

Abstract
Background
The relationship between various external agents such as pollen, food, and infectious agents and human sensitivity exists and is variable depending upon individual’s health conditions. For example, we believe that the pathogenetic potential of the Merkel cell polyomavirus (MCPyV), the resident virus in skin, is variable and depends from the degree of individual’s reactivity. MCPyV as well as Epstein-Barr virus, which are normally connected with humans under the form of subclinical infection, are thought to be involved at various degrees in several neoplastic and inflammatory diseases. In this review, we cover two types of Langerhans cell neoplasms, the Langerhans cell sarcoma (LCS) and Langerhans cell histiocytosis (LCH), represented as either neoplastic or inflammatory diseases caused by MCPyV.
Methods
We meta-analyzed both our previous analyses, composed of quantitative PCR for MCPyV-DNA, proteomics, immunohistochemistry which construct IL-17 endocrine model and interleukin-1 (IL-1) activation loop model, and other groups’ data.
Results
We have shown that there were subgroups associated with the MCPyV as a causal agent in these two different neoplasms. Comparatively, LCS, distinct from the LCH, is a neoplastic lesion (or sarcoma) without presence of inflammatory granuloma frequently observed in the elderly. LCH is a proliferative disease of Langerhans-like abnormal cells which carry mutations of genes involved in the RAS/MAPK signaling pathway. We found that MCPyV may be involved in the development of LCH.
Conclusion
We hypothesized that a subgroup of LCS developed according the same mechanism involved in Merkel cell carcinoma pathogenesis. We proposed LCH developed from an inflammatory process that was sustained due to gene mutations. We hypothesized that MCPyV infection triggered an IL-1 activation loop that lies beneath the pathogenesis of LCH and propose a new triple-factor model.
Keywords: Merkel cell polyomavirus, Langerhans cell neoplasm, Langerhans cell sarcoma, Langerhans cell histiocytosis, BRAF mutation, RAS/MAPK signaling pathway, Interleukin-1 loop model, Triple-factor model, ITIH4, Interleukin-17
Background
Langerhans cell neoplasms are divided into two distinct diseases, the Langerhans cell sarcoma (LCS) and Langerhans cell histiocytosis (LCH). Langerhans cells located in skin, function as sentinel or antigen-presenting cells that can capture invading viruses [1]. We discovered the relationship between Merkel cell polyomavirus (MCPyV) and these two diseases are similar to Epstein-Barr virus pathogenetic potential that by itself is involved in several neoplastic and inflammatory diseases (Table 1).
Table 1.
Proposed relationship between viruses and cigarette smoking and host
| Role | Cancer-causing | Inflammation-inducing |
|---|---|---|
| Epstein-Barr virus | Malignant lymphoma | Infectious mononucleosis |
| Gastric cancer | Hemophagocytic syndrome | |
| Burkitt lymphoma | Necrotizing lymphadenitis | |
| Nasopharyngeal cancer | ||
| Merkel cell polyomavirus | Merkel cell carcinoma | Langerhans cell histiocytosis (LCH) |
| Langerhans cell sarcoma | ||
| Cigarette smoking | Lung cancer, Pulmonary LCH | Chronic obstructive pulmonary disease, Pulmonary LCH |
In this review, we propose two distinct models for LCS and LCH pathogenesis (Fig. 1). Today, some LCS cases are considered as a malignant neoplasm initiated by MCPyV infection [2]. On the contrary, LCH is a reactive disorder with underlying neoplastic potential. In other words, LCH is an inflammatory process that is protracted by gene mutations, which we promote as an IL-1 loop model that was quoted in the WHO Classification of Tumours of Haematopoietic and Lymphoid tissues Revised 4th Edition in 2017 [2] as the major pathway in the development of Tumours derived from Langerhans cell.
Fig. 1.
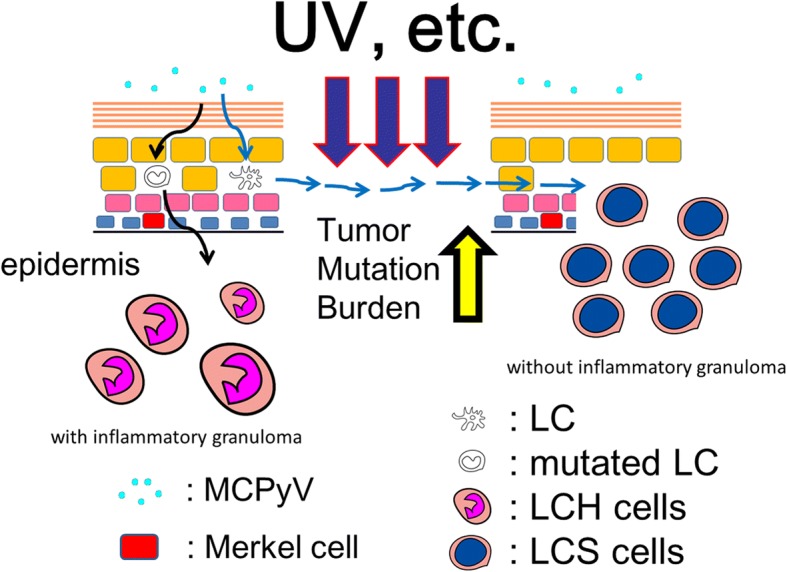
Proposed role of MCPyV in the development of LCH and LCS. We propose two distinct models for LCS and LCH pathogenesis. LCS is a malignant neoplasm initiated by MCPyV infection. On the contrary, LCH is a reactive disorder with underlying neoplastic potential. In other words, LCH is an inflammatory process that is prolonged by mutations. MCPyV: Merkel cell polyomavirus; LCH: Langerhans cell histiocytosis; LCS: Langerhans cell sarcoma; UV: ultraviolet; LC: Langerhans cell; LCH cells: CD1a-positive activated Langerhans (not atypical Langerhans cell, morphologically)-like cells in LCH lesion; LCS cells: sarcoma cells in LCS
We further propose a new triple-factor model for the pathogenesis of LCH.
LCS
MCPyV was discovered in 2008 and was linked to the pathogenesis of Merkel cell carcinoma (MCC), which is a rare and aggressive skin cancer occurring in the dermis of individuals aged 60 years or older [3, 4]. Approximately 80% MCC harbors MCPyV, indicating its prominent role in the development of the disease. Mechanistically, MCPyV-induced oncogenesis is considered to be induced by MCPyV large T (LT) antigen through molecular binding with the retinoblastoma protein [3]. Several tumorigenic pathways leading to MCC were proposed. One was that the induced mutations of MCPyV due to long exposure to ultraviolet light leads to integration of the cytoplasmic viral sequences into the DNA of originating MCC cells. MCPyV might primarily reside in the skin, which was confirmed by the presence of MCPyV-DNA sequences of cutaneous tissue in our studies [5–7].
Langerhans cells are present beyond the middle of the spinous epidermal layer [8], they have the capacity to capture external pathogens [9], and can act as antigen-presenting cells [10, 11]. We previously proposed that external pathogens may be initially recognized by Langerhans cells and may subsequently infect Merkel cells which are mostly located at the basal cell layer of the epidermis (Fig. 1). Therefore, we hypothesized that Langerhans cells serve as a reservoir for MCPyV and demonstrated this phenomenon by showing the presence of MCPyV-DNA sequences in microdissected Langerhans cells from dermatopathic lymphadenopathy [12].
We hypothesized the possibilities that some LCS cases develop from a long standing reservoir cell for MCPyV and showed higher frequency of MCPyV-DNA sequences in LCS tissues with high viral load compared with that in non-affected normal Langerhans cells [2, 13].
LCH
LCH is characterized by the proliferation of CD1a-positive activated Langerhans (not atypical Langerhans cell, morphologically)-like cells (LCH cells) generating inflammatory granuloma. LCH is classified by its involvement of either a single organ system (SS-LCH) or multiple organ systems (MS-LCH) [14]. The latter form is frequent in children younger than 2 years, whereas SS-LCH is more common in children older than 2 years [11, 15]. This rare disease affects 4–9 children per million each year [16–18]. The liver, spleen, and bone marrow (BM) are considered high-risk target organs for LCH [19, 20]. Therefore, LCH is also classified as involving at least one high-risk organ [LCH-RO (+)] or a no high-risk organ [LCH-RO (−)] [19] (Table 2) [21].
Table 2.
Comparison between the present and former classification of LCH [21]
| Classification | Prevalence | ||
|---|---|---|---|
| Present | Former | ||
| LCH-RO (+) | MS | Letterer-Siwe disease | 10% |
| LCH-RO (−) | MS | Hand–Schüller–Christian disease | 20% |
| SS | Eosinophilic granuloma | 70% | |
LCH-RO (+): LCH involving at least one high-risk organ; LCH-RO (−) LCH involving a no high-risk organ; SS-LCH: LCH involving a single organ system; MS-LCH: LCH involving multiple organ systems
The morphology of lesions is so unvarying that pathologists cannot determine whether a given biopsy originates from a patient with SS-LCH or MS-LCH, from a patient with LCH-RO (+) or LCH-RO (−), or from a child or an adult [22]. However, the clinical course of LCH is remarkably variable, ranging from lesions that spontaneously resolve, to a chronic disease that can be widespread and sometimes lethal [23–26].
Although LCH was first described a century ago, the etiology is still not understood [27]. For decades, it was thought that the disease is a reactive disorder rather than a neoplastic process [27]. As the former name, “eosinophilic granuloma” indicates that lesional LCH morphology is reminiscent of tissue reactions to an intracellular pathogen, where tuberculous granuloma is the prototype [22]. For example, scabies infections are reported to induce Langerhans cell hyperplasia, which mimics LCH [28]. However, recent studies indicate that LCH has a more neoplastic character [29–31]. While unexpected remission can rarely occur in neoplasms, spontaneous healing is more common in LCH, suggesting that there may be multiple pathological factors contributing to the LCH process [22, 32, 33]. In this context, an epidemiologic study revealed that risk factors for MS-LCH involve an increase in infections, the use of antibiotics in the first 6 months of life, and a family history of thyroid disease, whereas SS-LCH is significantly associated with symptoms like diarrhea and vomiting in the postnatal period [34].
Review
LCS: High viral load of MCPyV-DNA
In previous studies we specified the relationship between MCPyV infection and LCS [2, 13]. Thus, we suggest that MCPyV may play specific role as an oncogenic factor in certain subtypes of LCS. Based on the foregoing, we propose an LCS tumorigenesis model where MCPyV may be a cause of LCS. In this regard, the recent discovery of MCPyV as a causal agent opened new therapeutic avenues for MCC [35]. Although MCPyV-LT expression was not detected [13], some forms of LCS might originate from MCPyV-infected Langerhans cell (Fig. 1). When confirmed, these findings will also open novel possibilities for therapeutic interventions against LCS.
LCH: IL-17 endocrine model
Coury et al. found IL-17A to be elevated in the serum of patients with LCH and suggested that it might be involved in LCH pathogenesis according to the IL-17A autocrine model [36]. The IL-17A autocrine model in LCH and the IL-17A targeted therapies proposed by Coury et al. [36] have generated considerable controversy. Those authors showed high serum IL-17A levels in LCH and argued that serum IL-17A supported healthy monocyte-derived dendritic cell (DC) fusion capacities in vitro, rather than serum IL-17A levels, which is more correlated with LCH severity (i.e., the IL-17A paradox) [36]. On the contrary, Allen et al. [37–39] were unable to confirm the data presented in Coury et al. [36]. So started the controversy on the role of IL-17A [40, 41]. IL-17A is a proinflammatory cytokine produced by various cells including T helper type 17 cells (Th17), γδTcells, CD8+ T cells, natural killer T cells, lymphoid tissue inducer-like cells, neutrophils, monocytes, and natural killer cells [42–44]. IL-17A acts in both innate and acquired immunity [44]. Innate lymphoid populations can rapidly produce IL-17A [44], which is maintained at low levels in the absence of external stimulation [45]. Moreover, IL-17A/IL-17A receptor is highly important for host defense [46]. We approached the IL-17A controversy and the IL-17A paradox from a new perspective, i.e. considering the expression levels of IL-17A receptor, based upon what we propose an IL-17A endocrine model of LCH [47].
Our study about IL-17 [47] resulted in three major findings. First, the serum levels of IL-17A were higher in LCH as compared to controls with no significant differences among LCH subclasses. Second, higher levels of IL-17A receptor protein expression in MS-LCH were detected as compared to SS-LCH. Third, our results using LC/MS and LC/MRM-MS did not confirm the presence of IL-17A in LCH cells. An endocrine model supported our data: the IL-17A serum levels and expression levels of IL-17A receptor are higher in LCH tissue in patients with LCH. Accordingly, we postulate that the level of IL-17A receptor expression in LCH cells defines the LCH subclass [47]. We consider LCH as a reactive and neoplastic disorder that is influenced by environmental triggers such as pathogens or smoking. In this context, IL-17A is one of the proinflammatory cytokines acting against infective agents. A high serum IL-17A level might be considered to indicate the possibility of an infection in relation to LCH. Serum of patients with LCH can show an upregulation of IL-17A receptor in LCH cells but also in healthy monocyte-derived DCs. This hypothesis could elucidate the IL-17A paradox presented by Coury et al. [36]. In general, cytokines work throughout autocrine or paracrine mechanisms; however, IL-3 and some other proinflammatory cytokines exhibit endocrine mechanisms [47, 48]. In our analysis of LCH tissue, the results using LC/MS and LC/MRM-MS confirm the observation that the IL-17A reactivity in LCH cells observed by immunofluorescence [36] is due to nonspecific antibody binding as described by Allen et al. [37–39]. We resolved this problem by adding data of IL-17A RECEPTOR mRNA expression uploaded by Allen et al. [39]. Generally, stimuli are recognized by receptors such as Toll-like receptors (TLRs) in Langerhans cells [44, 49]. LCH cells also express these receptors in the GSE16395 dataset [39]. LCH cells that are in an active state [50] can induce IL-17A producers in a similar manner as activated Langerhans cells promote Th17 polarization [44]. In this context it would be useful to evaluate CD4/CD8 ratio and assess Th17 in peripheral blood of patients affected by LCH compared to healthy individuals. Though Allen et al. showed low serum levels of IL-17A [37, 38], Makras et al. showed high serum levels of IL-17A using the same enzyme-linked immunosorbent assay (ELISA) kit procedure in both: patients with LCH and controls without significant difference [41]. As IL-17A receptor is ubiquitously expressed [36, 44], it might be difficult to detect IL-17A in the blood as replied Delprat et al. to Allen et al. [37]. We analyzed patient’s sera using a Bio-Plex suspension array system (Bio-Rad), which is different from the other ELISA systems [37, 38, 41]. We found that the serum levels of IL-17A were higher in LCH as compared to controls with no significant differences among LCH subclasses. For host defense, IL-17A/IL-17A receptor complex is important [46], since IL-17A is commonly produced during viral infection [51]. In LCH, an overreaction by mutated LCH cells against stimuli such as viral agents might occur, including increased IL-17A receptor expression. In the context of infection, pathogens such as Epstein–Barr virus [52], human cytomegalovirus [53], and human herpes virus 6 [54, 55] were proven to exist in LCH cells. Although they were regarded as bystander in the LCH lesion in a case-controlled sero-epidemiological study and in situ analysis [11, 56], these investigations were done in 2008 before the discovery of BRAF gene mutations LCH cells in 2010 [31]. At present there is requirement to reexamine the health condition in patients with or without BRAF mutated precursor LCH cells. As reported using the LCH tissue [11, 57–60], serum levels of IL-1a and IL-6, which are known to stimulate Th17 [44], were also significantly higher as compared to controls. Our own analyses on LCH tissues using LC/MS and LC/MRM-MS could not confirm IL-17A positivity in LCH cells (i.e., the IL-17A autocrine model in LCH) [36]. Rather, we propose an IL-17A endocrine model and stress that alteratins in IL-17A receptor expression levels are important for defining LCH subclasses. Low IL-17A levels in sera are maintained by γδT cells in emergencies such as infection [45]. Allen et al. also showed that CD3-positive cells in tonsils produced IL-17A [37, 39]. In 2014, Lourda et al. investigated the presence of IL-17A-producing cells among peripheral blood mononuclear cells isolated from LCH patients and observed a high percentage of IL-17A(+) monocytes in peripheral blood of LCH patients compared to controls [61].
IL-17A/IL-17A receptor signaling pathways include matrix metalloproteinase-3 (MMP3) or MMP12 [62–64]. These MMP3 and MMP12 belong to a series of 1410 genes, the levels of which were more than twofold higher in LCH cells as compared to Langerhans cells in the re-analysis of GSE16395 mRNA data. These higher expression levels of MMP3 and MMP12 not only confirm IL-17A/IL-17A receptor signaling roles in LCH cells but also explain the inflammatory process of LCH such as bone absorption and accumulation of eosinophils [65–67]. In summary, LCH is a neoplastic disorder driven by abnormalities such as BRAF gene mutation [31] thus the severity of LCH might be driven by an inflammatory process under the form of a cytokine storm, especially involving IL-17A/IL-17A receptor signaling pathways. In the future, stimuli that govern IL-17A or IL-17A receptor production might serve as therapeutic targets to stop LCH progression, similar to cessation of smoking which induces pulmonary LCH regression [11, 68], which is almost always a disease of smokers [2].
LCH: IL-1 loop model
Patients with LCH often have dermal disorders such as seborrheic dermatitis [19] concomitant to LCH lesions [69], preceding [70–72], or following LCH lesions [73]. We recently described the possibility of a causal relationship between LCH and dermotropic MCPyV [12], which was discovered as the major pathogenic agent in MCC of the skin in 2008 [3]. Our data indicate that MCPyV-DNA sequences are present in LCH tissues excluding pulmonary LCH, with significant differences between LCH tissues and controls that included patients with dermatopathic lymphadenopathy and reactive lymphoid hyperplasia [12]. The numbers of MCPyV-DNA sequences in LCH tissues from patients younger than 2 years indicated a significant difference from tissues of non-LCH dermal disease patients of the same age [12]. Our data suggest that LCH is a reactive disorder with an underlying oncogenic potential. Thus, both LCH and pulmonary LCH harbor the BRAF V600E mutation [31, 74, 75] and NRAS mutation [76] and appear related to external stimuli such as viral infection [12, 77, 78] and cigarette smoking [79, 80]. In addition, the removal of such stimuli is reported to cause spontaneous healing of LCH [68, 81–83].
Expression of the constitutively active BRAF V600E mutant in LCH cells is predicted to bypass the requirement for mitogen-induced activation of RAF by RAS [31, 84]. The identification of activating BRAF mutations supports the hypothesis that LCH is a process with oncogenic potential [31]. A mouse LCH model using a BRAF V600E construct under the control of CD11c promoter and a BRAF V600E construct under control of the langerin promoter indicates that the BRAF V600E is not only a marker but also an essential driver of LCH pathogenesis [85]. Moreover, phosphorylated extracellular signal-regulated kinase (ERK) (pERK) is rapidly dephosphorylated by dual specificity phosphatase 6 (DUSP6) [86, 87], which is overexpressed in LCH cells [39]. However, BRAF V600E gene mutations are also detected in non-neoplastic disorders such as nevus cell nevus [88] and hyperplastic polyps of the colon [89]. Thus, LCH pathogenesis requires both limited proliferation of precursor LCH cells harboring the BRAF V600E mutation and the accumulation of gene mutations or an inflammatory trigger that activates the RAS/RAF/MEK/ERK signaling pathway [84].
MCPyV interferes with the function of DC towards evasion of the immune surveillance by targeting a NF-κB essential modulator [90] and down-regulating TLR9 [91]. Exposure to MCPyV as measured by serum antibodies against the viral capsid proteins appears to be widely prevalent among healthy subjects [92, 93]. Inapparent existence of MCPyV is indicated on the skin and environmental surface [94, 95]. Pancaldi et al. [96] indicated that buffy coats of healthy adult blood donors, which were examined for MCPyV-DNA tag sequences, showed a prevalence of 22%, with viral loads ranging from 10 to 100 molecules per 100,000 cells (0.0001 to 0.001 per cell). Mertz et al. [97] reported that CD14+ CD16− inflammatory monocytes are a reservoir for MCPyV, but CD14lowCD16+ resident monocytes, lymphocytes, or granulocytes are not. Our data from micro-dissected LC in both dermatopathic lymphadenopathy [12] and LCS [13] suggest that monocytes, precursor Langerhans cells, or Langerhans cells are one of the reservoir cells for MCPyV. In addition, members of the TLR/IL-1 receptor superfamily appear to play a fundamental role in the immune response [98]. Viral “pathogen-associated molecular patterns” are recognized by specific TLRs [99]. TLR agonists stimulate IL-1β production in DC [100], where TLR-triggered ERK activation play important roles [101]. IL-1α expression is induced by TLR-mediated NF-κB activation; such activation has been observed in some LCH cases [102, 103], with/without the presence of IL-1β [104]. All TLRs except TLR3 use the common MyD88-dependent pathway [105]. MyD88 is one of the adaptor proteins that links TLR/ IL-1 receptor [106] and binds to pERK via its D-domain, thereby preventing pERK-DUSP6 interaction and maintaining ERK in an active, phosphorylated state for a longer period [86]. This MyD88-dependent signal may lead to enhanced cell activation, proliferation, and eventually, accumulation and prolonged survival [86, 107] of a given LCH lesion [108].
LCH: ITIH-4
Interalpha-trypsin inhibitor heavy chain 4 (ITIH4, [PDB: Q14624]) is an acute-phase-related protein [109] and potential new biomarker for distinguishing MS-LCH and SS-LCH. Acute-phase proteins are involved in non-specific, physiological immune functions within the innate immune system [110]. The ITIH4 molecule has been detected in animals during experimental bacterial and viral infections [111].
Martel-Jantin et al. [112] reported seroprevalence rate of MCPyV antibodies of children 12 months or younger (49/105) in Cameroon and pointed out the presence of specific maternal antibodies in very young children. Their data indicated that MCPyV infections mostly occurred during early childhood, after the disappearance of specific maternal antibodies [112]. On the contrary Tolstov et al. [93] reported seroprevalence rate of MCPyV antibodies of children of 1 year or younger (0/6) in patients with LCH. We [12] identified a relationship between LCH and MCPyV. MCPyV-DNA in PBMC correlated with LCH-RO (+) [12]. Among patients with LCH-RO (−) (MS-LCH and SS-LCH), MCPyV-DNA was restricted to lesional LCH cells [12], thus we predicted that primary MCPyV infection may influence the LCH subtype involving cells in an early-activated state [27].
Generally, no response is observed after secondary viral infection [111]. For example, primary respiratory syncytial virus infection at 6 months or earlier often induces severe disease [113], although nearly all children are infected by 2–3 years of age [114]. Similarly, primary Epstein-Barr virus and cytomegalovirus infections in elderly individuals cause a severe condition called infectious mononucleosis; nonetheless, nearly all children are infected with these viruses [115]. Although no response is observed after MCPyV infection [94, 96], Kumar et al. [116], however, found that MCPyV-specific T helper cells (in vitro model of a secondary infection) secrete several cytokines, including IL-10. IL-10 is an anti-inflammatory cytokine and is one of cytokines to be produced in LCH [21]. ITIH4 production is up-regulated by IL-6 [109], which is known produced in LCH [21]. Innate immune function between newborns and elderly is extremely different and large quantities of IL-6 after stimulation of receptors, such as TLR, by term newborns are indicated [117]. In LCH, MCPyV infection may induce hyper-immunity in both LCH cells [108] and other inflammatory cells [11, 21].
LCH-RO (+) and LCH-RO (−)
We reported the presence of MCPyV-DNA in the peripheral blood cells of patients with LCH-RO (+) but not in the blood cells of patients with LCH-RO (−) [12]. Berres et al. [85] reported that patients with LCH-RO (+) carried the BRAF V600E mutation in circulating CD11c+ and CD14+ cellular fractions as well as in bone marrow CD34+ hematopoietic cell progenitors, whereas the mutation was restricted to lesional LCH cells in patients with LCH-RO (−). These findings (Table 3) specifically observed in LCH-RO (+) suggest the LCH pathogenetic pathway, though it needs further confirmation to conclude.
Table 3.
Detection of MCPyV-DNA and BRAF mutation in PBMC of patients with LCH
| Classification | MCPyV-DNA | BRAF mutation |
|---|---|---|
| LCH-RO (+) | (+) | (+) |
| LCH-RO (−) | (−) | (−) |
Pulmonary LCH
The incidence of BRAF mutation did not differ significantly [31] between pulmonary LCH that has been regarded as reactive to smoking [11, 74, 79] and non-pulmonary LCH that has been regarded as a neoplastic process [11, 29–31, 118]. Since smoking increases the number of Langerhans cells in chronic obstructive pulmonary disease [119], precursor LCH cells may overreact to smoking. Similarly in cutaneous LCH, overreaction to stimuli such as a dermotropic MCPyV infection may occur [12].
Spontaneous regression in LCH: Triple-factor model
Recently, congenital “self-healing” LCH (Hashimoto-Pritzker disease) condition was proposed as a model of LCH where Kansal et al. identified V600D mutation in Exon 15 of the BRAF gene [120]. As shown in Table 2 [21], patients with SS-LCH account for the majority of LCH patients. While unexpected remission can rarely occur in neoplasms, spontaneous healing is more common in LCH especially in SS-LCH, suggesting that there may be multiple pathogenetic influences to the LCH process [22, 32, 33].
In pulmonary LCH, the removal of stimuli, i.e. cessation of smoking, is well known cessation process for spontaneous healing [68, 81–83]. However, recent data indicate both LCH and pulmonary LCH harbor the BRAF V600E [31, 74, 75] and NRAS mutation [76] and appear linked to external stimuli such as viral infection [12, 77, 78] and cigarette smoking [79, 80].
We think that spontaneous healing in both LCH and pulmonary LCH suggest an oncogene-induced senescence [121] according to Chilosi, et al. who considered that oncogene-induced senescence distinguishes indolent from aggressive forms of pulmonary LCH and non-pulmonary LCH [122].
Using an in vitro model, Lipsky et al. [107] demonstrated that IL-1 production and signaling from the IL-1 receptor are necessary components of Raf-induced transformation of NIH 3 T3 cells, which exclude other factors involvement in the vivo model [85].
The only BRAF V600E mutation does not seem to affect prognosis [2, 123]. We proposed a triple-factor model for pathogenesis of LCH (Fig. 2). We think that balance between oncogene-induced senescence [121] and the requirement of IL-1 autocrine loop [2, 107, 108] of BRAF V600E mutation in pulmonary and non-pulmonary LCH indicates the clinical severity of the disease (Fig. 3).
Fig. 2.
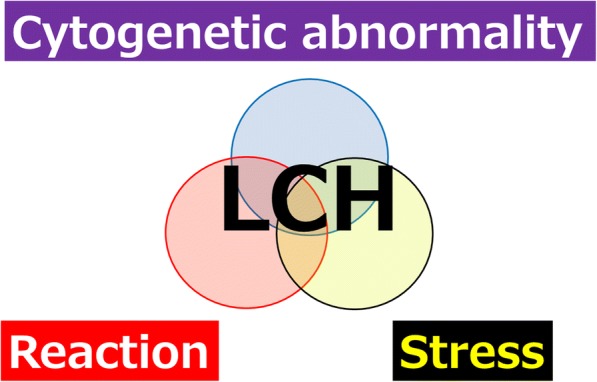
Proposed triple-factor model for LCH pathogenesis. We propose a triple-risk factor model for LCH pathogenesis. Triple-risk factor model is composed of three factors: cytogenetic abnormalities such as BRAF mutation, stress such as MCPyV infection and cigarette smoking, and reaction
Fig. 3.
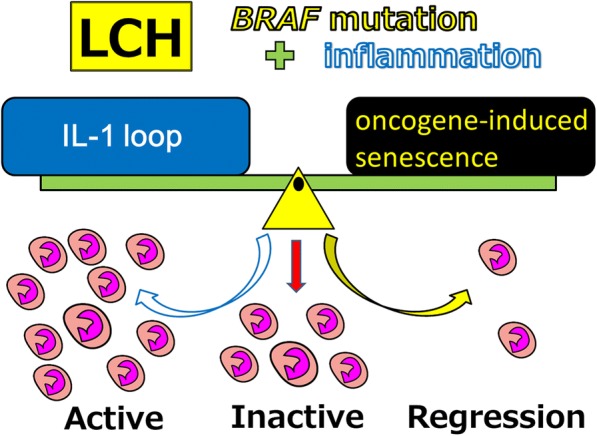
Proposed reaction model in LCH activity. We propose a triple-risk factor model for LCH pathogenesis. Triple-risk factor model is composed of three factors: cytogenetic abnormalities such as BRAF mutation, stress such as MCPyV infection and cigarette smoking, and reaction controlled by balance between an oncogene-induced senescence and an IL-1 loop. An oncogene-induced senescence is induced by cytogenetic abnormalities such as BRAF mutation and MEK mutation. An IL-1 loop is triggered by stress such as MCPyV infection, EBV infection, and cigarette smoking. An IL-1 loop may induce cytokine storm
Bone lesion in LCH
Approximately 80% of patients affected by LCH indicate presence of lesions in bone [124]. There are also bone lesions in recurrent LCH, even if it develops as MS-LCH, knowing that recurrent lesions may occur only in bone. In the LCH lesions, multinucleated giant cells coincide in bone as well as in skin and lymph nodes. In such conditions, tartrate-resistant acid phosphatase (osteoclast marker), vitronectin receptor, cathepsin K, and MMP9 are readily detected [125]. Bone homeostasis is a complex process controlled not only by bone metabolic cells but also by interaction with other distant tissues and cells. Both bone and immune system share many specific proteins such as cytokines that form a functional network [126, 127]. In this regards, Cathepsin K protease is important for bone resorption by osteoclasts, compare to DCs, where it acts to regulate signals from TLR9 and is involved in the Th17 cells differentiation [128]. Many immune system cytokines are involved in bone metabolism, such as involvement of IL-1, IL-6 in the heightening of bone resorption by osteoclasts also producing a cytokine storm. In particular, the receptor activator of nuclear factor kappa-B ligand (RANKL) is regarded as the most important protein [129]. RANKL is expressed on T cells and osteocytes [130] and is an essential protein in activation of DCs and differentiation of osteoclasts. In addition, the serum sRANKL value is significantly higher in LCH patients than in the control group [131]. We have found that MCPyV-DNA is also amplified in bone LCH [12]. MCPyV not only exists in the skin [5, 6, 94] but also exists in the blood of healthy people [96] and MCPyV is recognized by precursor LCH cells and appears to induce LCH lesion formation in bone which has so-called cytokine storm in order to keep homeostatic microenvironment. On the other hand, it has been pointed out that RANKL produced by regulatory T cells is involved in the metastasis of breast cancer [132]. In LCH, it is known that many regulatory T cells exist in lesions [23] and play a similar role in LCH bone lesion formation. Studies and treatments focusing on these relationships have also been realized [131, 133, 134].
Conclusions
We have proposed that there are subgroups of clinical conditions associated with MCPyV designated as two different Langerhans cell neoplasms, LCS and LCH in comparison to Epstein-Barr virus and cigarette smoking pathogens (Table 1, Fig. 1).
Although there is a case report indicating progression from LCH to LCS [135] and case reports of detection of BRAF mutation in LCS [136, 137], these two diseases are not in the same spectrum considering age distribution, neoplastic cell morphology, inflammatory granuloma, and cytokine storm release.
Finally, there is a correlation between LCH subtypes and inflammatory factors such as expression of ITIH4 molecule [138] and IL-17A receptor [47]. However, there are no specific pathological definitions between the LCH subtypes such as self-healing SS-LCH, LCH-RO (−), and life-threatening LCH-RO (+). The principal pathological characteristics of LCH include the morphologic aspects of activated Langerhans cells adjoined to inflammatory granuloma. Therefore, we propose that LCH entity is an inflammatory process that is protracted by gene mutations occurring in the LCH cells interacting with other immunologically competent cells (Fig. 4) [108].
Fig. 4.
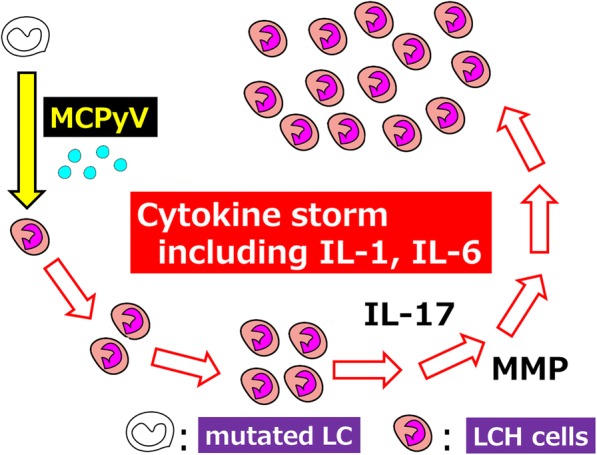
Proposed role of MCPyV in the development of LCH. We propose LCH is a reactive disorder with underlying neoplastic potential using a triple-risk factor model. Infection usually triggers cytokine production and antibody production. However, MCPyV infection triggers cytokine storm including IL-1 and IL-6 in LCH patients. IL-1 and IL-6 stimulate IL-17 producing cells. We found IL-17A receptor expression levels in LCH cells are important for defining LCH subclasses. IL-17/IL-17 receptor signaling pathways include matrix metalloproteinase-3 (MMP3) or MMP12 which induce bone absorption. MCPyV: Merkel cell polyomavirus; LCH: Langerhans cell histiocytosis; LC: Langerhans cell; LCH cells: CD1a-positive activated Langerhans (not atypical Langerhans cell, morphologically)-like cells in LCH lesion
Acknowledgments
The authors would like to thank all patients, parents, control subjects, and physicians who participated in the studies related to this review.
Funding
These works were partly supported by the Histiocytosis Association of America (HAA grant 2009); JSPS KAKENHI (Grants-in-aid for Scientific Research) Numbers (C) 23590426, (C) 26460451, and (C) 17 K08743 from the Japanese Ministry of Education, Science, Sports and Culture; and by 2010, 2011, and 2012 research grants from the Japan LCH Study Group.
Availability of data and materials
Not applicable.
Abbreviations
- DC
Dendritic cell
- DUSP6
Dual specificity phosphatase 6
- ELISA
Enzyme-linked immunosorbent assay
- ERK
Extracellular signal-regulated kinase
- ITIH4
Interalpha-trypsin inhibitor heavy chain 4
- LCH cells
CD1a-positive activated Langerhans (not atypical Langerhans cell, morphologically)-like cells
- LCH
Langerhans cell histiocytosis
- LCS
Langerhans cell sarcoma
- LT
large T
- MCC
Merkel cell carcinoma
- MCPyV
Merkel cell polyomavirus
- MMP
Matrix metalloproteinase
- RANKL
Receptor activator of nuclear factor kappa-B ligand
- Th17
T helper type 17 cells
- TLR
Toll-like receptor
Authors’ contributions
IM, KH, and JG wrote the manuscript and prepared all the Figs. YH, TH, MD, MM, TI, SK, KN, JG, FJ, and TO participated in experimental analyses. YH, TI, AM, and SI provided materials and clinical data from patients with LCH. NW, JN, MI, and MT participated in pathological analyses. KH, TY, SI, and FJ supervised the project. All authors read and approved the final manuscript.
Ethics approval and consent to participate
This study was approved by the Institutional Review Board of Okayama University Graduate School of Medicine, Dentistry, and Pharmaceutical Sciences, Okayama, Japan, Faculty of Medicine, Tottori University, Tottori, Japan, and Kochi Medical School, Kochi University, Kochi, Japan. All patients, parents, control subjects, and physicians participated in the studies related to this review with consent to participate.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Ichiro Murakami, Phone: +81-88-880-2329, Email: ichiro.murakami.09@gmail.com.
Noriko Wada, Email: rinko0106@gmail.co.
Junko Nakashima, Email: notch6015@gmail.com.
Mitsuko Iguchi, Email: iguchim@kochi-u.ac.jp.
Makoto Toi, Email: mtoi@kochi-u.ac.jp.
Yumiko Hashida, Email: hashida@kochi-u.ac.jp.
Tomonori Higuchi, Email: t.higuchi@kochi-u.ac.jp.
Masanori Daibata, Email: daibatam@kochi-u.ac.jp.
Michiko Matsushita, Email: michikohavefun@gmail.com.
Takeshi Iwasaki, Email: iwasakit-path@umin.ac.jp.
Satoshi Kuwamoto, Email: s.quamoto@gmail.com.
Yasushi Horie, Email: yhorie-ttr@umin.ac.jp.
Keiko Nagata, Email: nanamon@theia.ocn.ne.jp.
Kazuhiko Hayashi, Email: khayashi32@gmail.com.
Takashi Oka, Email: oka@md.okayama-u.ac.jp.
Tadashi Yoshino, Email: yoshino@md.okayama-u.ac.jp.
Toshihiko Imamura, Email: timamura@sa3.so-net.ne.jp.
Akira Morimoto, Email: akira@jichi.ac.jp.
Shinsaku Imashuku, Email: shinim95@mbox.kyoto-inet.or.jp.
Jean Gogusev, Email: jean.gogusev@gmail.com.
Francis Jaubert, Email: jaubert.francis@live.fr.
References
- 1.Cunningham AL, Abendroth A, Jones C, Nasr N, Turville S. Viruses and Langerhans cells. Immunol Cell Biol. 2010;88:416–423. doi: 10.1038/icb.2010.42. [DOI] [PubMed] [Google Scholar]
- 2.Weiss LM, Jaffe R, Facchetti F. Tumours derived from Langerhans cells. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues Revised 4th Edition. Lyon: International Agency for Research on Cancer (IARC); 2017. pp. 470–473. [Google Scholar]
- 3.Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096–1100. doi: 10.1126/science.1152586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuwamoto S. Recent advances in the biology of Merkel cell carcinoma. Hum Pathol. 2011;42:1063–1077. doi: 10.1016/j.humpath.2011.01.020. [DOI] [PubMed] [Google Scholar]
- 5.Matsushita M, Kuwamoto S, Iwasaki T, Higaki-Mori H, Yashima S, Kato M, Murakami I, Horie Y, Kitamura Y, Hayashi K. Detection of Merkel cell polyomavirus in the human tissues from 41 Japanese autopsy cases using polymerase chain reaction. Intervirology. 2013;56:1–5. doi: 10.1159/000338620. [DOI] [PubMed] [Google Scholar]
- 6.Hashida Y, Nakajima K, Nakajima H, Shiga T, Tanaka M, Murakami M, Matsuzaki S, Naganuma S, Kuroda N, Seki Y, et al. High load of Merkel cell polyomavirus DNA detected in the normal skin of Japanese patients with Merkel cell carcinoma. J Clin Virol. 2016;82:101–107. doi: 10.1016/j.jcv.2016.07.011. [DOI] [PubMed] [Google Scholar]
- 7.Hashida Y, Kamioka M, Tanaka M, Hosokawa S, Murakami M, Nakajima K, Kikuchi H, Fujieda M, Sano S, Daibata M. Ecology of Merkel cell Polyomavirus in healthy skin among individuals in an Asian cohort. J Infect Dis. 2016;213:1708–1716. doi: 10.1093/infdis/jiw040. [DOI] [PubMed] [Google Scholar]
- 8.Kanik A, Li M, Uramacher CD. Normal skin. In: Mills SE, editor. Histology for pathologist. 3. Philadelphia: Lippincott Williams & Wilkins; 2012. pp. 3–28. [Google Scholar]
- 9.Kubo A, Nagao K, Yokouchi M, Sasaki H, Amagai M. External antigen uptake by Langerhans cells with reorganization of epidermal tight junction barriers. J Exp Med. 2009;206:2937–2946. doi: 10.1084/jem.20091527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 11.Weitzman S, Egeler RM. Histiocytic disorders of children and adults: introduction to the problem, overview, historical perspective and epidemiology. In: Weitzman S, Egeler RM, editors. Histiocytic disorders of children and adults. Cambridge: Cambridge University Press; 2005. pp. 1–13. [Google Scholar]
- 12.Murakami I, Matsushita M, Iwasaki T, Kuwamoto S, Kato M, Horie Y, Hayashi K, Imamura T, Morimoto A, Imashuku S, et al. Merkel cell polyomavirus DNA sequences in peripheral blood and tissues from patients with Langerhans cell histiocytosis. Hum Pathol. 2014;45:119–126. doi: 10.1016/j.humpath.2013.05.028. [DOI] [PubMed] [Google Scholar]
- 13.Murakami I, Matsushita M, Iwasaki T, Kuwamoto S, Kato M, Horie Y, Hayashi K, Gogusev J, Jaubert F, Nakamoto S, et al. High viral load of Merkel cell polyomavirus DNA sequences in Langerhans cell sarcoma tissues. Infect Agent Cancer. 2014;9:15. doi: 10.1186/1750-9378-9-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chu T, D'Angio GJ, Favara B, Ladisch S, Nesbit M, Pritchard J. Histiocytosis syndromes in children. Lancet. 1987;1:208–209. doi: 10.1016/s0140-6736(87)93074-1. [DOI] [PubMed] [Google Scholar]
- 15.Hamre M, Hedberg J, Buckley J, Bhatia S, Finlay J, Meadows A, Nesbit M, Smithson A, Robison L. Langerhans cell histiocytosis: an exploratory epidemiologic study of 177 cases. Med Pediatr Oncol. 1997;28:92–97. doi: 10.1002/(SICI)1096-911X(199702)28:2<92::AID-MPO2>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 16.Salotti JA, Nanduri V, Pearce MS, Parker L, Lynn R, Windebank KP. Incidence and clinical features of Langerhans cell histiocytosis in the UK and Ireland. Arch Dis Child. 2009;94:376–380. doi: 10.1136/adc.2008.144527. [DOI] [PubMed] [Google Scholar]
- 17.Alston RD, Tatevossian RG, McNally RJ, Kelsey A, Birch JM, Eden TO Incidence and survival of childhood Langerhans cell histiocytosis in Northwest England from 1954 to 1998. Pediatr Blood Cancer. 2007;48:555–560. doi: 10.1002/pbc.20884. [DOI] [PubMed] [Google Scholar]
- 18.Guyot-Goubin A, Donadieu J, Barkaoui M, Bellec S, Thomas C, Clavel J. Descriptive epidemiology of childhood Langerhans cell histiocytosis in France, 2000-2004. Pediatr Blood Cancer. 2008;51:71–75. doi: 10.1002/pbc.21498. [DOI] [PubMed] [Google Scholar]
- 19.Donadieu J, Egeler RM, Pritchard J. Langerhans cell histiocytosis: a clinical update. In: Weitzman S, Egeler RM, editors. Histiocytic disorders of children and adults. Cambridge: Cambridge University Press; 2005. pp. 95–129. [Google Scholar]
- 20.Ronceray L, Potschger U, Janka G, Gadner H, Minkov M. German Society for Pediatric H, oncology LCHSG: pulmonary involvement in pediatric-onset multisystem Langerhans cell histiocytosis: effect on course and outcome. J Pediatr. 2012;161:129–133. doi: 10.1016/j.jpeds.2011.12.035. [DOI] [PubMed] [Google Scholar]
- 21.Morimoto A, Oh Y, Shioda Y, Kudo K, Imamura T. Recent advances in Langerhans cell histiocytosis. Pediatr Int. 2014;56:451–461. doi: 10.1111/ped.12380. [DOI] [PubMed] [Google Scholar]
- 22.Delprat C, Arico M. Blood spotlight on Langerhans cell histiocytosis. Blood. 2014;124:867–872. doi: 10.1182/blood-2014-02-556407. [DOI] [PubMed] [Google Scholar]
- 23.Senechal B, Elain G, Jeziorski E, Grondin V, Patey-Mariaud de Serre N, Jaubert F, Beldjord K, Lellouch A, Glorion C, Zerah M, et al. Expansion of regulatory T cells in patients with Langerhans cell histiocytosis. PLoS Med. 2007;4:e253. doi: 10.1371/journal.pmed.0040253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Donadieu J. A multicentre retrospective survey of Langerhans’ cell histiocytosis: 348 cases observed between 1983 and 1993. The French Langerhans’ cell Histiocytosis study group. Arch Dis Child. 1996;75:17–24. doi: 10.1136/adc.75.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bernard F, Thomas C, Bertrand Y, Munzer M, Landman Parker J, Ouache M, Colin VM, Perel Y, Chastagner P, Vermylen C, Donadieu J. Multi-Centre pilot study of 2-chlorodeoxyadenosine and cytosine arabinoside combined chemotherapy in refractory Langerhans cell histiocytosis with haematological dysfunction. Eur J Cancer. 2005;41:2682–2689. doi: 10.1016/j.ejca.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 26.Bernstrand C, Sandstedt B, Ahstrom L, Henter JI. Long-term follow-up of Langerhans cell histiocytosis: 39 years’ experience at a single Centre. Acta Paediatr. 2005;94:1073–1084. doi: 10.1080/08035250510028263. [DOI] [PubMed] [Google Scholar]
- 27.da Costa CE, Annels NE, Egeler RM. The immunological basis of Langerhans cell histiocytosis. In: Weitzman S, Egeler RM, editors. Histiocytic disorders of children and adults. Cambridge: Cambridge University Press; 2005. pp. 66–82. [Google Scholar]
- 28.Bhattacharjee P, Glusac EJ. Langerhans cell hyperplasia in scabies: a mimic of Langerhans cell histiocytosis. J Cutan Pathol. 2007;34:716–720. doi: 10.1111/j.1600-0560.2006.00723.x. [DOI] [PubMed] [Google Scholar]
- 29.Willman CL, Busque L, Griffith BB, Favara BE, McClain KL, Duncan MH, Gilliland DG. Langerhans’-cell histiocytosis (histiocytosis X)--a clonal proliferative disease. N Engl J Med. 1994;331:154–160. doi: 10.1056/NEJM199407213310303. [DOI] [PubMed] [Google Scholar]
- 30.Yu RC, Chu C, Buluwela L, Chu AC. Clonal proliferation of Langerhans cells in Langerhans cell histiocytosis. Lancet. 1994;343:767–768. doi: 10.1016/S0140-6736(94)91842-2. [DOI] [PubMed] [Google Scholar]
- 31.Badalian-Very G, Vergilio JA, Degar BA, MacConaill LE, Brandner B, Calicchio ML, Kuo FC, Ligon AH, Stevenson KE, Kehoe SM, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116:1919–1923. doi: 10.1182/blood-2010-04-279083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iwafuchi M, Watanabe H, Shiratsuka M. Primary benign histiocytosis X of the stomach. A report of a case showing spontaneous remission after 5 1/2 years. Am J Surg Pathol. 1990;14:489–496. doi: 10.1097/00000478-199005000-00010. [DOI] [PubMed] [Google Scholar]
- 33.Corbeel L, Eggermont E, Desmyter J, Surmont I, De Vos R, De Wolf-Peeters C, Cobbaert C, Eykens A. Spontaneous healing of Langerhans cell histiocytosis (histiocytosis X) Eur J Pediatr. 1988;148:32–33. doi: 10.1007/BF00441809. [DOI] [PubMed] [Google Scholar]
- 34.Bhatia S, Nesbit ME, Jr, Egeler RM, Buckley JD, Mertens A, Robison LL. Epidemiologic study of Langerhans cell histiocytosis in children. J Pediatr. 1997;130:774–784. doi: 10.1016/S0022-3476(97)80021-2. [DOI] [PubMed] [Google Scholar]
- 35.Schrama D, Ugurel S, Becker JC. Merkel cell carcinoma: recent insights and new treatment options. Curr Opin Oncol. 2012;24:141–149. doi: 10.1097/CCO.0b013e32834fc9fe. [DOI] [PubMed] [Google Scholar]
- 36.Coury F, Annels N, Rivollier A, Olsson S, Santoro A, Speziani C, Azocar O, Flacher M, Djebali S, Tebib J, et al. Langerhans cell histiocytosis reveals a new IL-17A-dependent pathway of dendritic cell fusion. Nat Med. 2008;14:81–87. doi: 10.1038/nm1694. [DOI] [PubMed] [Google Scholar]
- 37.Allen CE, McClain KL. Interleukin-17A is not expressed by CD207(+) cells in Langerhans cell histiocytosis lesions. Nat Med. 2009;15:483–484. doi: 10.1038/nm0509-483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peters TL, McClain KL, Allen CE. Neither IL-17A mRNA nor IL-17A protein are detectable in Langerhans cell histiocytosis lesions. Mol Ther. 2011;19:1433–1439. doi: 10.1038/mt.2011.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Allen CE, Li L, Peters TL, Leung HC, Yu A, Man TK, Gurusiddappa S, Phillips MT, Hicks MJ, Gaikwad A, et al. Cell-specific gene expression in Langerhans cell histiocytosis lesions reveals a distinct profile compared with epidermal Langerhans cells. J Immunol. 2010;184:4557–4567. doi: 10.4049/jimmunol.0902336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hogarty MD. IL-17A in LCH: systemic biomarker, local factor, or none of the above? Mol Ther. 2011;19:1405–1406. doi: 10.1038/mt.2011.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Makras P, Polyzos SA, Anastasilakis AD, Terpos E, Papatheodorou A, Kaltsas GA. Is serum IL-17A a useful systemic biomarker in patients with Langerhans cell histiocytosis? Mol Ther. 2012;20:6–7. doi: 10.1038/mt.2011.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol. 2009;9:556–567. doi: 10.1038/nri2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hamada H, Garcia-Hernandez Mde L, Reome JB, Misra SK, Strutt TM, McKinstry KK, Cooper AM, Swain SL, Dutton RW. Tc17, a unique subset of CD8 T cells that can protect against lethal influenza challenge. J Immunol. 2009;182:3469–3481. doi: 10.4049/jimmunol.0801814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Iwakura Y, Ishigame H, Saijo S, Nakae S. Functional specialization of interleukin-17 family members. Immunity. 2011;34:149–162. doi: 10.1016/j.immuni.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 45.DeFranco A, Locksley R, Robertson M. Immunity: the immune response in infectious and inflammatory disease. Oxford: Oxford University Press; 2007. [Google Scholar]
- 46.Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ, Ho AW, Hai JH, Yu JJ, Jung JW, et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med. 2009;206:299–311. doi: 10.1084/jem.20081463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Murakami I, Morimoto A, Oka T, Kuwamoto S, Kato M, Horie Y, Hayashi K, Gogusev J, Jaubert F, Imashuku S, et al. IL-17A receptor expression differs between subclasses of Langerhans cell histiocytosis, which might settle the IL-17A controversy. Virchows Arch. 2013;462:219–228. doi: 10.1007/s00428-012-1360-6. [DOI] [PubMed] [Google Scholar]
- 48.Simons PJ, Delemarre FG, Drexhage HA. Antigen-presenting dendritic cells as regulators of the growth of thyrocytes: a role of interleukin-1beta and interleukin-6. Endocrinology. 1998;139:3148–3156. doi: 10.1210/endo.139.7.6110. [DOI] [PubMed] [Google Scholar]
- 49.Aliahmadi E, Gramlich R, Grutzkau A, Hitzler M, Kruger M, Baumgrass R, Schreiner M, Wittig B, Wanner R, Peiser M. TLR2-activated human langerhans cells promote Th17 polarization via IL-1beta, TGF-beta and IL-23. Eur J Immunol. 2009;39:1221–1230. doi: 10.1002/eji.200838742. [DOI] [PubMed] [Google Scholar]
- 50.Barbey S, Gane P, Le Pelletier O, Nezelof C. Histiocytosis X Langerhans cells react with antiinterleukin-2 receptor monoclonal antibody. Pediatr Pathol. 1987;7:569–574. doi: 10.3109/15513818709161420. [DOI] [PubMed] [Google Scholar]
- 51.Ryzhakov G, Lai CC, Blazek K, To KW, Hussell T, Udalova I. IL-17 boosts proinflammatory outcome of antiviral response in human cells. J Immunol. 2011;187:5357–5362. doi: 10.4049/jimmunol.1100917. [DOI] [PubMed] [Google Scholar]
- 52.Shimakage M, Sasagawa T, Kimura M, Shimakage T, Seto S, Kodama K, Sakamoto H. Expression of Epstein-Barr virus in Langerhans’ cell histiocytosis. Hum Pathol. 2004;35:862–868. doi: 10.1016/j.humpath.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 53.Kawakubo Y, Kishimoto H, Sato Y, Yanagimoto K, Tsuruta T, Ogawa Y, Kameya T. Human cytomegalovirus infection in foci of Langerhans cell histiocytosis. Virchows Arch. 1999;434:109–115. doi: 10.1007/s004280050313. [DOI] [PubMed] [Google Scholar]
- 54.Leahy MA, Krejci SM, Friednash M, Stockert SS, Wilson H, Huff JC, Weston WL, Brice SL. Human herpesvirus 6 is present in lesions of Langerhans cell histiocytosis. J Invest Dermatol. 1993;101:642–645. doi: 10.1111/1523-1747.ep12371669. [DOI] [PubMed] [Google Scholar]
- 55.Glotzbecker MP, Dormans JP, Pawel BR, Wills BP, Joshi Y, Elkan M, Hodinka RL. Langerhans cell histiocytosis and human herpes virus 6 (HHV-6), an analysis by real-time polymerase chain reaction. J Orthop Res. 2006;24:313–320. doi: 10.1002/jor.20039. [DOI] [PubMed] [Google Scholar]
- 56.Jeziorski E, Senechal B, Molina TJ, Devez F, Leruez-Ville M, Morand P, Glorion C, Mansuy L, Gaudelus J, Debre M, et al. Herpes-virus infection in patients with Langerhans cell histiocytosis: a case-controlled sero-epidemiological study, and in situ analysis. PLoS One. 2008;3:e3262. doi: 10.1371/journal.pone.0003262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arenzana-Seisdedos F, Barbey S, Virelizier JL, Kornprobst M, Nezelof C. Histiocytosis X. Purified (T6+) cells from bone granuloma produce interleukin 1 and prostaglandin E2 in culture. J Clin Invest. 1986;77:326–329. doi: 10.1172/JCI112296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.de Graaf JH, Tamminga RY, Dam-Meiring A, Kamps WA, Timens W. The presence of cytokines in Langerhans’ cell histiocytosis. J Pathol. 1996;180:400–406. doi: 10.1002/(SICI)1096-9896(199612)180:4<400::AID-PATH701>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 59.Egeler RM, Favara BE, van Meurs M, Laman JD, Claassen E. Differential in situ cytokine profiles of Langerhans-like cells and T cells in Langerhans cell histiocytosis: abundant expression of cytokines relevant to disease and treatment. Blood. 1999;94:4195–4201. [PubMed] [Google Scholar]
- 60.Foss HD, Herbst H, Araujo I, Hummel M, Berg E, Schmitt-Graff A, Stein H. Monokine expression in Langerhans’ cell histiocytosis and sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease) J Pathol. 1996;179:60–65. doi: 10.1002/(SICI)1096-9896(199605)179:1<60::AID-PATH533>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 61.Lourda M, Olsson-Akefeldt S, Gavhed D, Bjornfot S, Clausen N, Hjalmars U, Sabel M, Tazi A, Arico M, Delprat C, et al. Detection of IL-17A-producing peripheral blood monocytes in Langerhans cell histiocytosis patients. Clin Immunol. 2014;153:112–122. doi: 10.1016/j.clim.2014.04.004. [DOI] [PubMed] [Google Scholar]
- 62.Sylvester J, Liacini A, Li WQ, Zafarullah M. Interleukin-17 signal transduction pathways implicated in inducing matrix metalloproteinase-3, −13 and aggrecanase-1 genes in articular chondrocytes. Cell Signal. 2004;16:469–476. doi: 10.1016/j.cellsig.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 63.Chen K, Pociask DA, McAleer JP, Chan YR, Alcorn JF, Kreindler JL, Keyser MR, Shapiro SD, Houghton AM, Kolls JK, Zheng M. IL-17RA is required for CCL2 expression, macrophage recruitment, and emphysema in response to cigarette smoke. PLoS One. 2011;6:e20333. doi: 10.1371/journal.pone.0020333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Raychaudhuri SP, Raychaudhuri SK, Genovese MC. IL-17 receptor and its functional significance in psoriatic arthritis. Mol Cell Biochem. 2012;359:419–429. doi: 10.1007/s11010-011-1036-6. [DOI] [PubMed] [Google Scholar]
- 65.Lanone S, Zheng T, Zhu Z, Liu W, Lee CG, Ma B, Chen Q, Homer RJ, Wang J, Rabach LA, et al. Overlapping and enzyme-specific contributions of matrix metalloproteinases-9 and -12 in IL-13-induced inflammation and remodeling. J Clin Invest. 2002;110:463–474. doi: 10.1172/JCI0214136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pouladi MA, Robbins CS, Swirski FK, Cundall M, McKenzie AN, Jordana M, Shapiro SD, Stampfli MR. Interleukin-13-dependent expression of matrix metalloproteinase-12 is required for the development of airway eosinophilia in mice. Am J Respir Cell Mol Biol. 2004;30:84–90. doi: 10.1165/rcmb.2003-0051OC. [DOI] [PubMed] [Google Scholar]
- 67.Hou P, Troen T, Ovejero MC, Kirkegaard T, Andersen TL, Byrjalsen I, Ferreras M, Sato T, Shapiro SD, Foged NT, Delaisse JM. Matrix metalloproteinase-12 (MMP-12) in osteoclasts: new lesson on the involvement of MMPs in bone resorption. Bone. 2004;34:37–47. doi: 10.1016/j.bone.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 68.Mogulkoc N, Veral A, Bishop PW, Bayindir U, Pickering CA, Egan JJ. Pulmonary Langerhans’ cell histiocytosis: radiologic resolution following smoking cessation. Chest. 1999;115:1452–1455. doi: 10.1378/chest.115.5.1452. [DOI] [PubMed] [Google Scholar]
- 69.Punia RS, Bagai M, Mohan H, Thami GP. Langerhans cell histiocytosis of skin: a clinicopathologic analysis of five cases. Indian J Dermatol Venereol Leprol. 2006;72:211–214. doi: 10.4103/0378-6323.25782. [DOI] [PubMed] [Google Scholar]
- 70.Erdem AP, Kasimoglu Y, Sepet E, Gencay K, Sahin S, Dervisoglu S. Oral manifestations may be the first sign of Langerhans cell histiocytosis. Oral Health Prev Dent. 2013;11:57–59. doi: 10.3290/j.ohpd.a29376. [DOI] [PubMed] [Google Scholar]
- 71.Mehta B, Venkatramani R. Images in clinical medicine. Langerhans’-cell histiocytosis N Engl J Med. 2014;371:1050. doi: 10.1056/NEJMicm1312532. [DOI] [PubMed] [Google Scholar]
- 72.Lai JY, Tang MM, Priya G, Rajasuriar JS, Suganthi T. Langerhans cell Histiocytosis in an adult - a rare, life-threatening and not to be missed. Med J Malaysia. 2014;69:95–97. [PubMed] [Google Scholar]
- 73.Wada R, Yagihashi S, Konta R, Ueda T, Izumiyama T. Gastric polyposis caused by multifocal histiocytosis X. Gut. 1992;33:994–996. doi: 10.1136/gut.33.7.994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yousem SA, Dacic S, Nikiforov YE, Nikiforova M. Pulmonary Langerhans cell histiocytosis: profiling of multifocal tumors using next-generation sequencing identifies concordant occurrence of BRAF V600E mutations. Chest. 2013;143:1679–1684. doi: 10.1378/chest.12-1917. [DOI] [PubMed] [Google Scholar]
- 75.Dimmler A, Geddert H, Werner M, Faller G. Molecular analysis of BRAF V600E mutation in multiple nodules of pulmonary Langerhans cell histiocytosis. Virchows Arch. 2017;470:429–435. doi: 10.1007/s00428-017-2092-4. [DOI] [PubMed] [Google Scholar]
- 76.Mourah S, How-Kit A, Meignin V, Gossot D, Lorillon G, Bugnet E, Mauger F, Lebbe C, Chevret S, Tost J, Tazi A. Recurrent NRAS mutations in pulmonary Langerhans cell histiocytosis. Eur Respir J. 2016;47:1785–1796. doi: 10.1183/13993003.01677-2015. [DOI] [PubMed] [Google Scholar]
- 77.Sakata N, Toguchi N, Kimura M, Nakayama M, Kawa K, Takemura T. Development of Langerhans cell histiocytosis associated with chronic active Epstein-Barr virus infection. Pediatr Blood Cancer. 2008;50:924–927. doi: 10.1002/pbc.21249. [DOI] [PubMed] [Google Scholar]
- 78.Chen CJ, Ho TY, Lu JJ, Sheu LF, Lee SY, Tien CH, Cheng SN. Identical twin brothers concordant for Langerhans’ cell histiocytosis and discordant for Epstein-Barr virus-associated haemophagocytic syndrome. Eur J Pediatr. 2004;163:536–539. doi: 10.1007/s00431-004-1493-y. [DOI] [PubMed] [Google Scholar]
- 79.Yousem SA, Colby TV, Chen YY, Chen WG, Weiss LM. Pulmonary Langerhans’ cell histiocytosis: molecular analysis of clonality. Am J Surg Pathol. 2001;25:630–636. doi: 10.1097/00000478-200105000-00010. [DOI] [PubMed] [Google Scholar]
- 80.Tazi A, Hiltermann JN, Vassallo R. Adult lung histiocytosis. In: Weitzman S, Egeler RM, editors. Histiocytic disorders of children and adults. Cambridge: Cambridge university press; 2005. pp. 187–207. [Google Scholar]
- 81.Von Essen S, West W, Sitorius M, Rennard SI. Complete resolution of roentgenographic changes in a patient with pulmonary histiocytosis X. Chest. 1990;98:765–767. doi: 10.1378/chest.98.3.765. [DOI] [PubMed] [Google Scholar]
- 82.Elia D, Torre O, Cassandro R, Caminati A, Harari S. Pulmonary Langerhans cell histiocytosis: a comprehensive analysis of 40 patients and literature review. Eur J Intern Med. 2015;26:351–356. doi: 10.1016/j.ejim.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 83.Tazi A, de Margerie C, Naccache JM, Fry S, Dominique S, Jouneau S, Lorillon G, Bugnet E, Chiron R, Wallaert B, et al. The natural history of adult pulmonary Langerhans cell histiocytosis: a prospective multicentre study. Orphanet J Rare Dis. 2015;10:30. doi: 10.1186/s13023-015-0249-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nichols KE, Arceci RJ. BRAF, a piece of the LCH puzzle. Blood. 2010;116:1825–1827. doi: 10.1182/blood-2010-06-289934. [DOI] [PubMed] [Google Scholar]
- 85.Berres ML, Lim KP, Peters T, Price J, Takizawa H, Salmon H, Idoyaga J, Ruzo A, Lupo PJ, Hicks MJ, et al. BRAF-V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups. J Exp Med. 2014;211:669–683. doi: 10.1084/jem.20130977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Coste I, Le Corf K, Kfoury A, Hmitou I, Druillennec S, Hainaut P, Eychene A, Lebecque S, Renno T. Dual function of MyD88 in RAS signaling and inflammation, leading to mouse and human cell transformation. J Clin Invest. 2010;120:3663–3667. doi: 10.1172/JCI42771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Muda M, Boschert U, Dickinson R, Martinou JC, Martinou I, Camps M, Schlegel W, Arkinstall S. MKP-3, a novel cytosolic protein-tyrosine phosphatase that exemplifies a new class of mitogen-activated protein kinase phosphatase. J Biol Chem. 1996;271:4319–4326. doi: 10.1074/jbc.271.8.4319. [DOI] [PubMed] [Google Scholar]
- 88.Yeh I, von Deimling A, Bastian BC. Clonal BRAF mutations in melanocytic nevi and initiating role of BRAF in melanocytic neoplasia. J Natl Cancer Inst. 2013;105:917–919. doi: 10.1093/jnci/djt119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chan TL, Zhao W, Leung SY, Yuen ST, Cancer Genome P. BRAF and KRAS mutations in colorectal hyperplastic polyps and serrated adenomas. Cancer Res. 2003;63:4878–4881. [PubMed] [Google Scholar]
- 90.Griffiths DA, Abdul-Sada H, Knight LM, Jackson BR, Richards K, Prescott EL, Peach AH, Blair GE, Macdonald A, Whitehouse A. Merkel cell polyomavirus small T antigen targets the NEMO adaptor protein to disrupt inflammatory signaling. J Virol. 2013;87:13853–13867. doi: 10.1128/JVI.02159-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shahzad N, Shuda M, Gheit T, Kwun HJ, Cornet I, Saidj D, Zannetti C, Hasan U, Chang Y, Moore PS, et al. The T antigen locus of Merkel cell polyomavirus downregulates human toll-like receptor 9 expression. J Virol. 2013;87:13009–13019. doi: 10.1128/JVI.01786-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kean JM, Rao S, Wang M, Garcea RL. Seroepidemiology of human polyomaviruses. PLoS Pathog. 2009;5:e1000363. doi: 10.1371/journal.ppat.1000363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tolstov YL, Pastrana DV, Feng H, Becker JC, Jenkins FJ, Moschos S, Chang Y, Buck CB, Moore PS. Human Merkel cell polyomavirus infection II. MCV is a common human infection that can be detected by conformational capsid epitope immunoassays. Int J Cancer. 2009;125:1250–1256. doi: 10.1002/ijc.24509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Foulongne V, Kluger N, Dereure O, Mercier G, Moles JP, Guillot B, Segondy M. Merkel cell polyomavirus in cutaneous swabs. Emerg Infect Dis. 2010;16:685–687. doi: 10.3201/eid1604.091278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Foulongne V, Courgnaud V, Champeau W, Segondy M. Detection of Merkel cell polyomavirus on environmental surfaces. J Med Virol. 2011;83:1435–1439. doi: 10.1002/jmv.22110. [DOI] [PubMed] [Google Scholar]
- 96.Pancaldi C, Corazzari V, Maniero S, Mazzoni E, Comar M, Martini F, Tognon M. Merkel cell polyomavirus DNA sequences in the buffy coats of healthy blood donors. Blood. 2011;117:7099–7101. doi: 10.1182/blood-2010-09-310557. [DOI] [PubMed] [Google Scholar]
- 97.Mertz KD, Junt T, Schmid M, Pfaltz M, Kempf W. Inflammatory monocytes are a reservoir for Merkel cell polyomavirus. J Invest Dermatol. 2010;130:1146–1151. doi: 10.1038/jid.2009.392. [DOI] [PubMed] [Google Scholar]
- 98.Loiarro M, Ruggiero V, Sette C. Targeting TLR/IL-1R signalling in human diseases. Mediat Inflamm. 2010;2010:674363. doi: 10.1155/2010/674363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- 100.He Y, Franchi L, Nunez G. TLR agonists stimulate Nlrp3-dependent IL-1beta production independently of the purinergic P2X7 receptor in dendritic cells and in vivo. J Immunol. 2013;190:334–339. doi: 10.4049/jimmunol.1202737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Brereton CF, Sutton CE, Lalor SJ, Lavelle EC, Mills KH. Inhibition of ERK MAPK suppresses IL-23- and IL-1-driven IL-17 production and attenuates autoimmune disease. J Immunol. 2009;183:1715–1723. doi: 10.4049/jimmunol.0803851. [DOI] [PubMed] [Google Scholar]
- 102.Brown RE. The NF-kappaB pathway and the successful application of anti-inflammatory and angiostatic therapy in Langerhans’ cell histiocytosis. Br J Haematol. 2005;130:147–148. doi: 10.1111/j.1365-2141.2005.05575.x. [DOI] [PubMed] [Google Scholar]
- 103.Brown RE. Brief communication: morphoproteomic analysis of osteolytic Langerhans cell histiocytosis with therapeutic implications. Ann Clin Lab Sci. 2005;35:131–136. [PubMed] [Google Scholar]
- 104.Fettelschoss A, Kistowska M, LeibundGut-Landmann S, Beer HD, Johansen P, Senti G, Contassot E, Bachmann MF, French LE, Oxenius A, Kundig TM. Inflammasome activation and IL-1beta target IL-1alpha for secretion as opposed to surface expression. Proc Natl Acad Sci USA. 2011;108:18055–18060. doi: 10.1073/pnas.1109176108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Akira S, Yamamoto M, Takeda K. Role of adapters in toll-like receptor signalling. Biochem Soc Trans. 2003;31:637–642. doi: 10.1042/bst0310637. [DOI] [PubMed] [Google Scholar]
- 106.Janssens S, Beyaert R. A universal role for MyD88 in TLR/IL-1R-mediated signaling. Trends Biochem Sci. 2002;27:474–482. doi: 10.1016/S0968-0004(02)02145-X. [DOI] [PubMed] [Google Scholar]
- 107.Vale T, Ngo TT, White MA, Lipsky PE. Raf-induced transformation requires an interleukin 1 autocrine loop. Cancer Res. 2001;61:602–607. [PubMed] [Google Scholar]
- 108.Murakami I, Matsushita M, Iwasaki T, Kuwamoto S, Kato M, Nagata K, Horie Y, Hayashi K, Imamura T, Morimoto A, et al. Interleukin-1 loop model for pathogenesis of Langerhans cell histiocytosis. Cell Commun Signal. 2015;13:13. doi: 10.1186/s12964-015-0092-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pineiro M, Alava MA, Gonzalez-Ramon N, Osada J, Lasierra P, Larrad L, Pineiro A, Lampreave F. ITIH4 serum concentration increases during acute-phase processes in human patients and is up-regulated by interleukin-6 in hepatocarcinoma HepG2 cells. Biochem Biophys Res Commun. 1999;263:224–229. doi: 10.1006/bbrc.1999.1349. [DOI] [PubMed] [Google Scholar]
- 110.Braciale TJ, Hahn YS, Burton DR: Adaptive immune response to viral infections. In Knipe DM, Howley PM, editors. Fields Virology. 6th ed. edition. Philadelphia: Lippincott Williams & Wilkins, a Wolter Kluwer business; 2013. p.214–253.
- 111.Pineiro M, Andres M, Iturralde M, Carmona S, Hirvonen J, Pyorala S, Heegaard PM, Tjornehoj K, Lampreave F, Pineiro A, Alava MA. ITIH4 (inter-alpha-trypsin inhibitor heavy chain 4) is a new acute-phase protein isolated from cattle during experimental infection. Infect Immun. 2004;72:3777–3782. doi: 10.1128/IAI.72.7.3777-3782.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Martel-Jantin C, Pedergnana V, Nicol JT, Leblond V, Tregouet DA, Tortevoye P, Plancoulaine S, Coursaget P, Touze A, Abel L, Gessain A. Merkel cell polyomavirus infection occurs during early childhood and is transmitted between siblings. J Clin Virol. 2013;58:288–291. doi: 10.1016/j.jcv.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 113.Hall CB, Weinberg GA, Iwane MK, Blumkin AK, Edwards KM, Staat MA, Auinger P, Griffin MR, Poehling KA, Erdman D, et al. The burden of respiratory syncytial virus infection in young children. N Engl J Med. 2009;360:588–598. doi: 10.1056/NEJMoa0804877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Glezen WP, Taber LH, Frank AL, Kasel JA. Risk of primary infection and reinfection with respiratory syncytial virus. Am J Dis Child. 1986;140:543–546. doi: 10.1001/archpedi.1986.02140200053026. [DOI] [PubMed] [Google Scholar]
- 115.Bravender T. Epstein-Barr virus, cytomegalovirus, and infectious mononucleosis. Adolesc Med State Art Rev. 2010;21:251–264. [PubMed] [Google Scholar]
- 116.Kumar A, Chen T, Pakkanen S, Kantele A, Soderlund-Venermo M, Hedman K, Franssila R. T-helper cell-mediated proliferation and cytokine responses against recombinant Merkel cell polyomavirus-like particles. PLoS One. 2011;6:e25751. doi: 10.1371/journal.pone.0025751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kollmann TR, Levy O, Montgomery RR, Goriely S. Innate immune function by toll-like receptors: distinct responses in newborns and the elderly. Immunity. 2012;37:771–783. doi: 10.1016/j.immuni.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Murakami I, Gogusev J, Fournet JC, Glorion C, Jaubert F. Detection of molecular cytogenetic aberrations in langerhans cell histiocytosis of bone. Hum Pathol. 2002;33:555–560. doi: 10.1053/hupa.2002.124035. [DOI] [PubMed] [Google Scholar]
- 119.Vassallo R, Walters PR, Lamont J, Kottom TJ, Yi ES, Limper AH. Cigarette smoke promotes dendritic cell accumulation in COPD; a lung tissue research consortium study. Respir Res. 2010;11:45. doi: 10.1186/1465-9921-11-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kansal R, Quintanilla-Martinez L, Datta V, Lopategui J, Garshfield G, Nathwani BN. Identification of the V600D mutation in exon 15 of the BRAF oncogene in congenital, benign langerhans cell histiocytosis. Genes Chromosomes Cancer. 2013;52:99–106. doi: 10.1002/gcc.22010. [DOI] [PubMed] [Google Scholar]
- 121.Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–724. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 122.Chilosi M, Facchetti F, Calio A, Zamo A, Brunelli M, Martignoni G, Rossi A, Montagna L, Piccoli P, Dubini A, et al. Oncogene-induced senescence distinguishes indolent from aggressive forms of pulmonary and non-pulmonary Langerhans cell histiocytosis. Leuk Lymphoma. 2014;55:2620–2626. doi: 10.3109/10428194.2014.887713. [DOI] [PubMed] [Google Scholar]
- 123.Mehes G, Irsai G, Bedekovics J, Beke L, Fazakas F, Rozsa T, Kiss C. Activating BRAF V600E mutation in aggressive pediatric Langerhans cell Histiocytosis: demonstration by allele-specific PCR/direct sequencing and immunohistochemistry. Am J Surg Pathol. 2014;38:1644–1648. doi: 10.1097/PAS.0000000000000304. [DOI] [PubMed] [Google Scholar]
- 124.Chellapandian D, Makras P, Kaltsas G, van den Bos C, Naccache L, Rampal R, Carret AS, Weitzman S, Egeler RM, Abla O. Bisphosphonates in Langerhans Cell Histiocytosis: An International Retrospective Case Series. Mediterr J Hematol Infect Dis. 2016;8:e2016033. doi: 10.4084/mjhid.2016.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.da Costa CE, Annels NE, Faaij CM, Forsyth RG, Hogendoorn PC, Egeler RM. Presence of osteoclast-like multinucleated giant cells in the bone and nonostotic lesions of Langerhans cell histiocytosis. J Exp Med. 2005;201:687–693. doi: 10.1084/jem.20041785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Takayanagi H. New developments in osteoimmunology. Nat Rev Rheumatol. 2012;8:684–689. doi: 10.1038/nrrheum.2012.167. [DOI] [PubMed] [Google Scholar]
- 127.Takayanagi H. Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat Rev Immunol. 2007;7:292–304. doi: 10.1038/nri2062. [DOI] [PubMed] [Google Scholar]
- 128.Asagiri M, Hirai T, Kunigami T, Kamano S, Gober HJ, Okamoto K, Nishikawa K, Latz E, Golenbock DT, Aoki K, et al. Cathepsin K-dependent toll-like receptor 9 signaling revealed in experimental arthritis. Science. 2008;319:624–627. doi: 10.1126/science.1150110. [DOI] [PubMed] [Google Scholar]
- 129.Theill LE, Boyle WJ, Penninger JM. RANK-L and RANK: T cells, bone loss, and mammalian evolution. Annu Rev Immunol. 2002;20:795–823. doi: 10.1146/annurev.immunol.20.100301.064753. [DOI] [PubMed] [Google Scholar]
- 130.Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, Bonewald LF, Kodama T, Wutz A, Wagner EF, et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med. 2011;17:1231–1234. doi: 10.1038/nm.2452. [DOI] [PubMed] [Google Scholar]
- 131.Ishii R, Morimoto A, Ikushima S, Sugimoto T, Asami K, Bessho F, Kudo K, Tsunematu Y, Fujimoto J, Imashuku S. High serum values of soluble CD154, IL-2 receptor, RANKL and osteoprotegerin in Langerhans cell histiocytosis. Pediatr Blood Cancer. 2006;47:194–199. doi: 10.1002/pbc.20595. [DOI] [PubMed] [Google Scholar]
- 132.Tan W, Zhang W, Strasner A, Grivennikov S, Cheng JQ, Hoffman RM, Karin M. Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling. Nature. 2011;470:548–553. doi: 10.1038/nature09707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kamizono J, Okada Y, Shirahata A, Tanaka Y. Bisphosphonate induces remission of refractory osteolysis in langerhans cell histiocytosis. J Bone Miner Res. 2002;17:1926–1928. doi: 10.1359/jbmr.2002.17.11.1926. [DOI] [PubMed] [Google Scholar]
- 134.Morimoto A, Shioda Y, Imamura T, Kanegane H, Sato T, Kudo K, Nakagawa S, Nakadate H, Tauchi H, Hama A, et al. Nationwide survey of bisphosphonate therapy for children with reactivated Langerhans cell histiocytosis in Japan. Pediatr Blood Cancer. 2011;56:110–115. doi: 10.1002/pbc.22703. [DOI] [PubMed] [Google Scholar]
- 135.Lee JS, Ko GH, Kim HC, Jang IS, Jeon KN, Lee JH. Langerhans cell sarcoma arising from Langerhans cell histiocytosis: a case report. J Korean Med Sci. 2006;21:577–580. doi: 10.3346/jkms.2006.21.3.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Mourah S, Lorillon G, Meignin V, Vercellino L, de Margerie-Mellon C, Pages C, Goldwirt L, How-Kit A, Tost J, Lebbe C, Tazi A. Dramatic transient improvement of metastatic BRAF(V600E)-mutated Langerhans cell sarcoma under treatment with dabrafenib. Blood. 2015;126:2649–2652. doi: 10.1182/blood-2015-06-650036. [DOI] [PubMed] [Google Scholar]
- 137.Chen W, Jaffe R, Zhang L, Hill C, Block AM, Sait S, Song B, Liu Y, Cai D. Langerhans cell sarcoma arising from chronic lymphocytic lymphoma/small lymphocytic leukemia: lineage analysis and BRAF V600E mutation study. N Am J Med Sci. 2013;5:386–391. doi: 10.4103/1947-2714.114172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Murakami I, Oh Y, Morimoto A, Sano H, Kanzaki S, Matsushita M, Iwasaki T, Kuwamoto S, Kato M, Nagata K, et al. Acute-phase ITIH4 levels distinguish multi-system from single-system Langerhans cell histiocytosis via plasma peptidomics. Clin Proteomics. 2015;12:16. doi: 10.1186/s12014-015-9089-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.