

ABSTRACT
Many neurodegenerative disorders feature the presence of misfolded polypeptide-containing intracellular inclusion bodies biochemically and morphologically analogous to cellular aggresomes. However, it is largely unknown how misfolded polypeptides form aggresomes and are eventually cleared by the aggresome-macroautophagy/autophagy pathway, so-called aggrephagy. Our recent study revealed that when the ubiquitin-proteasome system is impaired, the accumulated misfolded polypeptides are selectively recognized and transported to the aggresome by a CED complex. This complex is composed of CTIF, originally identified as a specific factor for nuclear cap-binding protein complex (a heterodimer of NCBP1/CBP80 and NCBP2/CBP20)-dependent translation (CT), and its associated factors EEF1A1 and DCTN1. Aggresomal targeting of a misfolded polypeptide via the CED complex is accompanied by CTIF release from the CT complex and thereby inhibits CT efficiency. Therefore, our study provides new mechanistic insights into the crosstalk between translational inhibition and aggresome formation under the influence of a misfolded polypeptide.
KEYWORDS: aggrephagy, aggresome, autophagy, CTIF, DCTN1, EEF1A1, misfolded polypeptide, NCBP1/CBP80, NCBP2/CBP20
CTIF (CBP80/20-dependent translation initiation factor) was originally identified as a specific factor involved in the first (or pioneer) round of translation driven by the nuclear cap-binding protein complex, a heterodimer of NCBP1/CBP80 and NCBP2/CBP20. Whereas translation driven by EIF4E, a cytoplasmic cap-binding protein, is largely in charge of bulk protein synthesis, nuclear cap-binding protein complex-dependent translation (CT) is thought to function in mRNA surveillance such as nonsense-mediated mRNA decay. Our recent study demonstrated that CTIF, a CT-specific factor, contributes to protein quality control as well as mRNA quality control.
A misfolded polypeptide can be generated in various ways, such as abnormal translation, the failure of quality control of a ribosome, and as a byproduct of translation-related mRNA quality control. Eukaryotic cells have evolved well-organized mechanisms to cope with misfolded polypeptides (Fig. 1). As one example, molecular chaperones such as heat shock proteins assist refolding of a misfolded polypeptide, which otherwise would be degraded by the ubiquitin-proteasome system (UPS) for minimizing accumulation of misfolded polypeptides. When these processes are overwhelmed or damaged, the misfolded polypeptides tend to form small cytoplasmic aggregates. Through dynein-mediated retrograde transport, small cytoplasmic aggregates are transported to the minus end of a microtubule, forming a larger aggregate, a so-called aggresome. The misfolded polypeptide-containing aggresome is eventually removed by aggrephagy, a selective form of autophagy. Because aggresome formation is interrelated with cell survival, aggresome-mediated protein surveillance is considered a cytoprotective mechanism against the accumulation of misfolded polypeptides when chaperone-mediated refolding and the UPS are impaired.
Figure 1.
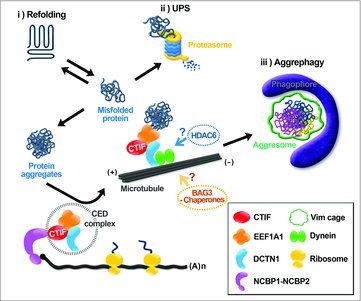
The proposed model of protein surveillance mechanisms. Once a misfolded polypeptide is generated within the cell, it can be subjected to various protein quality control pathways: (i) the misfolded polypeptide may be refolded into its native conformation by chaperones, (ii) it may be polyubiquitinated and degraded by the UPS, or (iii) the misfolded polypeptide may be recognized by CED, HDAC6, or BAG3-chaperones and transported to the aggresome and eventually degraded by autophagy.
In our study, we found that CTIF is localized to 1 or 2 punctate cytoplasmic bodies, which overlap with a TUBG/gamma-tubulin, one of the aggresome marker proteins. Next, confocal microscopy analyses revealed that the CTIF bodies overlap with previously characterized aggresomal components (VIM [vimentin], HDAC6, and ubiquitin) and aggresomal misfolded proteins (polypeptidyl-puromycin, CFTR-ΔF508, and SNCAIP/synphilin1). Notably, CTIF is also colocalized with inclusion bodies containing the G93A mutant of SOD1 and intracellular Lewy bodies containing aggregated SNCA/α-synuclein, which are histological hallmarks of amyotrophic lateral sclerosis and Parkinson disease, respectively. These data indicate that the observed CTIF-containing cytoplasmic bodies are aggresomes.
How does CTIF communicate with misfolded polypeptides for aggresome targeting? To address this question, we performed liquid chromatography with tandem mass spectrometry analysis and identified 2 CTIF-binding partners: DCTN1 (dynactin 1) and EEF1A1 (eukaryotic translation elongation factor 1 α 1). Previous studies have revealed that DCTN1 is involved in the retrograde transport of cargoes along a microtubule by interacting with the dynein motor protein and a microtubule. EEF1A1 directly interacts with a pre-existing and a newly synthesized misfolded protein, generating a signal for aggresome formation. Our GST affinity isolation and immunoprecipitation experiments revealed that CTIF is complexed with DCTN1 and EEF1A1 under normal conditions and during treatment with MG132. Each component is essential for the integrity of this complex. Therefore, we termed this ternary complex the CTIF-EEF1A1-DCTN1 (CED) complex.
When the UPS is impaired, small cytoplasmic aggregates containing misfolded polypeptides accumulate. Under these conditions, CTIF functions as a molecular adaptor that physically links the cytoplasmic aggregates, which are recognized by EEF1A1, to dynein motors via DCTN1. Consequently, the cytoplasmic aggregates are transported toward the aggresome via dynein-mediated retrograde movement. In support of this idea, we observed that when each CED component is downregulated, the aggresomes containing a misfolded polypeptide, either polypeptidyl-puromycin or CFTR-ΔF508, are dispersed into small cytoplasmic aggregates. Moreover, we noticed that the interaction between CTIF and a misfolded protein (polypeptidyl-puromycin) is abrogated by downregulation of EEF1A1.
Considering that CTIF is a common factor for CT and CED-mediated aggresome formation, the association between a misfolded polypeptide and the CED complex and consequent aggresomal targeting of the misfolded polypeptide may reduce the amount of CTIF in the CT complex. As a consequence, these events may reduce CT efficiency. Indeed, our immunoprecipitation data revealed that the misfolded polypeptides that accumulate after treatment of cells with MG132 (a potent proteasome inhibitor) bind to the CED complex and disrupt the associations of CTIF with CT components and mRNAs. In addition, polysome profile data indicate that significant amounts of NCBP1/CBP80 and wild-type CTIF are redistributed from the polysome fractions to the subpolysome fractions upon MG132 treatment. On the contrary, the relative distributions of an N-terminal deletion mutant of CTIF, CTIF (54–598), which fails to form the CED complex, is not affected by MG132 treatment, indicating that a misfolded polypeptide triggers the release of CTIF from the CT complex and, as a consequence, preferentially inhibits CT. Given that CT precedes EIF4E-dependent translation, which is involved in bulk protein synthesis, it may be beneficial for cells to detect the appearance of a misfolded polypeptide as soon as possible. In support of this notion, we found that downregulation of a component of the CED complex promotes apoptosis induced by accumulation of misfolded CFTR-ΔF508.
In summary, our study provides new mechanistic insights into how a misfolded polypeptide is selectively recognized and transported to the aggresome by a CED complex and how the aggresome formation pathway coordinately communicates with CT. Of note, previous data have shown that 2 other cellular adaptors, HDAC6 and the BAG3-containing chaperone complex, are also involved in the recognition of a misfolded polypeptide. How do these 3 adaptors coordinately act on a misfolded polypeptide? Does each of the 3 adaptors have unique substrate specificity? Further studies are required for addressing these questions.
Funding Statement
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korea Government (MSIP; NRF-2015R1A3A2033665) and by a Korea University Future Research Grant.
Disclosure of potential conflicts of Interest
No potential conflicts of interest were disclosed.