphingosine kinase 1 in breast cancer. Breast Cancer Res Treat. 2008;112(1):41–52. https://doi.org/ 10.1007/s10549-007-9836-9. PMID:18058224 [DOI] [PubMed] [Google Scholar]
- 10.Nagahashi M, Ramachandran S, Kim EY, Allegood JC, Rashid OM, Yamada A, Zhao R, Milstien S, Zhou H, Spiegel S, et al.. Sphingosine-1-phosphate produced by sphingosine kinase 1 promotes breast cancer progression by stimulating angiogenesis and lymphangiogenesis. Cancer Res. 2012;72(3):726–735. https://doi.org/ 10.1158/0008-5472.CAN-11-2167. PMID:22298596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Malavaud B, Pchejetski D, Mazerolles C, de Paiva GR, Calvet C, Doumerc N, Pitson S, Rischmann P, Cuvillier O. Sphingosine kinase-1 activity and expression in human prostate cancer resection specimens. Eur J Cancer. 2010;46(18):3417–3424. https://doi.org/ 10.1016/j.ejca.2010.07.053. PMID:20970322 [DOI] [PubMed] [Google Scholar]
- 12.Li W, Yu CP, Xia JT, Zhang L, Weng GX, Zheng HQ,Kong QL, Hu LJ, Zeng MS, Zeng YX, et al.. Sphingosine kinase 1 is associated with gastric cancer progression and poor survival of patients. Clin Cancer Res. 2009;15(4):1393–1399. https://doi.org/ 10.1158/1078-0432.CCR-08-1158. PMID:19228740 [DOI] [PubMed] [Google Scholar]
- 13.Bayerl MG, Bruggeman RD, Conroy EJ, Hengst JA, King TS, Jimenez M, Claxton DF, Yun JK. Sphingosine kinase 1 protein and mRNA are overexpressed in non-Hodgkin lymphomas and are attractive targets for novel pharmacological interventions. Leuk Lymphoma. 2008;49(5):948–954. https://doi.org/ 10.1080/10428190801911654. PMID:18452097 [DOI] [PubMed] [Google Scholar]
- 14.Marfe G, Di Stefano C, Gambacurta A, Ottone T,Martini V, Abruzzese E, Mologni L, Sinibaldi-Salimei P, de Fabritis P, Gambacorti-Passerini C, et al.. Sphingosine kinase 1 overexpression is regulated by signaling through PI3K, AKT2, and mTOR in imatinib-resistant chronic myeloid leukemia cells. Exp Hematol. 2011;39(6):653–665. https://doi.org/ 10.1016/j.exphem.2011.02.013. PMID:21392556 [DOI] [PubMed] [Google Scholar]
- 15.Li J, Guan HY, Gong LY, Song LB, Zhang N, Wu J,Yuan J, Zheng YJ, Huang ZS, Li M. Clinical significance of sphingosine kinase-1 expression in human astrocytomas progression and overall patient survival. Clin Cancer Res. 2008;14(21):6996–7003. https://doi.org/ 10.1158/1078-0432.CCR-08-0754. PMID:18980995 [DOI] [PubMed] [Google Scholar]
- 16.Van Brocklyn JR, Jackson CA, Pearl DK, Kotur MS,Snyder PJ, Prior TW. Sphingosine kinase-1 expression correlates with poor survival of patients with glioblastoma multiforme: roles of sphingosine kinase isoforms in growth of glioblastoma cell lines. J Neuropathol Exp Neurol. 2005;64(8):695–705. https://doi.org/ 10.1097/01.jnen.0000175329.59092.2c. PMID:16106218 [DOI] [PubMed] [Google Scholar]
- 17.Shi J, He YY, Sun JX, Guo WX, Li N, Xue J, Cheng SQ. The impact of sphingosine kinase 1 on the prognosis of hepatocellular carcinoma patients with portal vein tumor thrombus. Ann Hepatol. 2015;14(2):198–206. PMID:25671829 [PubMed] [Google Scholar]
- 18.Kunkel GT, Maceyka M, Milstien S, Spiegel S. Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat Rev Drug Discov. 2013;12(9):688–702. https://doi.org/ 10.1038/nrd4099. PMID:23954895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Evangelisti C, Evangelisti C, Buontempo F, Lonetti A, Orsini E, Chiarini F, Barata JT, Pyne S, Pyne NJ, Martelli AM. Therapeutic potential of targeting sphingosine kinases and sphingosine 1-phosphate in hematological malignancies. Leukemia. 2016;30(11):2142–2151. https://doi.org/ 10.1038/leu.2016.208. PMID:27461062 [DOI] [PubMed] [Google Scholar]
- 20.Truman JP, Garcia-Barros M, Obeid LM, Hannun YA. Evolving concepts in cancer therapy through targeting sphingolipid metabolism. Biochim Biophys Acta. 2014;1841(8):1174–1188. https://doi.org/ 10.1016/j.bbalip.2013.12.013. PMID:24384461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15(2):81–94. https://doi.org/ 10.1038/nrm3735. PMID:24401948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kruiswijk F, Labuschagne CF, Vousden KH. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol. 2015;16(7):393–405. https://doi.org/ 10.1038/nrm4007. PMID:26122615 [DOI] [PubMed] [Google Scholar]
- 23.Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8(4):275–283. https://doi.org/ 10.1038/nrm2147. PMID:17380161 [DOI] [PubMed] [Google Scholar]
- 24.Taha TA, Osta W, Kozhaya L, Bielawski J, Johnson KR, Gillanders WE, Dbaibo GS, Hannun YA, Obeid LM. Down-regulation of sphingosine kinase-1 by DNA damage: dependence on proteases and p53. J Biol Chem. 2004;279(19):20546–20554. https://doi.org/ 10.1074/jbc.M401259200. PMID:14988393 [DOI] [PubMed] [Google Scholar]
- 25.Heffernan-Stroud LA, Helke KL, Jenkins RW, De Costa AM, Hannun YA, Obeid LM. Defining a role for sphingosine kinase 1 in p53-dependent tumors. Oncogene. 2012;31(9):1166–1175. https://doi.org/ 10.1038/onc.2011.302. PMID:21765468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Green DR, Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature. 2009;458(7242):1127–1130. https://doi.org/ 10.1038/nature07986. PMID:19407794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A. 2005;102(23):8204–8209. https://doi.org/ 10.1073/pnas.0502857102. PMID:15928081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S,Lowe S, Levine AJ. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007;67(7):3043–3053. https://doi.org/ 10.1158/0008-5472.CAN-06-4149. PMID:17409411 [DOI] [PubMed] [Google Scholar]
- 29.Crighton D, Wilkinson S, O'Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126(1):121–134. https://doi.org/ 10.1016/j.cell.2006.05.034. PMID:16839881 [DOI] [PubMed] [Google Scholar]
- 30.Lavieu G, Scarlatti F, Sala G, Carpentier S, Levade T,Ghidoni R, Botti J, Codogno P. Regulation of autophagy by sphingosine kinase 1 and its role in cell survival during nutrient starvation. J Biol Chem. 2006;281(13):8518–8527. https://doi.org/ 10.1074/jbc.M506182200. PMID:16415355 [DOI] [PubMed] [Google Scholar]
- 31.Moruno Manchon JF, Uzor NE, Dabaghian Y, Furr-Stimming EE, Finkbeiner S, Tsvetkov AS. Cytoplasmic sphingosine-1-phosphate pathway modulates neuronal autophagy. Sci Rep. 2015;5:15213. https://doi.org/ 10.1038/srep15213. PMID:26477494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lepine S, Allegood JC, Park M, Dent P, Milstien S,Spiegel S. Sphingosine-1-phosphate phosphohydrolase-1 regulates ER stress-induced autophagy. Cell Death Differ. 2011;18(2):350–361. https://doi.org/ 10.1038/cdd.2010.104. PMID:20798685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiong Y, Lee HJ, Mariko B, Lu YC, Dannenberg AJ,Haka AS, Maxfield FR, Camerer E, Proia RL, Hla T. Sphingosine kinases are not required for inflammatory responses in macrophages. J Biol Chem. 2013;288(45):32563–32573. https://doi.org/ 10.1074/jbc.M113.483750. PMID:24081141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Young MM, Takahashi Y, Fox TE, Yun JK, Kester M, Wang HG. Sphingosine kinase 1 cooperates with Autophagy to maintain endocytic membrane trafficking. Cell Rep. 2016;17(6):1532–1545. https://doi.org/ 10.1016/j.celrep.2016.10.019. PMID:27806293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Y, Levine B. Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ. 2015;22(3):367–376. https://doi.org/ 10.1038/cdd.2014.143. PMID:25257169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arakawa S, Tsujioka M, Yoshida T, Tajima-Sakurai H, Nishida Y, Matsuoka Y, Yoshino I, Tsujimoto Y, Shimizu S. Role of Atg5-dependent cell death in the embryonic development of Bax/Bak double-knockout mice. Cell Death Differ. 2017;24(9):1598–1608. https://doi.org/ 10.1038/cdd.2017.84. PMID:28574506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dasari SK, Bialik S, Levin-Zaidman S, Levin-Salomon V, Merrill AH Jr, Futerman AH, Kimchi A. Signalome-wide RNAi screen identifies GBA1 as a positive mediator of autophagic cell death. Cell Death Differ. 2017;24(7):1288–1302. https://doi.org/ 10.1038/cdd.2017.80. PMID:28574511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goodall ML, Fitzwalter BE, Zahedi S, Wu M,Rodriguez D, Mulcahy-Levy JM, Green DR, Morgan M, Cramer SD, Thorburn A. The Autophagy Machinery Controls Cell Death Switching between Apoptosis and Necroptosis. Dev Cell. 2016;37(4):337–349. https://doi.org/ 10.1016/j.devcel.2016.04.018. PMID:27219062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dbaibo GS, Pushkareva MY, Rachid RA, Alter N,Smyth MJ, Obeid LM, Hannun YA. p53-dependent ceramide response to genotoxic stress. J Clin Invest. 1998;102(2):329–339. https://doi.org/ 10.1172/JCI1180. PMID:9664074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lopez-Marure R, Ventura JL, Sanchez L, Montaño LF, Zentella A. Ceramide mimics tumour necrosis factor-alpha in the induction of cell cycle arrest in endothelial cells. Induction of the tumour suppressor p53 with decrease in retinoblastoma/protein levels. Eur J Biochem. 2000;267(14):4325–4333. https://doi.org/ 10.1046/j.1432-1327.2000.01436.x. PMID:10880954 [DOI] [PubMed] [Google Scholar]
- 41.Oskouian B, Sooriyakumaran P, Borowsky AD, Crans A, Dillard-Telm L, Tam YY, Bandhuvula P, Saba JD. Sphingosine-1-phosphate lyase potentiates apoptosis via p53- and p38-dependent pathways and is down-regulated in colon cancer. Proc Natl Acad Sci U S A. 2006;103(46):17384–17389. https://doi.org/ 10.1073/pnas.0600050103. PMID:17090686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paugh SW, Paugh BS, Rahmani M, Kapitonov D,Almenara JA, Kordula T, Milstien S, Adams JK, Zipkin RE, Grant S, et al.. A selective sphingosine kinase 1 inhibitor integrates multiple molecular therapeutic targets in human leukemia. Blood. 2008;112(4):1382–1391. https://doi.org/ 10.1182/blood-2008-02-138958. PMID:18511810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodrigues NR, Rowan A, Smith ME, Kerr IB,Bodmer WF, Gannon JV, Lane DP. p53 mutations in colorectal cancer. Proc Natl Acad Sci U S A. 1990;87(19):7555–7559. https://doi.org/ 10.1073/pnas.87.19.7555. PMID:1699228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol. 2013;5(4):a008656. https://doi.org/ 10.1101/cshperspect.a008656. PMID:23545416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mitsudomi T, Steinberg SM, Nau MM, Carbone D,D'Amico D, Bodner S, Oie HK, Linnoila RI, Mulshine JL, Minna JD, et al.. p53 gene mutations in non-small-cell lung cancer cell lines and their correlation with the presence of ras mutations and clinical features. Oncogene. 1992;7(1):171–180. PMID:1311061 [PubMed] [Google Scholar]
- 46.Lim KG, Tonelli F, Li Z, Lu X, Bittman R, Pyne S,Pyne NJ. FTY720 analogues as sphingosine kinase 1 inhibitors: enzyme inhibition kinetics, allosterism, proteasomal degradation, and actin rearrangement in MCF-7 breast cancer cells. J Biol Chem. 2011;286(21):18633–18640. https://doi.org/ 10.1074/jbc.M111.220756. PMID:21464128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meek DW, Anderson CW. Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb Perspect Biol. 2009;1(6):a000950. https://doi.org/ 10.1101/cshperspect.a000950. PMID:20457558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y,Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18(3):283–293. https://doi.org/ 10.1016/j.molcel.2005.03.027. PMID:15866171 [DOI] [PubMed] [Google Scholar]
- 49.Romero Rosales K, Singh G, Wu K, Chen J, Janes MR, Lilly MB, Peralta ER, Siskind LJ, Bennett MJ, Fruman DA, et al.. Sphingolipid-based drugs selectively kill cancer cells by down-regulating nutrient transporter proteins. Biochem J. 2011;439(2):299–311. https://doi.org/ 10.1042/BJ20110853. PMID:21767261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim SM, Roy SG, Chen B, Nguyen TM, McMonigle RJ, McCracken AN, Zhang Y, Kofuji S, Hou J, Selwan E, et al.. Targeting cancer metabolism by simultaneously disrupting parallel nutrient access pathways. J Clin Invest. 2016;126(11):4088–4102. https://doi.org/ 10.1172/JCI87148. PMID:27669461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shamas-Din A, Brahmbhatt H, Leber B, Andrews DW. BH3-only proteins: Orchestrators of apoptosis. Biochim Biophys Acta. 2011;1813(4):508–520. https://doi.org/ 10.1016/j.bbamcr.2010.11.024. PMID:21146563 [DOI] [PubMed] [Google Scholar]
- 52.Graupner V, Alexander E, Overkamp T, Rothfuss O,De Laurenzi V, Gillissen BF, Daniel PT, Schulze-Osthoff K, Essmann F. Differential regulation of the proapoptotic multidomain protein Bak by p53 and p73 at the promoter level. Cell Death Differ. 2011;18(7):1130–1139. https://doi.org/ 10.1038/cdd.2010.179. PMID:21233848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sax JK, Fei P, Murphy ME, Bernhard E, Korsmeyer SJ, El-Deiry WS. BID regulation by p53 contributes to chemosensitivity. Nat Cell Biol. 2002;4(11):842–849. https://doi.org/ 10.1038/ncb866. PMID:12402042 [DOI] [PubMed] [Google Scholar]
- 54.Sionov RV, Vlahopoulos SA, Granot Z. Regulation of Bim in Health and Disease. Oncotarget. 2015;6(27):23058–23134. https://doi.org/ 10.18632/oncotarget.5492. PMID:26405162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87(1):99–163. https://doi.org/ 10.1152/physrev.00013.2006. PMID:17237344 [DOI] [PubMed] [Google Scholar]
- 56.Levine B, Abrams J. p53: the Janus of autophagy? Nat Cell Biol. 2008;10(6):637–639. https://doi.org/ 10.1038/ncb0608-637. PMID:18521069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moruno Manchon JF, Uzor NE, Finkbeiner S, Tsvetkov AS. SPHK1/sphingosine kinase 1-mediated autophagy differs between neurons and SH-SY5Y neuroblastoma cells. Autophagy. 2016;12(8):1418–1424. https://doi.org/ 10.1080/15548627.2016.1183082. PMID:27467777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kabeya Y, Mizushima N, Ueno T, Yamamoto A,Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19(21):5720–5728. https://doi.org/ 10.1093/emboj/19.21.5720. PMID:11060023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guo S, Liang Y, Murphy SF, Huang A, Shen H, Kelly DF, Sobrado P, Sheng Z. A rapid and high content assay that measures cyto-ID-stained autophagic compartments and estimates autophagy flux with potential clinical applications. Autophagy. 2015;11(3):560–572. https://doi.org/ 10.1080/15548627.2015.1017181. PMID:25714620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lima S, Milstien S, Spiegel S. Sphingosine and Sphingosine Kinase 1 Involvement in Endocytic Membrane Trafficking. J Biol Chem. 2017;292(8):3074–3088. https://doi.org/ 10.1074/jbc.M116.762377. PMID:28049734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mizugishi K, Yamashita T, Olivera A, Miller GF,Spiegel S, Proia RL. Essential role for sphingosine kinases in neural and vascular development. Mol Cell Biol. 2005;25(24):11113–11121. https://doi.org/ 10.1128/MCB.25.24.11113-11121.2005. PMID:16314531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mullen TD, Hannun YA, Obeid LM. Ceramide synthases at the centre of sphingolipid metabolism and biology. Biochem J. 2012;441(3):789–802. https://doi.org/ 10.1042/BJ20111626. PMID:22248339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tidhar R, Futerman AH. The complexity of sphingolipid biosynthesis in the endoplasmic reticulum. Biochim Biophys Acta. 2013;1833(11):2511–2518. https://doi.org/ 10.1016/j.bbamcr.2013.04.010. PMID:23611790 [DOI] [PubMed] [Google Scholar]
- 64.Gong YN, Guy C, Olauson H, Becker JU, Yang M,Fitzgerald P, Linkermann A, Green DR. ESCRT-III Acts Downstream of MLKL to Regulate Necroptotic Cell Death and Its Consequences. Cell. 2017;169(2):286–300. e16. https://doi.org/ 10.1016/j.cell.2017.03.020. PMID:28388412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18(4):571–580. https://doi.org/ 10.1038/cdd.2010.191. PMID:21311563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8(9):741–752. https://doi.org/ 10.1038/nrm2239. PMID:17717517 [DOI] [PubMed] [Google Scholar]
- 67.Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9(2):139–150. https://doi.org/ 10.1038/nrm2329. PMID:18216770 [DOI] [PubMed] [Google Scholar]
- 68.Jiang W, Ogretmen B. Autophagy paradox and ceramide. Biochim Biophys Acta. 2014;1841(5):783–792. https://doi.org/ 10.1016/j.bbalip.2013.09.005. PMID:24055889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cuvillier O, Pirianov G, Kleuser B, Vanek PG, Coso OA, Gutkind S, Spiegel S. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature. 1996;381(6585):800–803. https://doi.org/ 10.1038/381800a0. PMID:8657285 [DOI] [PubMed] [Google Scholar]
- 70.Mihaylova MM, Shaw RJ. The AMP-activated protein kinase (AMPK) signaling pathway coordinates cell growth, autophagy, & metabolism. Nat Cell Biol. 2011;13(9):1016–1023. https://doi.org/ 10.1038/ncb2329. PMID:21892142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Meley D, Bauvy C, Houben-Weerts JH, Dubbelhuis PF, Helmond MT, Codogno P, Meijer AJ. AMP-activated protein kinase and the regulation of autophagic proteolysis. J Biol Chem. 2006;281(46):34870–34879. https://doi.org/ 10.1074/jbc.M605488200. PMID:16990266 [DOI] [PubMed] [Google Scholar]
- 72.Nieminen AI, Eskelinen VM, Haikala HM, Tervonen TA, Yan Y, Partanen JI, Klefström J. Myc-induced AMPK-phospho p53 pathway activates Bak to sensitize mitochondrial apoptosis. Proc Natl Acad Sci U S A. 2013;110(20):E1839–E1848. https://doi.org/ 10.1073/pnas.1208530110. PMID:23589839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Young MM, Takahashi Y, Khan O, Park S, Hori T,Yun J, Sharma AK, Amin S, Hu CD, Zhang J, et al.. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J Biol Chem. 2012;287(15):12455–12468. https://doi.org/ 10.1074/jbc.M111.309104. PMID:22362782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Levine AJ, Oren M The first 30 years of p53: Growing ever more complex. Nat Rev Cancer. 2009;9:749–758.. https://doi.org/ 10.1038/nrc2723. PMID:19776744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA, Kroemer G. Autophagy regulation by p53. Curr Opin Cell Biol. 2010;22(2):181–185. https://doi.org/ 10.1016/j.ceb.2009.12.001. PMID:20044243 [DOI] [PubMed] [Google Scholar]
- 76.Tasdemir E, Maiuri MC, Galluzzi L, Vitale I,Djavaheri-Mergny M, D'Amelio M, Criollo A, Morselli E, Zhu C, Harper F, et al.. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10(6):676–687. https://doi.org/ 10.1038/ncb1730. PMID:18454141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Morselli E, Shen S, Ruckenstuhl C, Bauer MA,Mariño G, Galluzzi L, Criollo A, Michaud M, Maiuri MC, Chano T, et al.. p53 inhibits autophagy by interacting with the human ortholog of yeast Atg17, Cell Cycle. 2011;10(16):2763–2769. https://doi.org/ 10.4161/cc.10.16.16868. PMID:21775823 [DOI] [PubMed] [Google Scholar]
- 78.Galluzzi L, Vitale I, Abrams JM, Alnemri ES,Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, et al.. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19(1):107–120. https://doi.org/ 10.1038/cdd.2011.96. PMID:21760595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zakeri Z, Bursch W, Tenniswood M, Lockshin RA. Cell death: programmed, apoptosis, necrosis, or other? Cell Death Differ. 1995;2(2):87–96. PMID:17180070 [PubMed] [Google Scholar]
- 80.Zhang F, Wang W, Tsuji Y, Torti SV, Torti FM. Post-transcriptional modulation of iron homeostasis during p53-dependent growth arrest. J Biol Chem. 2008;283(49):33911–33918. https://doi.org/ 10.1074/jbc.M806432200. PMID:18819919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tavallai M, Hamed HA, Roberts JL, Cruickshanks N,Chuckalovcak J, Poklepovic A, Booth L, Dent P. Nexavar/Stivarga and viagra interact to kill tumor cells. J Cell Physiol. 2015;230(9):2281–2298. https://doi.org/ 10.1002/jcp.24961. PMID:25704960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lammering G, Valerie K, Lin PS, Mikkelsen RB,Contessa JN, Feden JP, Farnsworth J, Dent P, Schmidt-Ullrich RK. Radiosensitization of malignant glioma cells through overexpression of dominant-negative epidermal growth factor receptor. Clin Cancer Res. 2001;7(3):682–690. PMID:11297265 [PubMed] [Google Scholar]
- 83.Schindelin J, Rueden CT, Hiner MC, Eliceiri KW. The ImageJ ecosystem: An open platform for biomedical image analysis. Mol Reprod Dev. 2015;82(7-8):518–529. https://doi.org/ 10.1002/mrd.22489. PMID:26153368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hait NC, Allegood J, Maceyka M, Strub GM,Harikumar KB, Singh SK, Luo C, Marmorstein R, Kordula T, Milstien S, et al.. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science. 2009;325(5945):1254–1257. https://doi.org/ 10.1126/science.1176709. PMID:19729656 [DOI] [PMC free article] [PubMed] [Google Scholar]