

Abstract
Oxidation of biogenic volatile organic compounds (BVOC) by the nitrate radical (NO3) represents one of the important interactions between anthropogenic emissions related to combustion and natural emissions from the biosphere. This interaction has been recognized for more than 3 decades, during which time a large body of research has emerged from laboratory, field, and modeling studies. NO3-BVOC reactions influence air quality, climate and visibility through regional and global budgets for reactive nitrogen (particularly organic nitrates), ozone, and organic aerosol. Despite its long history of research and the significance of this topic in atmospheric chemistry, a number of important uncertainties remain. These include an incomplete understanding of the rates, mechanisms, and organic aerosol yields for NO3-BVOC reactions, lack of constraints on the role of heterogeneous oxidative processes associated with the NO3 radical, the difficulty of characterizing the spatial distributions of BVOC and NO3 within the poorly mixed nocturnal atmosphere, and the challenge of constructing appropriate boundary layer schemes and non-photochemical mechanisms for use in state-of-the-art chemical transport and chemistry–climate models.
This review is the result of a workshop of the same title held at the Georgia Institute of Technology in June 2015. The first half of the review summarizes the current literature on NO3-BVOC chemistry, with a particular focus on recent advances in instrumentation and models, and in organic nitrate and secondary organic aerosol (SOA) formation chemistry. Building on this current understanding, the second half of the review outlines impacts of NO3-BVOC chemistry on air quality and climate, and suggests critical research needs to better constrain this interaction to improve the predictive capabilities of atmospheric models.
1 Introduction
The emission of hydrocarbons from the terrestrial biosphere represents a large natural input of chemically reactive compounds to Earth’s atmosphere (Guenther et al., 1995; Goldstein and Galbally, 2007). Understanding the atmospheric degradation of these species is a critical area of current research that influences models of oxidants and aerosols on regional and global scales. Nitrogen oxides (NOx = NO + NO2) arising from combustion and microbial action on fertilizer are one of the major anthropogenic inputs that perturb the chemistry of the atmosphere (Crutzen, 1973). Nitrogen oxides have long been understood to influence oxidation cycles of biogenic volatile organic compounds (BVOC), especially through photochemical reactions of organic and hydroperoxy radical intermediates (RO2 and HO2) with nitric oxide (NO) (Chameides, 1978).
The nitrate radical (NO3) arises from the oxidation of nitrogen dioxide (NO2) by ozone (O3) and occurs principally in the nighttime atmosphere due to its rapid photolysis in sunlight and its reaction with NO (Wayne et al., 1991; Brown and Stutz, 2012). The nitrate radical is a strong oxidant, reacting with a wide variety of volatile organic compounds, including alkenes, aromatics, and oxygenates as well as with reduced sulfur compounds. Reactions of NO3 are particularly rapid with unsaturated compounds (alkenes) (Atkinson and Arey, 2003). BVOC such as isoprene, monoterpenes, and sesquiterpenes typically have one or more unsaturated functionalities such that they are particularly susceptible to oxidation by O3 and NO3.
The potential for NO3 to serve as a large sink for BVOC was recognized more than 3 decades ago (Winer et al., 1984). Field studies since that time have shown that in any environment with moderate to large BVOC concentrations, a majority of the NO3 radical oxidative reactions are with BVOC rather than VOC of anthropogenic origin (Brown and Stutz, 2012). This interaction gives rise to a mechanism that couples anthropogenic NOx emissions with natural BVOC emissions (Fry et al., 2009; Xu et al., 2015a). Although it is one of several such anthropogenic–biogenic interactions (Hoyle et al., 2011), reactions of NO3 with BVOC are an area of intense current interest and one whose study has proven challenging. These challenges arise from the more limited current database of laboratory data for NO3 oxidation reactions relative to those of other common atmospheric oxidants such as hydroxyl radical (OH) and O3. The mixing state of the night-time atmosphere and the limitations it imposes for characterization of nocturnal oxidation chemistry during field measurements and within atmospheric models present a second challenge to this field of research. Figure 1 illustrates these features of nighttime NO3-BVOC chemistry.
Figure 1.
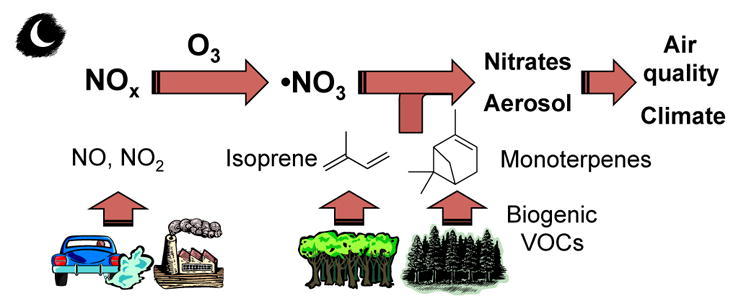
Schematic of nighttime NO3-BVOC chemistry.
Reactions of NO3 with BVOC have received increased attention in the recent literature as a potential source of secondary organic aerosol (SOA) (Pye et al., 2010; Fry et al., 2014; Boyd et al., 2015). This SOA source is intriguing for several reasons. First, although organics are now understood to comprise a large fraction of total aerosol mass, and although much of these organics are secondary, sources of SOA remain difficult to characterize, in part due to a large number of emission sources and potential chemical mechanisms (Zhang et al., 2007; Hallquist et al., 2009; Jimenez et al., 2009; Ng et al., 2010). Analysis of aerosol organic carbon shows that a large fraction is modern, arising either from biogenic hydrocarbon emissions or biomass burning sources (e.g., Schichtel et al., 2008; Hodzic et al., 2010). Conversely, field data in regionally polluted areas indicate strong correlations between tracers of anthropogenic emissions and SOA, which suggests that anthropogenic influences can lead to production of SOA from modern (i.e., non-fossil) carbon (e.g., Weber et al., 2007). Model studies confirm that global observations are best simulated with a biogenic carbon source in the presence of anthropogenic pollutants (Spracklen et al., 2011). Reactions of NO3 with BVOC are one such mechanism that may lead to anthropogenically influenced biogenic SOA (Hoyle et al., 2007), and it is important to quantify the extent to which such reactions can explain sources of SOA.
Second, some laboratory and chamber studies suggest that SOA yields from NO3 oxidation of common BVOC, such as isoprene and selected monoterpenes, are greater than that for OH or O3 oxidation (Hallquist et al., 1997b; Griffin et al., 1999; Spittler et al., 2006; Ng et al., 2008; Fry et al., 2009, 2011, 2014;; Rollins et al., 2009; Boyd et al., 2015). However, among the monoterpenes, the SOA yields may be much more variable for NO3 oxidation than for other oxidants, with anomalously low SOA yields in some cases and high SOA yields in others (Draper et al., 2015; Nah et al., 2016b).
Third, not only is NO3-BVOC chemistry a potentially efficient SOA formation mechanism, it is also a major pathway for the production of organic nitrates (von Kuhlmann et al., 2004; Horowitz et al., 2007), a large component of oxidized reactive nitrogen that may serve as either a NOx reservoir or NOx sink. Results from recent field measurements have shown that organic nitrates are important components of ambient OA (Day et al., 2010; Rollins et al., 2012; Fry et al., 2013; Ayres et al., 2015; Xu et al., 2015a, b; Kiendler-Scharr et al., 2016; Lee et al., 2016). Furthermore, within the last several years, the capability to measure both total and speciated gas-phase and particle-phase organic nitrates has been demonstrated (Fry et al., 2009, 2013, 2014; Rollins et al., 2010, 2013; Lee et al., 2016; Nah et al., 2016b). The life-times of organic nitrates derived from BVOC-NO3 reaction with respect to hydrolysis, photooxidation, and deposition play an important role in the NOx budget and formation of O3 and SOA. These processes appear to depend strongly on the parent VOC and oxidation conditions and must be better constrained for understanding organic nitrate lifetimes in the atmosphere (Darer et al., 2011; Hu et al., 2011; Liu et al., 2012b; Boyd et al., 2015; Pye et al., 2015; Rindelaub et al., 2015; Lee et al., 2016; Nah et al., 2016b).
Fourth, incorporation of SOA yields for NO3-BVOC reactions into regional and global models indicates that these reactions could be a significant, or in some regions even dominant, SOA contributor (Hoyle et al., 2007; Pye et al., 2010, 2015; Chung et al., 2012; Fry and Sackinger, 2012; Kiendler-Scharr et al., 2016). Model predictions of organic aerosol formation from NO3-BVOC until recently have been difficult to verify directly from field measurements. Recent progress in laboratory and field studies have provided some of the first opportunities to develop coupled gas and particle systems to describe mechanistically and predict SOA and organic nitrate formation from NO3-BVOC reactions (Pye et al., 2015).
Finally, analyses from several recent field studies examining diurnal variation in the organic and/or nitrate content of aerosols conclude that nighttime BVOC oxidation through NO3 radicals constitutes a large organic aerosol source (Rollins et al., 2012; Fry et al., 2013; Xu et al., 2015a, b; Kiendler-Scharr et al., 2016). Although such analyses may correct their estimates of aerosol production for the variation in boundary layer depth, field measurements at surface level are necessarily limited in their ability to accurately assess the atmospheric chemistry in the overlying residual layer or even the gradients that may exist within the relatively shallow nocturnal boundary layer (Stutz et al., 2004; Brown et al., 2007b). Thus, although there is apparent consistency between recent results from both modeling and field studies, the vertically stratified structure of the nighttime atmosphere makes such comparisons difficult to evaluate critically. There is a limited database of nighttime aircraft measurements that has probed this vertical structure with sufficient chemical detail to assess NO3-BVOC reactions (Brown et al., 2007a; Brown et al., 2009), and some of these data show evidence for an OA source related to this chemistry, especially at low altitude (Brown et al., 2013). A larger database of aircraft and/or vertically resolved measurements is required, however, for comprehensive comparisons to model predictions.
The purpose of this article is to review the current literature on the chemistry of NO3 and BVOC to critically assess the current state of the science. The review focuses on BVOC emitted from terrestrial vegetation. The importance of NO3 reactions with reduced sulfur compounds, such as dimethyl sulfide in marine ecosystems, is well known (Platt et al., 1990; Yvon et al., 1996; Allan et al., 1999, 2000; Vrekoussis et al., 2004; Stark et al., 2007; Osthoff et al., 2009) but is outside of the scope of this review. Key uncertainties include chemical mechanisms, yields of major reaction products such as SOA and organic nitrogen, the potential for NO3 and BVOC to interact in the ambient atmosphere, and the implications of that interaction for current understanding of air quality and climate. The review stems from an International Global Atmospheric Chemistry (IGAC) and US National Science Foundation (NSF) sponsored workshop of the same name held in June 2015 at the Georgia Institute of Technology (Atlanta, GA, USA). Following this introduction, Sect. 2 of this article reviews the current literature in several areas relevant to the understanding of NO3-BVOC atmospheric chemistry. Section 3 outlines perspectives on the implications of this chemistry for understanding climate and air quality, its response to current emission trends, and its relevance to implementation of control strategies. Finally, the review concludes with an assessment of the impacts of NO3-BVOC reactions on air quality, visibility, and climate.
2 Review of current literature
This section contains a literature review of the current state of knowledge of NO3-BVOC chemistry with respect to (1) reaction rate constants and mechanisms from laboratory and chamber studies; (2) secondary organic aerosol yields, speciation, and particle-phase chemistry; (3) heterogeneous reactions of NO3 and their implications for NO3-BVOC chemistry; (4) instrumental methods for analysis of reactive nitrogen compounds, including NO3, organic nitrates, and nitrogen-containing particulate matter; (5) field observations relevant to the understanding of NO3 and BVOC chemistry; and (6) models of NO3-BVOC chemistry.
2.1 NO3-BVOC reaction rates and chemical mechanisms
2.1.1 Reaction rates
Among the numerous BVOC emitted into the troposphere, kinetic data for NO3 oxidation have been provided for more than 40 compounds. The most emitted/important BVOC have been subject to several kinetic studies, using both absolute and relative methods, which are evaluated to determine rate constants by IUPAC (Table 1). This is the case for isoprene, α-pinene, β-pinene, and 2-methyl-3-buten-2-ol (MBO). However, for isoprene, β-pinene, and MBO, rate constants obtained by different studies range over a factor of 2. For some other terpenes, only few kinetic studies have been carried out, with at least one absolute rate determination. This is the case for sabinene, 2-carene, camphene, d-limonene, α-phellandrene, myrcene, γ -terpinene, and terpinolene. For these compounds, experimental data agree within 30–40 %, except α-phellandrene and terpinolene for which discrepancies are larger. For other BVOC, including other terpenes, sesquiterpenes, and oxygenated species, rate constants are mostly based on a single determination and highly uncertain. For these compounds, further rate constant determinations and end-product measurements are essential to better evaluate the role of NO3 in their degradation. The ability to predict the NO3-BVOC rate constants using structure–activity relationships (SARs) has been improved. A recent study (Kerdouci et al., 2010; Kerdouci, 2014) presented a new SAR parameterization based on 180 NO3-VOC reactions. The method is capable of predicting 90 % of the rate constants within a factor of 2.
Table 1.
Reaction rate constants of NO3+ BVOC.
| Compound | k(NO3+BVOC) (cm3 molecule−1 s−1)a | Temperature (K) | Technique/reference |
|---|---|---|---|
|
| |||
Isoprene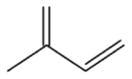
|
(5.94 ± 0.16)×10−13 | 295 | RR/(Atkinson et al., 1984) |
| (1.30 ± 0.14)×10−12 | 298 | DF-MS/(Benter and Schindler, 1988) | |
| (3.03 ± 0.45)×10−12exp[−(450 ± 70)/T] | 251–281 | F-LIF/(Dlugokencky and Howard, 1989) | |
| (6.52 ± 0.78)×10−13 | 297 | F-LIF/(Dlugokencky and Howard, 1989) | |
| (1.21 ± 0.20)×10−12 | 298 | RR/(Barnes et al., 1990) | |
| (7.30 ± 0.44)×10−13 | 298 | DF-MS/(Wille et al., 1991) | |
| (8.26 ± 0.60)×10−13 | 298 | DF-MS/(Wille et al., 1991) | |
| (1.07 ± 0.20)×10−12 | 295 | PR-A/(Ellermann et al., 1992) | |
| (6.86 ± 0.55)×10−13 | 298 | RR/(Berndt and Boge, 1997b) | |
| (7.3 ± 0.2)×10−13 | 298 | F-CIMS/(Suh et al., 2001) | |
| (6.24 ± 0.11)×10−13 | 295 | RR/(Zhao et al., 2011b) | |
| 6.5×10−13 (Δlog k: ± 0.15) | 298 | IUPAC | |
|
| |||
α-pinene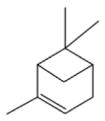
|
(5.82 ± 0.16)×10−12 | 295 | RR/(Atkinson et al., 1984) |
| (1.19 ± 0.31)×10−12exp[(490 ± 70)/T] | 261–383 | F-LIF/(Dlugokencky and Howard, 1989) | |
| (6.18 ± 0.94)×10−12 | 298 | F-LIF/(Dlugokencky and Howard, 1989) | |
| (6.56 ± 0.94)×10−12 | 298 | RR/(Barnes et al., 1990) | |
| (3.5 ± 1.4)×10−13exp[(841 ± 144)/T] | 298–423 | DF-LIF/(Martinez et al., 1998) | |
| (5.9 ± 0.8)×10−12 | 298 | DF-LIF/(Martinez et al., 1998) | |
| (5.82 ± 0.56)×10−12 | 298 | RR/(Kind et al., 1998) | |
| (4.88 ± 0.46)×10−12 | 298 | RR/(Stewart et al., 2013) | |
| 6.2×10−12 (Δlog k : ± 0.1) | 298 | IUPAC | |
|
| |||
β-pinene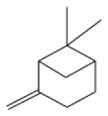
|
(2.36 ± 0.10)×10−12 | 295 | RR/(Atkinson et al., 1984) |
| (2.38 ± 0.05)×10−12 | 296 | RR/(Atkinson et al., 1988) | |
| (1.1 ± 0.4)×10−12 | 298 | RR/(Kotzias et al., 1989) | |
| (2.81 ± 0.47)×10−12 | 298 | RR/(Barnes et al., 1990) | |
| (1.6 ± 1.5)×10−10exp[(−1248 ± 36)/T] | 298–293 | DF-LIF/(Martinez et al., 1998) | |
| (2.1 ± 0.4)×10−12 | 298 | DF-LIF/(Martinez et al., 1998) | |
| (2.81 ± 0.56)×10−12 | 298 | RR/(Kind et al., 1998) | |
| 2.5×10−12 (Δlog k : ± 0.12) | 298 | IUPAC | |
|
| |||
Sabinene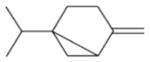
|
(1.01 ± 0.03)×10−11 | 296 | RR/(Atkinson et al., 1990) |
| (1.07 ± 0.16)×10−11 | 298 | DF-LIF/(Martinez et al., 1999) | |
| (2.3 ± 1.3)×10−10exp[(−940 ± 200)/T] | 298–393 | DF-LIF/(Martinez et al., 1999) | |
| 1.0×10−11 (Δlog k : ± 0.15) | 298 | IUPAC | |
|
| |||
Camphene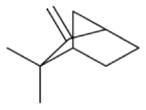
|
(6.54 ± 0.16)×10−13 | 296 | RR/(Atkinson et al., 1990) |
| (3.1 ± 0.5)×10−12exp[(−481 ± 55)/T] | 298–433 | DF-LIF/(Martinez et al., 1998) | |
| (6.2 ± 2.1)×10−13 | 298 | DF-LIF/(Martinez et al., 1998) | |
|
| |||
2-carene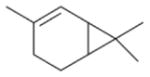
|
(1.87 ± 0.11)×10−11 | 295 | RR/(Corchnoy and Atkinson, 1990) |
| (2.16 ± 0.36)×10−11 | 295 | RR/(Corchnoy and Atkinson, 1990) | |
| (1.66 ± 0.18)×10−11 | 298 | DF-LIF/(Martínez et al., 1999) | |
| (1.4 ± 0.7)×10−12exp[(741 ± 190)/T] | 298–433 | DF-LIF/(Martínez et al., 1999) | |
| 2.0×10−11 (Δlog k : ± 0.12) | 298 | IUPAC | |
|
| |||
3-carene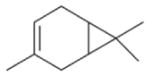
|
(1.01 ± 0.02)×10−11 | 295 | RR/(Atkinson et al., 1984) |
| (8.2 ± 1.2)×10−11 | 298 | RR/(Barnes et al., 1990) | |
| 9.1×10−11 (Δlog k : ± 0.12) | 298 | IUPAC | |
|
| |||
Δ-limonene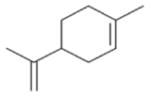
|
(1.31 ± 0.04)×10−11 | 295 | RR/(Atkinson et al., 1984) |
| (1.12 ± 0.17)×10−11 | 298 | RR/(Barnes et al., 1990) | |
| (9.4 ± 0.9)×10−12 | 298 | DF-LIF/(Martínez et al., 1999) | |
| 1.2×10−11 (Δlog k : ± 0.12) | 298 | IUPAC | |
|
| |||
α-phellandrene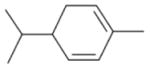
|
(8.52 ± 0.63)×10−11 | 294 | RR/(Atkinson et al., 1985) |
| (5.98 ± 0.20)×10−11 | 298 | RR/(Berndt et al., 1996) | |
| (4.2 ± 1.0)×10−11 | 298 | DF-LIF/(Martínez et al., 1999) | |
| (1.9 ± 1.3)×10−9exp[−(1158 ± 270)/T] | 298–433 | DF-LIF/(Martínez et al., 1999) | |
| 7.3×10−11 (Δlog k : ± 0.15) | 298 | IUPAC | |
|
| |||
β-phellandrene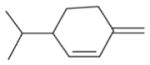
|
(7.96 ± 2.82)×10−12 | 297 | RR/(Shorees et al., 1991) |
|
| |||
α-terpinene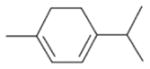
|
(1.82 ± 0.07)×10−10 | 294 | RR/(Atkinson et al., 1985) |
| (1.03 ± 0.06)×10−10 | 298 | RR/(Berndt et al., 1996) | |
| 1.8×10−10 (Δlog k : ± 0.25) | 298 | IUPAC | |
|
| |||
γ-terpinene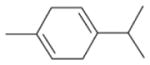
|
(2.94 ± 0.05)×10−11 | 294 | RR/(Atkinson et al., 1985) |
| (2.4 ± 0.7)×10−11 | 298 | DF-LIF/(Martínez et al., 1999) | |
| 2.9×10−11 (Δlog k : ± 0.12) | 298 | IUPAC | |
|
| |||
Terpinolene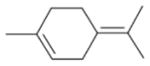
|
(9.67 ± 0.51)×10−11 | 295 | RR/(Corchnoy and Atkinson, 1990) |
| (5.2 ± 0.9)×10−11 | 298 | DF-LIF/(Martínez et al., 1999) | |
| (6.12 ± 0.52)×10−11 | 298 | RR/(Stewart et al., 2013) | |
| 9.7×10−11 (Δlog k : ± 0.25) | 298 | IUPAC | |
|
| |||
Ocimene (cis, trans)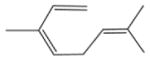
|
(2.23 ± 0.06)×10−11 | 294 | RR/(Atkinson et al., 1985) |
| 2.2×10−11 (Δlog k : ± 0.15) | 298 | IUPAC | |
|
| |||
Myrcene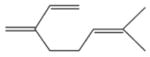
|
(1.06 ± 0.02)×10−11 | 294 | RR/(Atkinson et al., 1985) |
| (1.28 ± 0.11)×10−11 | 298 | DF-LIF/(Martínez et al., 1999) | |
| (2.2± 0.2)×10−12exp[(523 ± 35)/T] | 298–433 | DF-LIF/(Martínez et al., 1999) | |
| 1.1×10−11 (Δlog k : ± 0.12) | 298 | IUPAC | |
|
| |||
α-cedrene
|
(0.82 ± 0.30)×10−11 | 296 | RR/(Shu and Atkinson, 1995) |
|
| |||
α-copaene
|
(1.6 ± 0.6)×10−11 | 296 | RR/(Shu and Atkinson, 1995) |
|
| |||
β-caryophyllene
|
(1.9 ± 0.8)×10−11 | 296 | RR/(Shu and Atkinson, 1995) |
|
| |||
α-humulene
|
(3.5 ± 1.3)×10−11 | 296 | RR/(Shu and Atkinson, 1995) |
|
| |||
Longifolene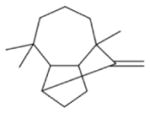
|
(6.8 ± 2.1)×10−13 | 296 | RR/(Shu and Atkinson, 1995) |
|
| |||
Isolongifolene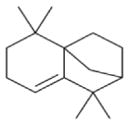
|
(3.9 ± 1.6)×10−12 | 298 | RR/(Canosa-Mas et al., 1999b) |
|
| |||
Alloisolongifolene
|
(1.4 ± 0.7)×10−12 | 298 | RR/(Canosa-Mas et al., 1999b) |
|
| |||
α-neoclovene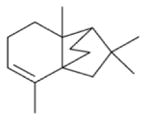
|
(8.2 ± 4.6)×10−12 | 298 | RR/(Canosa-Mas et al., 1999b) |
|
| |||
2-methyl-3-buten-2-ol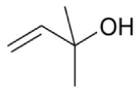
|
4.6×10−14exp[−(400 ± 35)/T] | 267–400 | F-A/(Rudich et al., 1996) |
| (1.21 ± 0.09)×10−14 | 298 | F-A/(Rudich et al., 1996) | |
| (2.1 ± 0.3)×10−14 | 294 | DF-A/(Hallquist et al., 1996) | |
| (1.55 ± 0.55)×10−14 | 294 | RR/(Hallquist et al., 1996) | |
| (8.7 ± 3.0)×10−14 | 298 | RR/(Fantechi et al., 1998b) | |
| (1.0 ± 0.2)×10−14 | 297 | RR/(Noda et al., 2002) | |
| (1.1 ± 0.1)×10−14 | 297 | RR/(Noda et al., 2002) | |
| 1.2×10−14 (Δlog k : ± 0.2) | 298 | IUPAC | |
|
| |||
3-methyl-2-buten-1-ol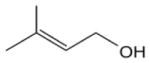
|
(1.0 ± 0.1)×10−12 | 297 | RR/(Noda et al., 2002) |
|
| |||
3-methyl-3-buten-1-ol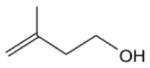
|
(2.7 ± 0.2)×10−13 | 297 | RR/(Noda et al., 2002) |
|
| |||
|
cis-3-hexen-1-ol |
(2.72 ± 0.83)×10−13 | 296 | RR/(Atkinson et al., 1995) |
| (2.67 ± 0.42)×10−13 | 298 | DF-CEAS/(Pfrang et al., 2006) | |
|
| |||
|
trans-3-hexen-1-ol |
(4.43 ± 0.91)×10−13 | 298 | DF-CEAS/(Pfrang et al., 2006) |
|
| |||
cis-4-hexen-1-ol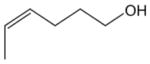
|
(2.93 ± 0.48)×10−13 | 298 | DF-CEAS/(Pfrang et al., 2006) |
|
| |||
|
trans-2-hexen-1-ol |
(1.30 ± 0.24)×10−13 | 298 | DF-CEAS/(Pfrang et al., 2006) |
|
| |||
|
cis-2-hexen-1-ol |
(1.56 ± 0.24)×10−13 | 298 | DF-CEAS/(Pfrang et al., 2006) |
|
| |||
|
trans-2-hexenal |
(1.21 ± 0.44)×10−14 | 296 | RR/(Atkinson et al., 1995) |
| (1.36 ± 0.29)×10−14) | 295 | RR/(Zhao et al., 2011b) | |
| (4.7 ± 1.5)×10−15 | 294 | AR/(Kerdouci et al., 2012) | |
|
| |||
| 4-methylenehex-5-enal |
(4.75 ± 0.35)×10−13 | 296 | RR/(Baker et al., 2004) |
|
| |||
| (3Z)-4-methylhexa-3,5-dienal |
(2.17 ± 0.30)×10−12 | 296 | RR/(Baker et al., 2004) |
|
| |||
(3E)-4-methylhexa-3,5-dienal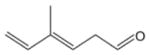
|
(1.75 ± 0.27)×10−12 | 296 | RR/(Baker et al., 2004) |
|
| |||
4-methylcyclohex-3-en-1-one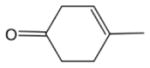
|
(1.81 ± 0.35)×10−12 | 296 | RR/(Baker et al., 2004) |
|
| |||
|
cis-3-hexenyl acetate |
(2.46 ± 0.75)×10−13 | 296 | RR/(Atkinson et al., 1995) |
|
| |||
Methyl vinyl ketone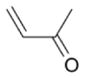
|
< 1.2×10−16 | 298 | F-A/(Rudich et al., 1996) |
| < 6×10−16 | 296 | DF- RR/(Kwok et al., 1996) | |
| (3.2 ± 0.6)×10−16 | 296 | LIF/(Canosa-Mas et al., 1999a) | |
| (5.0 ± 1.2)×10−16 | 296 | RR/(Canosa-Mas et al., 1999a) | |
| < 6×10−16 | 298 | IUPAC | |
|
| |||
Methacrolein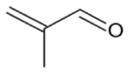
|
(4.46 ± 0.58)×10−15 | 296 | RR/(Kwok et al., 1996) |
| (3.08 ± 0.18)×10−15 | 298 | RR/(Chew et al., 1998) | |
| (3.50 ± 0.15)×10−15 | 298 | RR/(Chew et al., 1998) | |
| (3.72 ± 0.47)×10−15 | 296 | RR/(Canosa-Mas et al., 1999a) | |
| 3.4×10−15 (Δlog k : ± 0.15) | 298 | IUPAC | |
|
| |||
Pinonaldehyde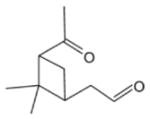
|
(2.40 ± 0.38)×10−14 | 299 | RR/(Hallquist et al., 1997a) |
| (6.0 ± 2.0)×10−14 | 300 | RR/(Glasius et al., 1997) | |
| (2.0 ± 0.9)×10−14 | 296 | RR/(Alvarado et al., 1998) | |
| 2.0×10−14 (Δlog k : ± 0.25) | 298 | IUPAC | |
|
| |||
Linalool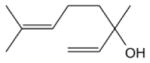
|
(1.12 ± 0.40)×10−11 | 296 | RR/(Atkinson et al., 1995) |
|
| |||
α-terpineol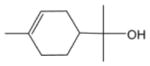
|
(1.6 ± 0.4)×10−11 | 297 | RR/(Jones and Ham, 2008) |
|
| |||
Sabinaketone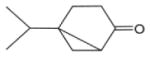
|
(3.6 ± 2.3)×10−16 | 296 | RR/(Alvarado et al., 1998) |
|
| |||
Caronaldehyde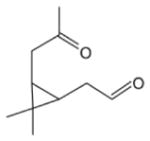
|
(2.5 ± 1.1)×10−14 | 296 | RR/(Alvarado et al., 1998) |
Given uncertainties are those provided by the authors of the kinetic studies. The procedures used to calculate them are not detailed here, as they often differ from one study to another. Readers are referred to the original papers for more information on the uncertainties’ determination.
RR: relative rate; DF-MS: discharge flow–mass spectrometry; DF-LIF: discharge flow–laser–induced fluorescence; DF-A: discharge flow–absorption; DF-CEAS: discharge flow–cavity–enhanced absorption spectroscopy; F-LIF: flow system–laser–induced fluorescence; F-CIMS: flow system–chemical ionization mass spectrometry; F-A: flow system–absorption; PR-A: pulse radiolysis–absorption; AR: absolute rate in simulation chamber.
2.1.2 Mechanisms
In general, NO3 reacts with unsaturated VOC by addition to a double bond (Wayne et al., 1991), though hydrogen abstraction may occur, most favorably for aldehydic species (Zhang and Morris, 2015). The location and likelihood of the NO3 addition to a double bond depends on the substitution on each end of the double bond, with the favored NO3 addition position being the one resulting in the most substituted carbon radical. In both cases, molecular oxygen adds to the resulting radical to form a peroxy radical (RO2). For example, the major RO2 isomers produced from isoprene and β-pinene oxidation via NO3 are shown in Fig. 2. The RO2 distribution for isoprene oxidation by OH has been shown to be dependent on the RO2 lifetime (Peeters et al., 2009, 2014), but no similar theoretical studies have been conducted on the NO3 system. Schwantes et al. (2015) determined the RO2 isomer distribution at an RO2 lifetime of ~ 30 s for isoprene oxidation via NO3. More theoretical and experimental studies are needed to understand the influence of RO2 lifetime, which is long at night (~ 50–200 s for isoprene; Schwantes et al., 2015), on the RO2 isomer distribution, as this distribution influences the formation of all subsequent products (Fig. 2).
Figure 2.

Condensed reaction mechanism for isoprene and β-pinene oxidation via NO3 (adapted from Schwantes et al., 2015 and Boyd et al., 2015). For brevity, only products generated from the dominant peroxy radicals (RO2) are shown. R′ represents an alkoxy radical, carbonyl compound, or hydroxy compound. Two of the largest uncertainties in β-pinene oxidation are shown in red: (1) quantification of product yields from the RO2+ HO2 channel and (2) identification of carbonyl products formed from RO2 reaction with NO3, RO2, or HO2 (see text for more details).
The fate of RO2 determines the subsequent chemistry. During the nighttime in the ambient atmosphere, RO2 will isomerize or react with another RO2, NO3, or HO2. In order to monitor RO2 isomerization reaction products, RO2 life-times must be long in laboratory studies similar to the ambient atmosphere (e.g., Peeters et al., 2009; Crounse et al., 2011). The NO3 plus BVOC (NO3+ BVOC) reaction can be a source of nighttime HO2 and OH radicals (Platt et al., 1990). Reaction with NO is a minor peroxy radical fate at night (Pye et al., 2015; Xiong et al., 2015). Few laboratory studies have contrasted the fates of RO2 and their impacts on gas-phase oxidation and aerosol formation (Ng et al., 2008; Boyd et al., 2015; Schwantes et al., 2015). Boyd et al. (2015) examined how RO2 fate influences SOA formation and yields, and studied the competition between the RO2-NO3 and RO2-HO2 channels for β-pinene. Boyd et al. (2015) determined that the SOA yields for both channels are comparable, indicating that the volatility distribution of products may not be very different for the different RO2 fates. In contrast, the results from NO3 oxidation of smaller BVOC, such as isoprene, show large differences in SOA yields depending on the RO2 fate (Ng et al., 2008), with larger SOA yields for second-generation NO3 oxidation (Rollins et al., 2009).
The well-established gas-phase first-generation products from the major β- and δ-RO2 isomers formed from isoprene oxidation are shown in Fig. 2 (adapted from Schwantes et al., 2015). Some of the products are common between all the pathways, such as methyl vinyl ketone for the dominant β-RO2 isomer. However, some products are unique to only one channel (e.g., hydroxy nitrates form from RO2-RO2 reactions and nitrooxy hydroperoxides form from RO2-HO2 reactions). In this case, the overall nitrate yield and the specific nitrates formed from isoprene depend on the initial RO2 isomer distribution and the fate of the RO2. Furthermore, the distribution of gas-phase products will then influence the formation of SOA. For isoprene, the SOA yields from RO2-RO2 reactions are ~ 2 times greater than the yield from RO2-NO3 reactions (Ng et al., 2008). The less well-established first-generation products from β-pinene oxidation are also shown in Fig. 2 (adapted from Boyd et al., 2015). There are still lingering uncertainties (shown in red) in the first-generation products formed from β-pinene oxidation. The product yields from the RO2+ HO2 channel are not well constrained, largely due to the unavailability of authentic standards. In Fig. 2, a carbonyl product is assumed to form directly from the RO2 + HO2 reaction instead of proceeding through an alkoxy intermediate consistent with theoretical calculations from different compounds (Hou et al., 2005a, b; Praske et al., 2015). This is also uncertain, as few theoretical studies have been conducted on large molecules like β-pinene. The identification of the carbonyl compound(s) produced from RO2 reaction with NO3, RO2, or HO2 is unknown. Hallquist et al. (1999) detected a low molar yield (0–2 %; Table 2) of nopinone from β-pinene NO3 oxidation. Further laboratory studies identifying other carbonyl products are recommended.
Table 2.
Oxidation products and SOA yields observed in previous studies of NO3-BVOC reactions. Except where noted, carbonyl and organic nitrate molar yields represent initial gas-phase yields measured by FTIR spectroscopy (carbonyl and organic nitrate) or thermal desorption laser-induced fluorescence (TD-LIF) (organic nitrate only; Rollins et al., 2010; Fry et al., 2013). In some cases, the ranges reported correspond to wide ranges of organic aerosol loading, listed in the rightmost column. Where possible, the mass yield at 10 μg m−3 is reported for ease of comparison.
| BVOC | Carbonyl molar yield | Organic nitrate molar yield | SOA mass yield | Corresponding OA loading or other relevant information |
|---|---|---|---|---|
| Isoprene | 62–78 % (Rollins et al., 2009) | 2 % (14 % after further oxidation) (Rollins et al., 2009) | Nucleation (1 μg m−3) | |
| 4–24 % (Ng et al., 2008) | 3–70 μg m−3; 12 % at 10 μg m −3 | |||
|
| ||||
| α-pinene | 58–66 % (Wangberg et al., 1997); 69–81 % (Berndt and Boge, 1997a); 65–72 % (Hallquist et al., 1999); 39–58 % (Spittler et al., 2006) | 14 % (Wangberg et al., 1997); 12–18 % (Berndt and Boge, 1997b); 18–25 % (Hallquist et al., 1999); 11–29 % (Spittler et al., 2006); 10 % (Fry et al., 2014) | 0.2–16 % (Hallquist et al., 1999) | Nucleation; 0.5 % at 10 ppt N2O5 reacted, 7 % at 100 ppt N2O5 reacteda |
| 4 or 16 % (Spittler et al., 2006) | Values for 20 % RH and dry conditions, respectively, at M∞b | |||
| 1.7–3.6 % (Nah et al., 2016a) | 1.2–2.5 μg m−3 | |||
| 0 % (Fry et al., 2014) | Both nucleation and ammonium sulfate seeded | |||
| 9 % (Perraud et al., 2010) | Nucleation at 1 ppm N2O5 and 1 ppm α-pinene; OA is 480 μg m−3 assuming density is1.235 g cm−3 | |||
|
| ||||
| β-pinene | 0–2 % (Hallquist et al., 1999) | 51–74 % (Hallquist et al., 1999); 40 % (Fry et al., 2009); 22 % (Fry et al., 2014); 45–74 % of OA mass (Boyd et al., 2015) | 32–89 % (Griffin et al., 1999) | 32–470 μg m−3 ; low end closest to 10 μg m− 3 |
| 7–40 % (Moldanova and Ljungstrom, 2000) using new model to reinterpret data from Hallquist et al. (1999) (10–52 %) | 7–10 % at 7 ppt N2O5 reacted; 40–52 % at 39 ppt N2O5 reacted | |||
| 50 % (Fry et al., 2009) | 40 μg m−3; same yield at both 0 and 60 % RH | |||
| 33–44 % (Fry et al., 2014) | 10 μg m−3 c | |||
| 27–104 % (Boyd et al., 2015) | 5–135 μg m−3, various seeds and RO2 fate regimes; 50 % for experiments near 10 μg m −3 | |||
|
| ||||
| Δ-carene | 0–3 % (Hallquist et al., 1999) | 68–74 % (Hallquist et al., 1999); 77 % (Fry et al., 2014) | 13–72 % (Griffin et al., 1999) | 24–310 μg m−3 ; low end closest to 10 μg m− 3 |
| 12–49 % (Moldanova and Ljungstrom, 2000) using new model to reinterpret data from Hallquist et al. (1999) (15–62 %) | 7–395 ppt N2O 5 reacted; 12–15 % at 6.8 ppt N2O5 reacted | |||
| 38–65 % (Fry et al., 2014) | 10 μg m−3 c | |||
|
| ||||
| Limonene | 69 % (Hallquist et al., 1999); 25–33 % (Spittler et al., 2006) | 48 % (Hallquist et al., 1999); 63–72 % (Spittler et al., 2006); 30 % (Fry et al., 2011); 54 % (Fry et al., 2014) | 14–24 % (Moldanova and Ljungstrom, 2000) using new model to reinterpret data from Hallquist et al. (1999) (17 %) | 10 ppt N2O5 reacted; higher number in Moldanova and Ljungstrom (2000) from an additional injection of 7 ppt N2O5 and accounting for secondary reactions |
| 21 or 40 % (Spittler et al., 2006) | Ammonium sulfate or organic seed, respectively, at M∞b | |||
| 25–40 % (Fry et al., 2011) | Nucleation to 10 μg m−3 (second injection of oxidant) | |||
| 44–57 % (Fry et al., 2014) | 10 μg m−3 c | |||
|
| ||||
| Sabinene | 14–76 % (Griffin et al., 1999) | 24–277 μg m−3 ; low end closest to 10 μg m− 3 | ||
| 25–45 % (Fry et al., 2014) | 10 μg m−3 c | |||
|
| ||||
| β-caryophyllene | 91–146 % (Jaoui et al., 2013) | 60–130 μg m−3 ; low end closest to 10 μg m− 3 | ||
| 86 % (Fry et al., 2014) | 10 μg m−3 | |||
The authors assume that N2O5 reacted is equal to BVOC reacted. The anomalously low 0.2 % yield observed at 390 ppt N2O5 reacted is a lower limit; Hallquist et al. note that the number–size distribution for that experiment fell partly outside the measured range.
M∞ corresponds to extrapolated value at highest mass loading. Organic seed aerosol in these experiments was generated from O3 + BVOC. Full dataset was shown only for limonene, where asymptote is 400 μg m−3.
Yield range corresponds to two different methods of calculating ΔBVOC.
Given the limited number of studies that have considered the fate of the peroxy radical, generalizations cannot yet be made for all VOC. Indeed, more studies are needed to determine systematically how gas-phase products and SOA yields are influenced by reactions of RO2. More specifically, for all chamber experiments, constraining the fate and lifetime of RO2 is required to attribute product and SOA yields to a specific pathway. As shown in Table 2 in Sect. 2.2, the nitrate yields and SOA yields for NO3-induced degradation of many VOC vary significantly between different studies. This is likely, in part, a result of each experiment having a different distribution of RO2 fates, but may also arise from vapor wall losses.
In general, there are very few mechanistic studies for NO3 relative to other oxidants. Furthermore, the elucidation of mechanisms is limited by the fact that most studies provide overall yields of organic nitrates (without individual identification of the species) and/or identification (without quantification) due to the lack of standards.
2.2 Organic aerosol yields, speciation, and particle-phase chemistry
Several papers have reported chamber studies to measure the organic aerosol yield and/or gaseous and aerosol-phase oxidation product distribution from NO3-BVOC reactions. These are summarized in Table 2. In general, these experimental results show that monoterpenes are efficient sources of SOA, with reported yields variable but consistently above 20 %, with the notable exception of α-pinene (yields 0–15 %). This anomalous monoterpene also has a much larger product yield of carbonyls instead of organic nitrates compared to the others. This difference among monoterpenes was investigated in the context of the competition between O3 and NO3 oxidation (Draper et al., 2015), in which shifting from O3-dominated to NO3-dominated oxidation was observed to suppress SOA formation from α-pinene, but not from β-pinene, Δ-carene, or limonene. The smaller isoprene has substantially lower SOA yields (2–24 %), and the only sesquiterpene studied, β-caryophyllene, has a much larger yield (86–150 %) than the monoterpenes.
In general, these chamber experiments are conducted under conditions that focus on first-generation oxidation only, but further oxidation can continue to change SOA loadings in the real atmosphere (e.g., Rollins et al., 2009; Chacon-Madrid et al., 2013). Recent experiments showed that particulate organic nitrates formed from β-pinene-NO3 are resilient to photochemical aging, while those formed from α-pinene-NO3 evaporate more readily (Nah et al., 2016b).
Other chamber studies have not reported SOA mass yields or gas-phase product measurements but have otherwise demonstrated the importance of NO3-BVOC reactions to SOA production. These studies have identified β-pinene and Δ-carene as particularly efficient sources of SOA upon NO3 oxidation (Hoffmann et al., 1997), confirmed the greater aerosol-forming potential from β-pinene versus α-pinene (Bonn and Moortgat, 2002), and reported Fourier transform infrared spectroscopy (FTIR) and aerosol mass spectrometry (AMS) measurements of the composition of organic nitrates detected in aerosol formed from NO3-isoprene, α-pinene, β-pinene, Δ-carene, and limonene reactions (Bruns et al., 2010).
Relative humidity (RH) can be an important parameter, as it affects the competition between NO3-BVOC reactions and heterogeneous uptake of N2O5. Among existing laboratory studies, only a few have focused on the effect of RH on SOA formation from NO3-initiated oxidation (Bonn and Moortgat, 2002; Spittler et al., 2006; Fry et al., 2009; Boyd et al., 2015). The impact of RH might be important, especially at night and during the early morning when RH near the surface is high and NO3 radical chemistry is competitive with O3 and OH reactions. However, observations of the effect of water on SOA formation originating from NO3 oxidation hint at a varied role. Spittler et al. (2006) reported lower SOA yields under humid conditions, but other studies did not observe a significant effect (Bonn and Moortgat, 2002; Fry et al., 2009; Boyd et al., 2015). Among the important effects of water is its role as a medium for hydrolysis. In laboratory studies, primary and secondary organic nitrates were found to be less prone to aqueous hydrolysis than tertiary organic nitrates (Darer et al., 2011; Hu et al., 2011). First-generation organic nitrates retaining double bonds may also hydrolyze relatively quickly, especially in the presence of acidity (Jacobs et al., 2014; Rindelaub et al., 2015). Depending on the relative amount of these different types of organic nitrates, the overall hydrolysis rate could be different for organic nitrates formed from NO3 oxidation and photooxidation in the presence of NOx (Boyd et al., 2015). Recently, there has been increasing evidence from field measurements that organic nitrates hydrolyze in the particle phase, producing HNO3 (Liu et al., 2012b; Browne et al., 2013). This has been only a limited focus of chamber experiments to date (Boyd et al., 2015). In addition to the effect of RH, particle-phase acidity is known to affect SOA formation from ozonolysis and OH reaction (e.g., Gao et al., 2004; Tolocka et al., 2004). Thus far, only one study has examined the effect of acidity on NO3-initiated SOA formation and found a negligible effect (Boyd et al., 2015). Notably, an effect of acidity was observed for the hydrolysis of organic nitrates produced in photochemical reactions (Szmigielski et al., 2010; Rindelaub et al., 2015). While much organic nitrate aerosol is formed via NO3+ BVOC reactions, some fraction can also form from RO2+ NO chemistry. Rollins et al. (2010) observed the organic nitrate moiety in 6–15 % of total SOA mass generated from high-NOx photooxidation of limonene, α-pinene, Δ3-carene, and tridecane. A very recent study of Berkemeier et al. (2016) showed that organic nitrates accounted for ~ 40 % of SOA mass during initial particle formation in α-pinene oxidation by O3 in the presence of NO, decreasing to ~ 15 % upon particle growth to the accumulation-mode size range. They also observed a tight correlation (R2 = 0.98) between organic nitrate content and SOA particle number concentrations. This implies that organic nitrates may be among the extremely low volatility organic compounds (ELVOC) (Ehn et al., 2014; Tröstl et al., 2016) that play a critical role in nucleation and nanoparticle growth.
2.3 Heterogeneous and aqueous-phase NO3 processes
The NO3 radical is not only a key nighttime oxidant of organic (and especially biogenic) trace gases but it can also play an important role in the aqueous phase of tropospheric clouds and deliquesced particles (Chameides, 1978; Wayne et al., 1991; Herrmann and Zellner, 1998; Rudich et al., 1998). Whilst the reaction of NO3 with organic particles and aqueous droplets in the atmosphere is believed to represent only an insignificant fraction of the overall loss rate for NO3, it can have a substantial impact on the chemical and physical properties of the particle by modifying its lifetime, oxidation state, viscosity, and hygroscopic properties and thus its propensity to act as a cloud condensation nucleus (Rudich, 2003).
Biogenic VOC include, but are not limited to the isoprenoids (isoprene, mono-, and sesquiterpenes) as well as alkanes, alkenes, carbonyls, alcohols, esters, ethers, and acids (Kesselmeier and Staudt, 1999). Recent measurements indicate that biogenic emissions of aromatic trace gases are also significant (Misztal et al., 2015). The gas-phase degradation of BVOC leads to the formation of a complex mixture of organic trace gases including hydroxyl- and nitrate-substituted oxygenates which can transfer to the particle phase by condensation or dissolution. Our present understanding is that non-anthropogenic SOA has a large contribution from isoprenoid degradation.
As is generally the case for laboratory studies of heterogeneous processes, most of the experimental investigations on heterogeneous uptake of NO3 to organic surfaces have dealt with single-component systems that act as surrogates for the considerably more complex mixtures found in atmospheric SOA. A further level of complexity arises when we consider that initially reactive systems, e.g., containing condensed or dissolved unsaturated hydrocarbons, can become deactivated as SOA ages, single bonds replace double bonds, and the oxygen-to-carbon ratio increases.
We summarize the results of the laboratory studies to provide a rough guide to NO3 reactivity on different classes of organics which may be present in SOA and note that further studies of NO3 uptake to biogenic SOA which was either generated and aged under well-defined conditions (Fry et al., 2011) or sampled from the atmosphere are required to confirm predictions of uptake efficiency based on the presently available database.
2.3.1 Heterogeneous processes
For some particle-phase organics, the reaction with NO3 is at least as important as other atmospheric oxidants such as O3 and OH (Shiraiwa et al., 2009; Kaiser et al., 2011). The lifetime (τ) of a single component, liquid organic particle with respect to loss by reaction with NO3 at concentration [NO3] is partially governed by the uptake coefficient (γ) (Robinson et al., 2006; Gross et al., 2009):
| (1) |
where Dp is the particle diameter, ρorg and Morg are the density and molecular weight of the organic component, respectively, c̄ is the mean molecular velocity of gas-phase NO3, and NA is Avogadro number. Thus, defined, τ is the time required for all the organic molecules in a spherical (i.e., liquid) particle to undergo a single reaction with NO3.
Recent studies have shown that organic aerosols can adopt semi-solid (highly viscous) or amorphous solid (crystalline or glass) phase states, depending on the composition and ambient conditions (Virtanen et al., 2010; Koop et al., 2011; Renbaum-Wolff et al., 2013). Typically, the bulk phase diffusion coefficients of NO3 are ~ 10−7–10−9 cm2 s−1 in semi-solid and ~ 10−10 cm2 s−1 in solids (Shiraiwa et al., 2011). Slow bulk diffusion of NO3 in a viscous organic matrix can effectively limit the rate of uptake (Xiao and Bertram, 2011; Shiraiwa et al., 2012). Similarly, the solubility may be different in a concentrated, organic medium. If bulk diffusion is slow, the reaction may be confined to the near-surface layers of the particle or bulk substrate. The presence of organic coatings on aqueous aerosols was found to suppress heterogeneous N2O5 hydrolysis by providing a barrier through which N2O5 needs to diffuse to undergo hydrolysis (Alvarado et al., 1998; Cosman et al., 2008; Grifiths et al., 2009). Reactive uptake by organic aerosols is expected to exhibit a pronounced decrease at low RH and temperature, owing to a phase transition from viscous liquid to semi-solid or amorphous solid (Arangio et al., 2015). Therefore, the presence of a semi-solid matrix may effectively shield reactive organic compounds from chemical degradation in long-range transport in the free troposphere.
To get an estimate of the processing rate of BVOC-derived SOA, we have summarized the results of several laboratory studies to provide a rough guide to NO3 reactivity on different classes of organics that may be present in SOA (Fig. 3). A rough estimate of the reactivity of NO3 to freshly generated, isoprenoid-derived SOA, which still contains organics with double bonds (e.g., from diolefinic monoterpenes such as limonene), may be obtained by considering the data on alkenes and unsaturated acids, where the uptake coefficient is generally close to 0.1.
Figure 3.
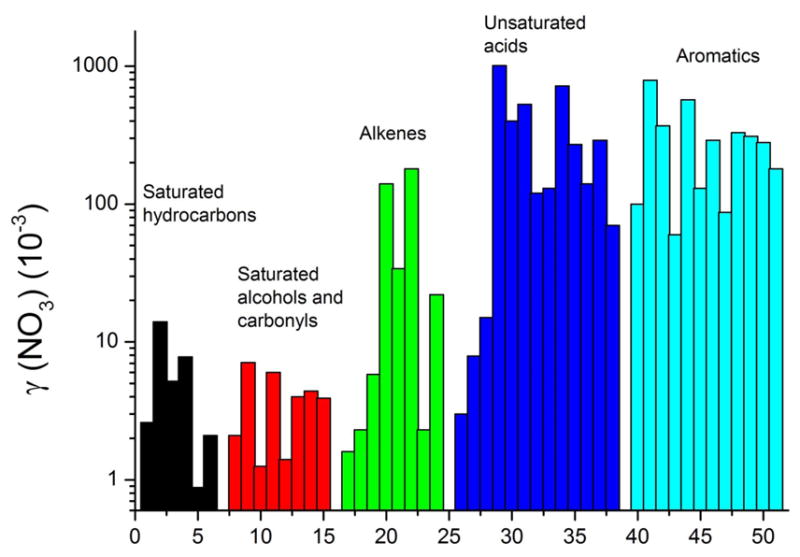
Uptake coefficients, γ (NO3), for the interaction of NO3 with single-component organic surfaces. Details of the experiments and the references (corresponding to the x-axis numbers) are given in Table S1 in the Supplement.
The classes of organics for which heterogeneous reactions with NO3 have been examined are alkanoic/alkenoic acids, alkanes and alkenes, alcohols, aldehydes, polyaromatic hydrocarbons (PAHs), and secondary organic aerosols. Laboratory studies have used either pure organic substrates, with the organic of interest internally mixed in an aqueous particle; as a surface coating, with the reactive organic mixed in a nonreactive organic matrix; or in the form of self-assembling monolayers. The surrogate surface may be available as a macroscopic bulk liquid (or frozen liquid) or in particulate form and both gas-phase and particle-phase analyses have been used to derive kinetic parameters and investigate products formed.
In the gas phase, the NO3 radical reacts slowly (by H-abstraction) with alkanes, more rapidly with aldehydes due to the weaker C-H bond of the carbonyl group, and most readily with alkenes and aromatics via electrophilic addition. This trend in reactivity is also observed in the condensed-phase reactions of NO3 with organics so that long-chain organics, for which non-sterically hindered addition to a double bond is possible, and aromatics are the most reactive. In very general terms, uptake coefficients are in the range of 1–10 × 10−3 for alkanes, alcohols, and acids without double bonds, 2–200 × 10−3 for alkenes with varying numbers of double bonds, 3–1000 × 10−3 for acids with double bonds again depending on the number of double bonds, and 100–500 × 10−3 for aromatics. These trends are illustrated in Fig. 3 which plots the experimental data for the uptake of NO3 to single-component organic surfaces belonging to different classes of condensable organics. Condensed-phase organic nitrates have been frequently observed following interaction of NO3 with organic surfaces (see below).
Saturated hydrocarbons
Uptake of NO3 to saturated hydrocarbons is relatively slow, with uptake coefficients close to 10−3. Moise et al. (2002) found that (for a solid sample) uptake to a branched-chain alkane was more efficient than for a straight-chain alkane, which is consistent with known trends in gas-phase reactivity of NO3. The slow surface reaction with alkanes enables both surface and bulk components of the reaction to operate in parallel. The observation of RONO2 as product is explained (Knopf et al., 2006; Gross and Bertram, 2009) by processes similar to those proceeding in the gas phase, i.e., abstraction followed by formation of peroxy and alkoxy intermediates which react with NO2 and NO3 to form the organic nitrate.
Unsaturated hydrocarbons
With exception of the data of Moise et al. (2002), the up-take of NO3 to an unsaturated organic surface is found to be much more efficient than to the saturated analogue. The NO3 uptake coefficient for, e.g., squalene, is at least an order of magnitude more efficient than for squalane (Xiao and Bertram, 2011; Lee et al., 2013). The location of the double bond is also important and the larger value for γ found for a self-assembling monolayer of NO3+ undec-10-ene-1-thiol compared to liquid, long-chain alkenes is due to the fact that the terminal double bond is located at the interface and is thus more accessible for a gas-phase reactant (Gross and Bertram, 2009). NO3 uptake to mixtures of unsaturated methyl oleate in a matrix of saturated organic was found to be consistent with either a surface or bulk reaction (Xiao and Bertram, 2011). The formation of condensed-phase organic nitrates and simultaneous loss of the vinyl group indicates that the reaction proceeds, as in the gas phase, by addition of NO3 to the double bond followed by reaction of NO3 (or NO2) with the resulting alkyl and peroxy radicals formed (Zhang et al., 2014b).
Saturated alcohols and carbonyls
Consistent with reactivity trends for NO3 in the gas phase, the weakening of some C-H bonds in oxidized, saturated organics results in a more efficient interaction of NO3 than for the non-oxidized counterparts although, as far as the limited dataset allows trends to be deduced, the gas-phase reactivity trend of polyalcohol being greater than alkanoate appears to be reversed in the liquid phase (Gross et al., 2009). For multicomponent liquid particles, the uptake coefficient will also depend on the particle viscosity (Iannone et al., 2011) though it has not been clearly established if the reaction proceeds predominantly at the surface or throughout the particle (Iannone et al., 2011). The reaction products are expected to be formed via similar pathways as seen in the gas phase, i.e., abstraction of the aldehydic-H atom for aldehydes and abstraction of an H atom from either the O-H or adjacent α-CH2 group for alcohols prior to reaction of NO2 and NO3 with the ensuing alkyl and peroxy radicals (Zhang and Morris, 2015).
Organic acids
The efficiency of uptake of NO3 to unsaturated acids is comparable to that found with other oxidized, saturated organics (Moise et al., 2002) suggesting that the reaction proceeds, as in the gas phase, via abstraction rather than addition. Significantly larger uptake coefficients have been observed for a range of unsaturated, long-chain acids, with γ often between 0.1 and 1 (Gross et al., 2009; Knopf et al., 2011; Zhao et al., 2011a). γ depends on the number and position (steric factors) of the double bond. For example, the uptake coefficient for abietic acid is a factor of 100 lower than for linoleic acid (Knopf et al., 2011). The condensed-phase products formed in the interaction of NO3 with unsaturated acids are substituted carboxylic acids, including hydroxy nitrates, carbonyl nitrates, dinitrates, and hydroxy dinitrates (Hung et al., 2005; Docherty and Ziemann, 2006; McNeill et al., 2007; Zhao et al., 2011a).
Aromatics
The interaction of NO3 with condensed-phase aromatics and PAHs results in the formation of a large number of nitrated aromatics and nitro PAHs. Similar to the gas-phase mechanism, the reaction is initiated by addition of NO3 to the aromatic ring, followed by breaking of an N-O bond to release NO2 to the gas phase and forming a nitrooxy-cyclohexadienyl-type radical which can further react with O2, NO2, or undergo internal rearrangement to form hydroxyl species (Gross and Bertram, 2008; Lu et al., 2011). The uptake coefficients are large and comparable to those derived for the unsaturated fatty acids.
The literature results on the interaction of NO3 with organic substrates are tabulated in Table S1 in the Supplement, in which the uptake coefficient is listed (if available) along with the observed condensed- and gas-phase products.
2.3.2 Aqueous-phase reactions
The in situ formation of NO3 (e.g., electron transfer reactions between nitrate anions and other aqueous radical anions such as , sulfur-containing radical anions, or ) is generally of minor importance and the presence of NO3 in aqueous particles is largely a result of transfer from the gas phase (Herrmann et al., 2005; Tilgner et al., 2013). Concentrations of NO3 in tropospheric aqueous solutions cannot be measured in situ, and literature values are based on multiphase model predictions (Herrmann et al., 2010). Model studies with the chemical aqueous-phase radical mechanism (CAPRAM; Herrmann et al., 2005; Tilgner et al., 2013) predict [NO3] between 1 × 6 × 10−16 and 2.7 × 10−13 mol L−1. High NO3 concentration levels are associated with urban clouds, while in rural and marine clouds these levels are an order of magnitude lower. Since the NO3 concentrations are related to the NOx budget, typically higher NO3 concentrations are present under urban cloud conditions compared to rural and marine cloud regimes.
NO3 radicals react with dissolved organic species via three different pathways: (i) by H-atom abstraction from saturated organic compounds, (ii) by electrophilic addition to double bonds within unsaturated organic compounds, and (iii) by electron transfer from dissociated organic acids (Huie, 1994; Herrmann and Zellner, 1998). For a detailed overview on aqueous-phase NO3 radical kinetics, the reader is referred to several recent summaries (Neta et al., 1988; Herrmann and Zellner, 1998; Ross et al., 1998; Herrmann, 2003; Herrmann et al., 2010, 2015). Compared to the highly reactive and non-selective OH radical, the NO3 radical is characterized by a lower reactivity and represents a more selective aqueous-phase oxidant. The available kinetic data indicate that the reactivity of NO3 radicals with organic compounds in comparison to the two other key radicals (OH, ) is as follows: (Herrmann et al., 2015).
In Table S2, we list kinetic parameters for reaction of NO3 with aliphatic organic compounds as presently incorporated in the CAPRAM database (Bräuer et al., 2017). Typical ranges of rate constants (in M−1 s−1) for reactions of NO3 in the aqueous phase are 106–107 for saturated alcohols, carbonyls, and sugars; 104–106 for protonated aliphatic mono- and dicarboxylic acids, with higher values for oxygenated acids; 106–108 for deprotonated aliphatic mono- and dicarboxylic acids (higher values typically for oxygenated acids); 107–109 for unsaturated aliphatic compounds; and 108–2 × 109 for aromatic compounds (without nitro/acid functionality). The somewhat larger rate constants for deprotonated aliphatic mono- and dicarboxylic acids, unsaturated aliphatic compounds and aromatic compounds is related to the occurrence of electron transfer reactions and addition reaction pathways, which are often faster than H-abstraction reactions.
Many aqueous-phase NO3 reaction rate constants, even for small oxygenated organic compounds, are not available in the literature and have to be estimated. In the absence of SARs for NO3 radical reactions with organic compounds, Evans–Polanyi-type reactivity correlations are used to predict kinetic data for H-abstraction NO3 radical reactions. The latest correlation for NO3 reactions in aqueous solution based on 38 H-abstraction reactions of aliphatic alcohols, carbonyl compounds and carboxylic acids was published by Hoffmann et al. (2009):
| (2) |
where BDE is the bond dissociation energy (in kJ mol−1). The correlation is quite tight, with a correlation coefficient of R = 0.9.
A direct comparison of the aqueous-phase OH and NO3 radical rate constants (k298 K) of organic compounds from different compound classes is presented in Fig. 4, which shows that the NO3 radical reaction rate constants for many organic compounds are about 2 orders of magnitude smaller than respective OH rate constants. In contrast, deprotonated dicarboxylic acids can react with NO3 via electron transfer and have similar rate constants for OH reaction. Rate constants for OH and NO3 with alcohols and diols/polyols are well correlated (R2 values are given in Table S3), whereas those rate constants for carbonyl compounds and diacids have a lower degree of correlation.
Figure 4.

(a) Correlation of OH versus NO3 radical rate constants in the aqueous phase for the respective compound classes. The linear regression fits for the different compound classes are presented in the same color as the respective data points. The black line represents the correlation of the overall data. (b) Comparison of modeled, aqueous-phase reaction fluxes (mean chemical fluxes in mol cm−3 s−1 over a simulation period of 4–5 days) of organic compounds with hydroxyl (OH) versus nitrate (NO3) radicals distinguished by different compound classes (urban CAPRAM summer scenario).
Figure 4b shows a comparison of the modeled chemical turnovers of reactions of organic compounds with OH versus NO3 radicals distinguished for different compound classes. The simulations were performed with the SPACCIM model (Wolke et al., 2005) for the urban summer CAPRAM scenario (see Tilgner et al., 2013 for details) using the master chemical mechanism (MCM) 3.2/CAPRAM 4.0 mechanism (Rickard, 2015; Bräuer et al., 2017) which has in total 862 NO3 radical reactions with organic compounds.
Most of the data lie under the 1 : 1 line, indicating that, for most of the organic compounds considered, chemical degradation by OH is more important than by NO3, with a significant fraction of the data lying close to a 10 : 1 line, though OH fluxes sometimes exceed NO3 fluxes by a factor of 103 – 104. Approximate relative flux ratios (NO3 / OH) for different classes of organic are 10−1 – 10−2 for alcohols (including diols and polyols) and carbonyl compounds, 10−1 – 10−4 for undissociated monoacids and diacids, ~ 1 (or larger) for dissociated monoacids, 10−2 – > 10 for dissociated diacids, and 10−2–1 for organic nitrates. For carboxylate ions, NO3-initiated electron transfer is thus the dominant oxidation pathway. As OH-initiated oxidation proceeds via an H-abstraction, high NO3-OH flux ratios can be observed for carboxylate ions but not for protonated carboxylic acids.
Overall, Fig. 4b shows that, over a 4-day summer cycle, NO3 radical reactions can compete with OH radical reactions in particular for protonated carboxylic acids and multifunctional compounds. Nevertheless, aqueous NO3 radical reactions with organics will become more important during winter or at higher latitudes, where photochemistry as the main source of OH is less important. Finally, it should be noted that NO3 aqueous-phase nighttime chemistry will influence the concentration levels of many aqueous-phase reactants available for reaction during the next day.
2.4 Instrumental methods
Atmospheric models of the interaction of NO3 with BVOC rely on experimental data gathered in both the laboratory and the field. These experimental data are used to define model parameters and to evaluate model performance by comparison to observed quantities. Instrumentation for measurements of nitrogen-containing species, oxidants, and organic compounds, including NOx, O3, NO3, BVOC, and oxidized reactive nitrogen compounds, are all important to understand the processes at work. Of particular importance to the subject of this review is the characterization of organic nitrates, which are now known to exist in both the gas and particle phases and whose atmospheric chemistry is complex. This section reviews historical and current experimental methods used for elucidating NO3-BVOC atmospheric chemistry.
2.4.1 Nitrate radical measurements
Optical absorption spectroscopy has been the primary measurement technique for NO3. It usually makes use of two prominent absorption features of NO3 near 623 and 662 nm. Note that the dissociation limit of the NO3 molecule lies between the two absorption lines (Johnston et al., 1996); thus, illumination by measurement radiation at the longer wave-length band does not lead to photolysis of NO3. The room temperature absorption cross section of NO3 at 662 nm is ~ 2 × 10−17 cm2 molec−1 and increases at lower temperature (Yokelson et al., 1994; Osthoff et al., 2007). Thus, at a typical minimum detectable optical density (reduction of the intensity compared to no absorption) and a light-path length of 5 km, a detection limit of 107 molec cm−3 or ~ 0.4 ppt (under standard conditions) is achieved.
Initial measurements of NO3 in the atmosphere were long-path averages using light paths between either the sun or the moon (e.g., Noxon et al., 1978) and the receiving spectrometer (also called passive techniques because natural light sources were used) or between an artificial light source and spectrometer over a distance of several kilometers (active techniques; e.g., Platt et al., 1980). Passive techniques were later extended to yield NO3 vertical profiles (e.g., Weaver et al., 1996). In recent years, resonator cavity techniques allowed construction of very compact instruments capable of performing in situ measurements of NO3 with absorption spectroscopy (see in situ measurement techniques below).
An important distinction between the techniques is whether NO3 can be deliberately or inadvertently removed from the absorption path as part of the observing strategy. Long-path absorption spectroscopy does not allow control over the sample for obtaining a zero background by removing NO3 (Category 1). Resonator techniques (at least as long as the resonator is encased) allow deliberate removal of NO3 from the absorption path as part of the measurement sequence and may also result in inadvertent removal during sampling (Category 2).
For instruments of Category 1, the intensity without absorber (I0) cannot be easily detected. Therefore, the information about the absorption due to NO3 (and any other trace gas) has to be determined from the structure of the absorption, which is usually done by using differential optical absorption spectroscopy (DOAS) (Platt and Stutz, 2008), which relies on the characteristic fingerprint of the NO3 absorption structure in a finite wavelength range (about several 10 nm wide). Thus, a spectrometer of sufficient spectral range and resolution (around 0.5 nm) is required.
Instruments of Category 2 can determine the NO3 concentration from the difference (or rather log of the ratio) of the intensity with and without NO3 in the measurement volume. In this case, only an intensity measurement at a single wavelength (typically of a laser) is necessary, and specificity can be achieved through chemical titration with NO (Brown et al., 2001). However, enhanced specificity without chemical titration can be gained by combining resonator techniques with DOAS detection. It should be noted that the advantage of a closed cavity to be able to remove (or manipulate) NO3 comes at the expense of potential wall losses, which have to be characterized. Such instruments have the advantage of being able to also detect N2O5, which is in thermal equilibrium with NO3 and can be quantitatively converted to NO3 by thermal dissociation (Brown et al., 2001, 2002).
Another complication arises from the presence of water vapor and oxygen lines in the wavelength range of strong NO3 absorptions. To compensate for these potential interferences in open-path measurements (where NO3 cannot easily be removed), daytime measurements are frequently used as reference because NO3 levels are typically very low (but not necessarily negligibly low) (Geyer et al., 2003). Thus, a good fraction of the reported NO3 data (in particular, older data) represents day–night differences.
Passive long-path remote sensing techniques
Measurements of the NO3 absorption structure using sunlight take advantage of the fact that NO3 is very quickly photolyzed by sunlight (around 5 s lifetime during the day) allowing for vertically resolved measurements during twilight (e.g., Aliwell and Jones, 1998; Allan et al., 2002; Coe et al., 2002; von Friedeburg et al., 2002). The fact that the NO3 concentration is nearly zero due to rapid photolysis in the directly sunlit atmosphere, while it is largely undisturbed in a shadowed area, can be used to determine NO3 vertical concentration profiles during sunrise using the moon as a light source (Smith and Solomon, 1990; Smith et al., 1993; Weaver et al., 1996). Alternatively, the time series of the NO3 column density derived from scattered sunlight originating from the zenith (or from a viewing direction away from the sun) during sunrise can be evaluated to yield NO3 vertical profiles (Allan et al., 2002; Coe et al., 2002; von Friedeburg et al., 2002).
Nighttime NO3 total column data have been derived by spectroscopy of moonlight and starlight (Naudet et al., 1981), the intensity of which is about 4–5 orders of magnitude lower than that of sunlight. Thus, photolysis of NO3 by moonlight is negligible. A series of moonlight NO3 measurements have been reported (Noxon et al., 1980; Noxon, 1983; Sanders et al., 1987; Solomon et al., 1989, 1993; Aliwell and Jones, 1996a, b; Wagner et al., 2000). These measurements yield total column data of NO3, the sum of tropospheric and stratospheric partial columns. Separation between stratospheric and tropospheric NO3 can be accomplished (to some extent) by the Langley plot method (Noxon et al., 1980), which takes advantage of the different dependence of tropospheric and stratospheric NO3 slant column density on the lunar zenith angle.
Active long-path techniques
A large number of NO3 measurements have been made using the active long-path DOAS technique (Platt et al., 1980, 1981, 1984; Pitts et al., 1984; Heintz et al., 1996; Allan et al., 2000; Martinez et al., 2000; Geyer et al., 2001a, b, 2003; Gölz et al., 2001; Stutz et al., 2002, 2004, 2010; Asaf et al., 2009; McLaren et al., 2010; Crowley et al., 2011; Sobanski et al., 2016). Here, a searchlight-type light source is used to transmit a beam of light across a kilometer-long light path in the open atmosphere to a receiving telescope–spectrometer combination. The light source typically is a broadband thermal radiator (incandescent lamp, Xe arc lamp, laser-driven light source). More recently, LED light sources were also used (Kern et al., 2006). The telescope (around 0.2 m diameter) collects the radiation and transmits it, usually through an optical fiber, into the spectrometer, which produces the absorption spectrum. Modern instruments now almost exclusively use transmitter/receiver combinations at one end of the light path and retro-reflector arrays (e.g., cat-eye-like optical devices) at the other end. The great advantage of this approach is that power and optical adjustment is only required at one end of the light path while the other end (with the retro-reflector array) is fixed. In this way, several retro-reflector arrays, for instance, mounted at different altitudes, can be used sequentially with the same transmitter/receiver unit allowing determination of vertical profiles of NO3 (and other species measurable by DOAS) (Stutz et al., 2002, 2004, 2010).
In situ measurement techniques
Cavity ring-down spectroscopy (CRDS) and cavity-enhanced absorption spectroscopy (CEAS) are related techniques for in situ quantification of atmospheric trace gases such as NO3. These methods are characterized by high sensitivity, specificity, and acquisition speed (Table 3a), and they allow for spatially resolved measurements on mobile platforms.
Table 3.
(a)Selected CRDS and CEAS instruments used to quantify NO3 mixing ratios in ambient air. (b) Selected instruments used to quantify NO3 and N2O5 mixing ratios in ambient air other than by cavity-enhanced absorption spectroscopy.
| (a)
| ||
|---|---|---|
| Principle of measurement (laser pulse rate) | LOD or precision (integration time) | Reference |
| BB-CEAS | 2.5 pptv (8.6 min) | Ball et al. (2004) |
| BB-CRDS | 1 pptv (100 s) | Bitter et al. (2005) |
| Off-axis cw CRDS (500 Hz) | 2 pptv (5 s) | Ayers and Simpson (2006) |
| On-axis pDL-CRDS (33 Hz) | <1 pptv (1 s) | Dubé et al. (2006) |
| BB-CEAS | 4 pptv (60 s) | Venables et al. (2006) |
| pDL-CRDS (10 Hz) | 2.2 pptv (100 s) | Nakayama et al. (2008) |
| Off-axis cw CRDS (200 Hz) | 2 pptv (5 s) | Schuster et al. (2009), Crowley et al. (2010) |
| CE-DOAS | 6.3 pptv (300 s) | Platt et al. (2009), Meinen et al. (2010) |
| BB-CEAS | 2 pptv (15 s) | Langridge et al. (2008), Benton et al. (2010) |
| BB-CEAS | < 2 pptv (1s) | Kennedy et al. (2011) |
| On-axis cw-CRDS (500 Hz) | <1 pptv (1 s) | Wagner et al. (2011) |
| On-axis cw-CRDS (300 Hz) | 8 pptv (10 s) | Odame-Ankrah and Osthoff (2011) |
| BB-CEAS | 1 pptv (1 s) | Le Breton et al. (2014) |
| BB-CEAS | 7.9 pptv (60 s) | Wu et al. (2014) |
| (b)
| |||
|---|---|---|---|
| Principle of measurement | LOD or precision e(integration time) | Species detected | Reference |
| MIESR | < 2 pptv (30 min) | NO3 | Geyer et al. (1999) |
| CIMS | 12 pptv (1 s) | NO3 + N2O5 | Slusher et al. (2004) |
| LIF | 11 pptv (10 min) | NO3 | Matsumoto et al. (2005a), Matsumoto et al. (2005b) |
| LIF | 28 pptv (10 min) | NO3 | Wood et al. (2005) |
| CIMS | 30 pptv (30 s) | N2O5 | Zheng et al. (2008) |
| CIMS | 5 pptv (1 min) | N2O5 | Kercher et al. (2009) |
| CIMS | 7.4 pptv (1 s) | N2O5 | Le Breton et al. (2014) |
| CIMS | 39 pptv (6 s) | N2O5 | Wang et al. (2014) |
CEAS = cavity-enhanced absorption spectroscopy; CRDS = cavity ring-down spectroscopy; BB = broadband; pDL = pulsed dye laser; CE-DOAS = cavity-enhanced differential optical absorption spectroscopy; cw = continuous-wave diode laser. MIESR = matrix isolation electron spin resonance; CIMS = chemical ionization mass spectrometry; LIF = laser-induced fluorescence; LOD = limit of detection.
In CRDS, laser light is “trapped” in a high-finesse stable optical cavity, which usually consists of a pair of highly reflective spherical mirrors in a near-confocal arrangement. The concentrations of the optical absorbers present within the resonator are derived from the Beer–Lambert law and the rate of light leaking from the cavity after the input beam has been switched off (O’Keefe and Deacon, 1988). CRDS instruments are inherently sensitive as they achieve long effective optical absorption paths (up to, or in some cases exceeding, 100 km) as the light decay is monitored for several 100 μs, and the absorption measurement is not affected by laser intensity fluctuations. For detection of NO3 at 662 nm, pulsed laser sources such as Nd:YAG pumped dye lasers have been used because of the relative ease of coupling the laser beam to the optical cavity (Brown et al., 2002, 2003; Dubé et al., 2006). Relatively lower cost continuous-wave (cw) diode laser modules that are easily modulated also have been popular choices (e.g., King et al., 2000; Simpson, 2003; Ayers et al., 2005; Odame-Ankrah and Osthoff, 2011; Wagner et al., 2011).
In a CEAS instrument (also referred to as integrated cavity output spectroscopy, ICOS, or cavity-enhanced DOAS, CE-DOAS), the spectrum transmitted through a high-finesse optical cavity is recorded. Mixing ratios of the absorbing gases are derived using spectral retrieval routines similar to those used for open-path DOAS (e.g., O’Keefe, 1998, 1999; Ball et al., 2001; Fiedler et al., 2003; Platt et al., 2009; Schuster et al., 2009).
CRDS and CEAS are, in principle, absolute measurement techniques and do not need to rely on external calibration. In practice, however, chemical losses can occur on the inner walls of the inlet (even when constructed from inert materials such as Teflon) or at the aerosol filters necessary for CRDS instruments. Hence, the inlet transmission efficiencies have to be monitored for measurements to be accurate (Fuchs et al., 2008, 2012; Odame-Ankrah and Osthoff, 2011). On the other hand, a key advantage of in situ instruments over open-path instruments is that the sampled air can be manipulated. Deliberate addition of excess NO to the instrument’s inlet titrates NO3 and allows measurement of the instrument’s zero level and separation of contributions to optical extinction from other species, such as NO2, O3, and H2O. Adding a heated section to the inlet (usually in a second detection channel) enables (parallel) detection of N2O5 via the increase in the NO3 signal (Brown et al., 2001; Simpson, 2003).
In addition, non-optical techniques have been used to detect and quantify NO3. Chemical ionization mass spectrometry (CIMS) is a powerful method for sensitive, selective, and fast quantification of a variety of atmospheric trace gases (Huey, 2007). NO3 is readily detected after reaction with iodide reagent ion as the nitrate anion at m/z 62; at this mass, however, there are several known interferences, including dissociative generation from N2O5, HNO3, and HO2NO2 (Slusher et al., 2004; Abida et al., 2011; Wang et al., 2014). There has been more success with the quantification of N2O5, usually as the iodide cluster ion at m/z 235 (Kercher et al., 2009), though accurate N2O5 measurement at m/z 62 has been reported from recent aircraft measurements with a large N2O5 signal (Le Breton et al., 2014).
Two groups have used laser-induced fluorescence (LIF) to quantify NO3 (and N2O5 through thermal dissociation) in ambient air (Wood et al., 2003; Matsumoto et al., 2005a, b). The major drawback of this method is the relatively low fluorescence quantum yield of NO3, and hence the method has not gained wide use.
Another technique that was demonstrated to be capable of measuring NO3 radicals at atmospheric concentration is matrix isolation electron spin resonance (MIESR) (Geyer et al., 1999). Although the technique allows simultaneous detection of other radicals (including HO2 and NO2), it has not been used extensively, probably because of its complexity.
Recently, a variety of in situ NO3 (Dorn et al., 2013) and N2O5 (Fuchs et al., 2012) measurement techniques were compared at the SAPHIR chamber in Jülich, Germany. All instruments measuring NO3 were optically based (absorption or fluorescence). N2O5 was detected as NO3 after thermal decomposition in a heated inlet by either CRDS or LIF. Generally, agreement within the accuracy of instruments was found for all techniques detecting NO3 and/or N2O5 in this comparison exercise. This study showed excellent agreement between the instruments on the single-digit ppt NO3 and N2O5 levels with no noticeable interference due to NO2 and water vapor for instruments based on cavity ring-down or cavity-enhanced spectroscopy. Because of the low sensitivity of LIF instruments, N2O5 measurements by these instruments were significantly noisier compared to the measurements by cavity-enhanced methods. The agreement between instruments was less good in experiments with high aerosol mass loadings, specifically for N2O5, presumably due to enhanced, unaccounted loss of NO3 and N2O5 demonstrating the need for regular filter changes in closed-cavity instruments. Whereas differences between N2O5 measurements were less than 20 % in the absence of aerosol, measurements differed up to a factor of 2.5 for the highest aerosol surface concentrations of 5 × 108 nm2 cm−3. Also, differences between NO3 measurements showed an increasing trend (up to 50 %) with increasing aerosol surface concentration for some instruments.
2.4.2 Gas-phase organic nitrate measurements
Analytical techniques to detect gaseous organic nitrates have been documented in a recent review by Perring et al. (2013). Sample collection techniques for organic nitrates include preconcentration on solid adsorbents (Atlas and Schauffler, 1991; Schneider and Ballschmiter, 1999; Grossenbacher et al., 2001), cryogenic trapping (Flocke et al., 1991) or collection in stainless steel canisters (Flocke et al., 1998; Blake et al., 1999), or direct sampling (Day et al., 2002; Beaver et al., 2012).
The approaches to the analysis of the organic nitrates fall into three broad categories. First, one or more chemically speciated organic nitrates are measured by a variety of techniques including liquid chromatography (LC) (Kastler et al., 2000) or gas chromatography (GC) with electron capture detection (Fischer et al., 2000), GC with electron impact or negative-ion chemical ionization mass spectrometry (GC-MS) (Atlas, 1988; Luxenhofer et al., 1996; Blake et al., 1999, 2003a, b; Worton et al., 2008), GC followed by conversion to NO and chemiluminescent detection (Flocke et al., 1991, 1998), GC followed by photoionization mass spectrometry (Takagi et al., 1981), GC followed by conversion of organic nitrates to NO2 and luminol chemiluminescent detection (Hao et al., 1994), CIMS (Beaver et al., 2012; Paulot et al., 2012), and proton transfer reaction MS (PTR-MS) (Perring et al., 2009). Second, the sum of all organic nitrates can be measured directly by thermal dissociation to NO2, which is subsequently measured by LIF (TD-LIF) (Day et al., 2002), CRDS (TD-CRDS) (Paul et al., 2009; Thieser et al., 2016), or cavity-attenuated phase shift spectroscopy (TD-CAPS) (Sadanaga et al., 2016). Finally, the sum of all organic nitrates can be measured indirectly as the difference between all reactive NOx except for organic nitrates and total oxidized nitrogen (NOy ) (Parrish et al., 1993).
Recent advances in adduct ionization utilize detection of the charged cluster of the parent reagent ion with the compound of interest. This scheme is then coupled to high-resolution time-of-flight (HR-ToF) mass spectrometry. The combination of these methods allows the identification of molecular composition due to the soft ionization approach that minimizes fragmentation. Multifunctional organic nitrates resulting from the oxidation of BVOC have been detected using CF3O− (Bates et al., 2014; Nguyen et al., 2015; Schwantes et al., 2015; Teng et al., 2015) and iodide as reagent ions (Lee et al., 2014a, 2016; Xiong et al., 2015, 2016; Nah et al., 2016b).
2.4.3 Online analysis of particulate matter
Total (organic plus inorganic) mass of particulate nitrates is routinely quantified using online AMS (Jayne et al., 2000; Allan et al., 2004), from which the mass of organic nitrates can be obtained by three techniques. First, the ratio (or ratio) in the mass spectra is used to distinguish organic from inorganic nitrates (Fry et al., 2009, 2013; Farmer et al., 2010; Xu et al., 2015b; Kiendler-Scharr et al., 2016). It is noted that the approaches zero in the case of low or nonexistent signal, while gives large numbers. Second, positive matrix factorization (PMF) of data matrices including the NO+ and ions in addition to organic ions (Sun et al., 2012; Hao et al., 2014; Xu et al., 2015b) is used. Third, the particulate inorganic nitrate concentration, as measured by an independent method such as ion chromatography, is subtracted from the total particulate nitrate concentration (Schlag et al., 2016; Xu et al., 2015a, b). A detailed comparison of these three methods is presented in Xu et al. (2015b). As the ratio in AMS data is dependent on instruments and the types of nitrates (inorganic and organic nitrates from different VOC oxidations), different strategies were developed when using this method to estimate particulate organic nitrates (Fry et al., 2013; Xu et al., 2015b).
A specialized inlet that selectively scrubs gaseous organic nitrates or collects particulate mass on a filter has been coupled to some of the techniques summarized in this section and utilized to observe particulate organic nitrates in the ambient atmosphere and laboratory studies. A TD-LIF equipped with a gas-scrubbing denuder (Rollins et al., 2010, 2012) and the filter inlet for gases and aerosols (FIGAERO) (Lopez-Hilfiker et al., 2014) at the front end of an iodide adduct HR-ToF-CIMS are examples (Lee et al., 2016; Nah et al., 2016b).
2.4.4 Offline analysis of particulate matter
Owing to its ability to analyze polar organic compounds without a prior derivatization step, liquid chromatography coupled to MS (HPLC/MS) is well suited for the characterization of SOA compounds originating from the reactions of BVOC and NO3. Unlike in GC/MS methods, a soft ionization technique such as electrospray ionization (ESI) is utilized to ionize target analytes in the LC/MS technique. In the ESI/MS, target analytes are detected as a cation adduct of a target analyte (e.g., [M + H]+ or [M + Na]+) for a positive mode or a deprotonated form of a target analyte ([M − H]−) for a negative mode. As a biogenic SOA compound typically bears a functional group, such as a carboxylic group or a sulfate group, that easily loses a proton, the negative-mode ESI ((−)ESI) is commonly applied to detect SOA compounds. High-resolution MS such as TOF or Fourier transform ion cyclotron (FTICR) MS is commonly used to assign chemical formulas for SOA compounds unambiguously.
The LC/(−)ESI-MS technique played a crucial role in relating the formation of organosulfates (OS) and nitrooxy-organosulfates (NOS) to NO3-initiated oxidation of BVOC in laboratory-generated and ambient SOA. Since these earlier works, a number of studies have reported the presence of OS and/or NOS compounds in ambient samples (Table S4), though most studies do not connect these compounds explicitly to the NO3 oxidation of BVOC. It should be noted that the direct infusion (−)ESI-MS technique rather than LC/(−)ESI-MS is often used for the analysis of fog, rainwater, and cloud water samples as diluted liquid water samples can be injected into the ion source directly without a sample pretreatment procedure. However, caution is warranted for the direct infusion technique because it cannot separate isobaric isomers and it is susceptible to ion suppression, especially from the presence of inorganic ions in the samples.
Whilst the LC or direct infusion (−)ESI-MS techniques have been successfully applied for the detection of the oxidation products from NO3-BVOC reactions, the techniques have been less successful in quantifying these compounds, mainly due to the lack of authentic standard compounds. The synthesis of these compounds should be a priority for future studies.
Finally, total organic nitrate functional groups within the particle phase have been quantified in ambient air using FTIR of particles collected on ZnSe impaction disks (low-pressure cascade impactor, size segregated) or Teflon filters (PM1) (Mylonas et al., 1991; Garnes and Allen, 2002; Day et al., 2010). The organic nitrate content of particles can be quantified offline as well by collection on quartz fiber filters, extraction into solution (e.g., with water–acetonitrile mixtures), and analysis using standard wet chemistry techniques such as high-pressure liquid chromatography coupled to electrospray ionization mass spectrometry (HPLC-ESI-MS) (Angove et al., 2006; Perraud et al., 2010; Draper et al., 2015).
2.5 Field observations
This section surveys the current literature on field observations of nitrate radicals and BVOC (Sect. 2.5.1), and organic nitrate aerosol attributable to NO3-BVOC chemistry (Sect. 2.5.2).
2.5.1 Nitrate radicals and BVOC
A few years after the first measurement of tropospheric NO3 (Noxon et al., 1980; Platt et al., 1980), it was recognized that the nitrate radical is a significant sink for BVOC, especially monoterpenes in terrestrial ecosystems and dimethyl sulfide (DMS) in maritime air influenced by continental NOx sources (Winer et al., 1984). The conclusion was based upon computer simulations using NO3 concentrations measured in field studies in the western US and Europe, and measured rate constants of NO3 with olefins. The scenarios in these simulations showed very low monoterpene concentrations in the early morning that were directly attributable to BVOC reactions with NO3. An analysis of NO3 formation rates at several urban and rural sites in Scandinavia (Ljungström and Hallquist, 1996) resulted in the conclusion that while night-time urban loss of NO3 is dominated by reaction with NO, the loss in rural regions is likely dominated by reactive hydrocarbons, especially monoterpenes.
Due to the fast reactions of NO3 with BVOC, lifetimes of NO3 in biogenically influenced environments can be very short, making simultaneous detection of VOC and NO3 in biogenic regions very difficult. For this reason, several studies have inferred levels of NO3 and its role in processing BVOC using observational analysis and supporting modeling. In particular, the rapid decay of isoprene after sunset has received considerable attention. Measurements of BVOC ~ 1–2 m above canopy level in a loblolly pine plantation in Alabama during the 1990 ROSE program (Goldan et al., 1995) were used to infer a nighttime NO3 mixing ratio of only 0.2 ppt and NO3 lifetime of only 7 s due to high levels of monoterpenes. The 4 h decay time of isoprene after sun-set could not be accounted for by gas reactions with NO3 and O3 although the decrease in the α- / β-pinene ratio at night was consistent with known NO3 and O3 chemistry. As part of the North American Research Strategy for Tropospheric Ozone – Canada East (NARSTO-CE) campaign, measurements of BVOC were made in Nova Scotia in a heavily forested region (Biesenthal et al., 1998). A box-model simulation based on the observational analysis found that the short lifetime of isoprene at night (τ = 1–3 h) could not be explained by the NO3 radical, which was estimated to be 0.1 ppt maximum at night due to low NOx and O3 levels and high monoterpene emissions. When OH yields from ozonolysis of BVOC were included in the model, this nighttime OH oxidant could partially account for the isoprene decay. During the Southern Oxidants Study (SOS) campaign in Nashville, TN (Starn et al., 1998), a chemical box model was used to show that rapid nighttime decays of isoprene were consistent with simulated NO3 but only when the site was impacted by urban NOx emissions. During the PROPHET study, measurements of VOC were made in a mixed forest approximately 10 m above the canopy surface (Hurst et al., 2001). Isoprene decays at night had an average lifetime of ~ 2.7 h. Box modeling showed that O3 reactions as well as dry deposition were insufficient to account for the decay, and that the NO3 radical was a significant sink only after the majority of isoprene had already decayed. On some nights, oxidation by OH could account for all the decay but the decay rates were overpredicted. The authors concluded that vertical transport of isoprene-depleted air aloft contributes to the fast initial decay of isoprene, followed by nighttime OH, NO3, and O3 chemistry decay. Steinbacher et al. (2005) reported on surface measurements in the Po Valley at a site 200–300 m from the closest edge of a deciduous forest. Bimodal diurnal cycles of isoprene were observed with morning and evening maxima that were reproduced by a Eulerian model. Isoprene decay lifetimes of 1–3 h were partially explained by NO3 decay, although a dynamic influence on isoprene decrease seemed to be likely including horizontal and vertical dispersion. During the HOHenpeissenberg Photochemistry Experiment (HOHPEX) field campaign, BVOC were analyzed via 2-D GC at a site located on a hilltop above adjacent rural agricultural/forested area that is frequently in the residual layer at night (Bartenbach et al., 2007). For the reactive monoterpenes, a significant non-zero dependency of the concentration variability on lifetime was found, indicating that chemistry (as well as transport) was playing a role in determining the ambient VOC concentrations. The night-time analysis gave an estimate of the NO3 mixing ratio of 6.2 ± 4.2 ppt, indicating it was a significant chemical factor in depletion of monoterpenes.
While the studies above made indirect conclusions about the role of NO3 in BVOC processing, field studies including direct measurements of NO3 are key to confirming the above findings. Golz et al. (2001) reported measurements of NO3 by long-path DOAS at an eucalyptus forest site in Portugal during the FIELDVOC94 campaign in 1994. The DOAS beam passed directly over the canopy at heights of 15 and 25 m, and as a result, they were unable to measure NO3 above the 6 ppt instrumental detection limit despite NO3 production rates of 0.4 ppt s−1. Rapid reaction with BVOC limited the NO3 lifetime to approximately 20 s such that NO3 reactions dominated other indirect losses, such as heterogeneous N2O5 uptake. Simultaneous measurements of NO3 and VOC during the Berliner Ozonexperiment (BERLIOZ) campaign in 1998 allowed one of the first assessments of the NO3 budget in comparison to OH and O3 oxidants (Geyer et al., 2001b). Surface measurements at this semi-rural location close to forests found the NO3 radical above detection limit (2.4 ppt) on 15 of 19 nights with a maximum of 70 ppt, a steady-state lifetime ranging from 20 to 540 s and N2O5 ranging from 2 to 900 ppt. The two most significant losses of NO3 were found to be its direct reaction with olefins (monoterpenes dominating) and indirect loss due to heterogeneous hydrolysis of N2O5. Over the study, it was possible for the first time to quantify the relative contribution of the NO3 radical to oxidation of VOC as 28 (24 h) and 31 % for olefinic VOC (24 h) compared to the total oxidation via NO3, OH, and O3. As part of the 1999 SOS study, NO3, isoprene, and its oxidation products were measured at a suburban forested site in Nashville, TN (Stroud et al., 2002). The nitrate radical measured at multiple beam heights by DOAS had maximum mixing ratios of 100 ppt that were generally found to anticorrelate with isoprene levels with significant vertical gradients on some nights. Early evening losses of isoprene were attributable to reaction with the NO3 radical. During the Pacific 2001 Air Quality Study (PACIFIC 2001) field campaign, NO3 was measured by long-path DOAS at an elevated forested site in the lower Fraser Valley of British Columbia with beam-path nighttime NO3 levels up to a maximum of 50 ppt (average of nighttime boundary layer and residual layer) (McLaren et al., 2004). Simultaneous analysis of carbonyl compounds in aerosol samples (Liggio and Mclaren, 2003) during the study found that only monoterpene oxidation products pinonaldehyde and nopinone (not reported) were enhanced in aerosol filters collected at night, evidence of the role of NO3 in nighttime oxidation of BVOC in the valley. In 2004 measurements of NO3 and N2O5 by CRDS, isoprene and its oxidation products were made on board the NOAA P-3 aircraft as part of the New England Air Quality Study (NEAQS) and International Consortium for Atmospheric Research on Transport and Transformation (ICARTT) campaigns in the northeast US (Brown et al., 2009). These studies found a very clear anticorrelation between isoprene levels after dark and NO3 mixing ratios, which varied as high as 350 ppt when isoprene was absent from the air mass. The loss frequencies (i.e., first-order loss rate constants) of NO3 were strongly correlated with the loss rate constant of NO3 with isoprene for lifetimes less than 20 min, clearly showing that isoprene was the most important factor determining the lifetime of NO3. It was also shown that more than 20 % of emitted isoprene was oxidized at night and that 1–17 % of SOA was contributed by NO3-isoprene oxidation. A number of recent studies have also investigated the role of NO3+ BVOC chemistry in more polluted areas. In many urban areas, the NO3+ BVOC chemistry occurs in parallel to heterogeneous NO3 / N2O5 chemistry and reactions of NO3 with anthropogenic VOC. Examples of such environments have been discussed in Brown et al. (2011, 2013) and Stutz et al. (2010) who presented observations in Houston, TX. Brown et al. (2011) and Stutz et al. (2010) found that up to 50 % of the NO3+ VOC reactions in Houston are driven by isoprene, with the other VOC emitted by industrial sources. Surprisingly, heterogeneous NO3 / N2O5 chemistry plays a minor role in Houston. Brown et al. (2011) also point out that the nocturnal VOC oxidation by NO3 dominates over that by ozone. Nocturnal NO3 formation rates were rapid and comparable to those of OH during the day. Crowley et al. (2011) compared NO3 chemistry in air masses of marine, continental, and urban origin at a field site in southern Spain. Under all conditions, NO3+ BVOC reactions (predominately α-pinene and limonene) contributed to the overall NO3 reactivity, confirming other observations that concluded that this chemistry is important in all environments where BVOC sources are present. In the southeastern US summer, this importance extends even through the daytime, when photolysis and NO reactions compete (Ayres et al., 2015). The NO3+ BVOC reaction rates observed in these studies imply a high production rate of SOA and organic peroxy radicals.
2.5.2 Organic nitrate aerosols
There are many factors that motivate understanding organic nitrate in the particulate phase through field deployment of a variety of instrumentation, much of which is described in other sections of this review. Nitrogen-containing organic fragments (not necessarily organic nitrates) have been identified in atmospheric particles using mass spectrometric techniques (Reemtsma et al., 2006; Farmer et al., 2010; O’Brien et al., 2014). Total atmospheric organic nitrates, as well as organic nitrates segregated by phase, also have been measured in the atmosphere using techniques such as TD-LIF, CIMS, etc. (Day et al., 2003; Beaver et al., 2012). Given these observations and the propensity of organic nitrate compounds to partition to the condensed phase to create SOA (Rollins et al., 2013), it is critical to determine the level of organic nitrates that reside specifically in the atmospheric aerosol phase under typical ambient conditions and to identify the chemical and physical processes that determine their concentrations. It is also important to note that formation of SOA that contains organic nitrate groups has the potential to sequester NOx, thereby influencing the cycling of atmospheric oxidants.
Organic nitrates in urban PM that were identified using functional group analyses such as FTIR spectroscopy have been attributed to emission of nitrogen-containing primary organic aerosol or to involvement of reactive nitrogen compounds in SOA formation chemistry (Mylonas et al., 1991; Garnes and Allen, 2002; Day et al., 2010). Other more advanced techniques, such as TD-LIF enhanced with the ability to separate phases or techniques to obtain high-resolution mass spectra (HR-ToF-AMS), have been utilized to quantify the amount of organic nitrate in particles in areas less likely to be influenced strongly by BVOC emissions, such as urban areas or areas influenced by oil and gas operations (Lee et al., 2015). Of specific interest here, however, are observations of organic nitrate PM in areas with a significant influence of BVOC, especially if co-located measurements allow for insight into the role that NO3 plays in the initial BVOC oxidation step. As such, we focus here on online measurements and on measurements that allow specific attribution to BVOC-NO3 reactions. Such measurements broadly can be categorized by region of sampling: the eastern United States (US), the western US, and Europe. Figure 5a summarizes average mass concentrations of submicrometer particulate organic nitrates (NO3, org) and particulate inorganic nitrates (NO3, inorg) in different months at multiple sites around the world. Figure 5b summarizes the corresponding percentage (by mass) of submicrometer particulate organic nitrate aerosols in ambient organic aerosols. Detailed information and measurements for each site are provided in Table S5.
Figure 5.

(a) Average mass concentrations (in μg m−3, ambient temperature and pressure) of submicrometer particulate organic nitrates (NO3, org) and particulate inorganic nitrates (NO3, inorg) in different months at multiple sites. The concentrations correspond to mass concentrations of –ONO2 functionality. Note that the y axis is different for sites with total nitrates greater than 1 μg m−3 (shaded). Detailed information and measurements for each site are provided in Table S5. (b) Percentage (by mass; cyan) of submicrometer particulate organic nitrate aerosols in ambient organic aerosols in different months at multiple sites. Detailed information and measurements for each site are provided in Table S5.
Eastern United States
The first reports of aerosol organic nitrates in the southeastern (SE) US resulted from composition analysis of four daily PM filter samples from four Southeastern Aerosol Research and Characterization (SEARCH) network sites during summer 2004. Filters were analyzed for polar compounds, with particular focus on organosulfates, using offline chromatographic–MS methods (Gao et al., 2006; Surratt et al., 2007, 2008). Several nitrooxy organosulfates were identified, but the only one quantified (1–2 % of organic mass) was associated with α-pinene photooxidation or reaction with NO3. Several of the nitrooxy organosulfates were likely the same as products from BVOC-oxidant–NOx -seed systems based on comparison to spectra collected from chamber studies.
Brown et al. (2013) examined several nighttime aircraft vertical profiles in Houston (October 2006 during the Texas Air Quality Study 2006) that showed increases of total nitrate aerosol (and increases in AMS m/z 30 to m/z 46 ratio, the unit mass resolution approximation for , indicative of organic nitrates; Farmer et al., 2010) and oxygenated organic aerosol (OOA). The OA versus carbon monoxide (CO) slopes at lower altitudes were consistent with SOA sources from NO3-BVOC reactions, with a combination of observations and zero-dimensional modeling showing 1 to 2 μg m−3 SOA formation from NO3-BVOC oxidation overnight with formation rates of 0.05 to 1 μg m−3 h−1.
More recently, during the summer Southern Oxidant and Aerosol Study (SOAS; mixed, semi-polluted forest) in Alabama (2013), an unprecedented suite of instruments quantified particle-phase organic nitrates using five different online methods: HR-ToF-AMS ( ), HR-ToF-AMS – PiLS (particle-into-liquid sampler) ion chromatography (PiLS-IC), HR-ToF-AMS (PMF), TD-LIF (denuded), and iodide CIMS. Total particle-phase nitrates increased throughout the night and peaked in early/mid-morning. Xu et al. (2015b) systematically evaluated the three AMS-related methods in estimating ambient particulate organic nitrate concentrations. Analysis presented in Xu et al. (2015a, b) using the HR-ToF-AMS – PiLS-IC method showed that organic nitrate functional groups comprised ~ 5–12 % of OA mass and correlated with PMF-derived less-oxidized oxygenated OA (LO-OOA). Two-thirds of the LO-OOA was estimated to be formed via NO3-BVOC chemistry (dominantly monoterpenes, ~ 80 %), with the balance due to ozone (O3)-BVOC chemistry. Organic nitrates were calculated to comprise 20–30 % of the LO-OOA factor. Ayres et al. (2015) used a measurement-constrained model for nighttime that compared NO3 production/loss to total organic nitrate (HR-ToF-AMS , TD-LIF) formation to calculate a molar yield of aerosol-phase organic nitrates of 23–44 % (organic nitrate formed per NO3-BVOC reaction) that was dominated by monoterpene oxidation. They noted that the estimated yield was low compared to aggregated aerosol-phase organic nitrate yields, possibly due to rapid nitrate losses not considered in the model. Organic nitrate hydrolysis in the particle phase is one potential loss pathway, although recent laboratory studies suggest this process is slow for NO3 + β-pinene SOA (Boyd et al., 2015). Also, particle-phase organic nitrates were observed to contribute 30–45 % to the total NOy budget. Lee et al. (2016) quantified speciated particle-phase organic nitrates using iodide CIMS (88 individual C4-C17 mono/dinitrates). A large fraction was highly functionalized, with six to eight oxygen atoms per molecule. Diurnal cycles of isoprene-derived organic nitrates generally peaked during daytime, and monoterpene-derived organic nitrates peaked at night or during early/mid-morning. Using an observationally constrained diurnal zero-dimensional model, they showed that the observations were consistent with fast gas–particle equilibrium and a short particle-phase lifetime (2–4 h), again possibly due to hydrolysis if the field-derived lifetimes for particle-phase organic nitrates can be reconciled with recent laboratory studies (Boyd et al., 2015). The sum of the CIMS particle-phase organic nitrates (mass of nitrate functional groups only) was correlated with the two total aerosol organic nitrate AMS-based methods (R2 = 0.52, 0.67) with slopes of 0.63 and 0.90 (Lee et al., 2016). The CIMS sum was also correlated with the total measured with the TD-LIF method (R2 = 0.55); however, since the TD-LIF measurements were ~ 2–4 times higher (depending on period) than the AMS-based methods, the CIMS versus TD-LIF slope was substantially lower (0.19). Reasons for the differences between the total organic nitrate measured by different methods have been investigated but remain unclear.
A seasonal and regional survey of particle-phase organic nitrates is reported by Xu et al. (2015b) using a HR-ToF-AMS and an aerosol chemical speciation monitor (ACSM) (Ng et al., 2011) at four rural and urban sites in the greater Atlanta area (2012–2013) and in Centreville, AL (summer 2013 only, SOAS). They show strong diurnal cycles during summer, peaking early/mid-morning, and cycles with similar timing but smaller magnitude during winter. The concentrations were slightly higher in summer, which was attributed to compensating effects of source strength and gas–particle partitioning. Shallower boundary layers during winter also may have played a role in making the summer and winter concentrations more similar (Kim et al., 2015).
Fisher et al. (2016) report a broad regional survey of particle-phase (and gas-phase) organic nitrates (HR-ToF-AMS ) during summertime for the Studies of Emissions and Atmospheric Composition, Clouds and Climate Coupling by Regional Surveys (SEAC4RS) aircraft campaign (August–September, 2013, SE US only) as well as the ground-based SOAS measurements. A substantial vertical gradient was observed in particle-phase organic nitrates, with concentrations decreasing by several-fold from the boundary/residual layer into the free troposphere. Consistent with SOAS ground observations, 10–20 % of observed boundary layer total (gas plus particle) organic nitrates were in the particle phase for the aircraft measurements.
In addition to the measurements made in the SE US, characterization of aerosol organic nitrates has been performed in New England. As part of the New England Air Quality Study (NEAQS) in summer 2002, Zaveri et al. (2010) observed evolution of aerosols in the nocturnal residual layer with an airborne quadrupole (Q)-AMS in the Salem Harbor power plant plume. The aerosols were acidic and internally mixed, suggesting that the observed nitrate was in the form of organic nitrate and that the enhanced particulate organics in the plume were possibly formed from NO3-initiated oxidation of isoprene present in the residual layer.
Western United States
Significant work on understanding ambient organic nitrate formation from BVOC-NO3 has been performed in California. During the California Research at the Nexus of Air Quality and Climate Change (CalNex) field campaign from mid-May through June 2010, Rollins et al. (2012, 2013) measured particulate total alkyl and multifunctional nitrates (pΣANs) with TD-LIF at a ground site in Bakersfield, California. They attributed the increase in pΣAN concentrations at night to oxidation of BVOC by NO3 forming SOA, with an estimated 27 to 40 % of the OA growth due to molecules with nitrate functionalities. On average, 21 % of ΣANs were in the particle phase and increased with OA, which was fit to a volatility basis set in which pΣANs / ΣANs increased from ~ 10 % at < 1 μg m−3 and plateaued at ~ 30 % by ~ 5 μg m−3. At the same site, using PMF analysis of FTIR and HR-ToF-AMS measurements, Liu et al. (2012a) showed that the organic nitrate-containing biogenic SOA condensed onto 400 to 700 nm sized primary particles at night. As part of the Carbonaceous Aerosol and Radiative Effects Study (CARES) in June 2010, Setyan et al. (2012) observed enhanced SOA formation due to interactions between anthropogenic and biogenic emissions at a forest site in the foothills of the Sierra Nevada mountains, approximately 40 km downwind of Sacramento. While nitrate accounted for only ~ 4 % of the particle mass measured by a HR-ToF-AMS, it was attributed potentially to organic nitrates based on the much higher ion ratio than observed in pure ammonium nitrate.
During the Rocky Mountain Biogenic Aerosol Study field campaign in Colorado’s Front Range (rural coniferous montane forest) (BEACHON-RoMBAS) from July to August 2011, Fry et al. (2013) observed aerosol-phase organic nitrates by optical spectroscopic (denuded TD-LIF) and mass spectrometric (HR-ToF-AMS, ) instruments. The two methods agreed well on average (AMS/TD-LIF slope of 0.94–1.16, depending on averaging method) with a fair correlation (R2 = 0.53). Similar to studies in other forested environments, the organic nitrate concentration was found to peak at night. The organic nitrate concentration was positively correlated with the product of the nitrogen dioxide and O3 mixing ratios but not with that of O3 alone; this suggested nighttime NO3-initiated oxidation of monoterpenes as a significant source of nighttime aerosol organic nitrates. The gas–particle partitioning also showed a strong diurnal cycle, with the fraction in the particle phase peaking at ~ 30 % at night and decreasing to a broad minimum of ~ 5 % during daytime, which suggests a change in composition in addition to thermodynamic partitioning effects.
Europe
Iinuma et al. (2007) analyzed ambient aerosol samples collected on filters in a Norway spruce forest in northeastern Germany during the BEWA campaign (Regional biogenic emissions of reactive volatile organic compounds from forests: process studies, modeling, and validation experiments) and compared the results to those from chamber studies. The filter extracts were analyzed using LC-ESI-ToF-MS in parallel to ion trap MS. Several nitrooxy organosulfates with significant mass in the BEWA ambient samples were enhanced in the nighttime samples relative to the daytime samples. Their abundance in the nighttime samples strongly suggests that NO3-monoterpene chemistry in the presence of sulfate aerosols has an important role in the formation of these nitrooxy organosulfate aerosols.
A similar study by Gómez-González et al. (2008) focused on isoprene through LC-multidimensional MS (MSn) analysis of filter samples from both chamber studies and ambient summer day/night PM2.5 samples from K-Puszta, Hungary, a mixed deciduous/coniferous forest site. Although not the focus of the study, they confirmed the presence of significant quantities of nitrooxy organosulfates that were enhanced in the nighttime samples over the daytime samples.
Initial online evidence of the production of organic nitrate aerosols in Europe was provided by Allan et al. (2006) when studying nucleation events driven by BVOC oxidation in Hyytiälä, a (boreal) forested region in Finland. The Q-AMS m/z 30 to m/z 46 ratio (the unit mass resolution approximation for ratio) was frequently found to be very high, ~ 10, for a distinct organic Aitken mode that became apparent late in the afternoon and increased at night. They hypothesized that the excess m/z 30 (NO+) signal was associated with organic nitrates, although could not rule out amine contributions. During the same field study, Vaattovaara et al. (2009) applied two tandem differential mobility analyzer methods to study the evolution of the nucleation-and Aitken-mode particle compositions at this boreal forest site. The results showed a clear anthropogenic influence on the nucleation- and Aitken-mode-particle compositions during the events and suggested organic nitrate and organosulfate aerosol was generated from monoterpene oxidation. Also, it was shown that organic nitrate was enhanced in aerosol exposed to elevated temperatures, implying low volatility of organic nitrates (Häkkinen et al., 2012).
More recently, Hao et al. (2014) used a HR-ToF-AMS on a tower in Kuopio, Finland, 224 m above a lake surrounded by a mixed forest of mostly coniferous (pine and spruce) mixed with deciduous trees (mostly birch) to measure submicron aerosol composition. The site also was influenced by urban emissions. A particular focus of the study was to separate organic and inorganic nitrate using PMF. They found that ~ 37 % of the nitrate mass at this location and time could be allocated to organic nitrate factors, the rest being inorganic nitrate. The organic nitrate aerosol was segregated into two organic factors, less-oxidized OOA (LO-OOA), and more-oxidized OOA (MO-OOA) (previously called SV- and LV-OOA, respectively); the majority (74 %) of the organic nitrate was found to be in the more volatile LO-OOA factor. Based on meteorology, the air mass source of the organic nitrate aerosol was from a sector with residential and forested areas. Again, the organic nitrate aerosol showed a diurnal trend that was highest at night.
An analysis of AMS data taken across Europe within EU-CAARI and EMEP intensive measurement campaigns (Kulmala et al., 2011; Crippa et al., 2014) has recently shown high organic nitrate contributions to total measured PM1 nitrate (Kiendler-Scharr et al., 2016). The spatial distribution and diurnal pattern of particulate organic nitrate indicate a gradient of concentration. High concentrations are found in source regions with NOx emissions and during the night. Low concentrations are found in remote regions and during the day. EURAD-IM simulations for Europe show an increase of SOA by 50 to 70 % when considering SOA formation by NO3 oxidation with maximum ground-level concentrations of SOA from NO3 oxidation in the range of 2 to 4 μg m−3 (Li et al., 2013; Kiendler-Scharr et al., 2016).
Summary of organic nitrate aerosol observations
Taken together, the observations of particle-phase organic nitrates in the US and Europe suggest that particle-phase organic nitrates (formed substantially via NO3-BVOC chemistry) are ubiquitous, especially in, but not limited to, summer. Their formation appears to play an important role in SOA formation, which can potentially be underestimated due to short particle-phase lifetimes. Regions with widespread NOx and BVOC emissions and a humid climate may create optimal conditions for a rapid life cycle of particle-phase organic nitrates.
2.6 Models of NO3-BVOC chemistry
To understand the implications of NO3-BVOC chemistry on atmospheric chemistry as a whole, under both current and future scenarios, the physical and chemical processes, such as those reported in Sect. 2.1 through Sect. 2.3, must be parameterized in numerical models. In this section, we summarize how these reactions are represented in current air quality models (AQMs).
2.6.1 Chemical mechanisms
Organic nitrates are produced from the reactions of VOC with OH followed by NO as well as with NO3, and both of these pathways are represented in chemical mechanisms albeit at varying levels of detail. The use of the term “model” below refers to the treatment of BVOC + NO3 chemistry in lumped chemical mechanisms. The products formed from the OH-initiated (typically daytime) versus NO3-initiated (typically nighttime) chemistry may or may not be treated separately.
The NO3-BVOC reactions result in an RO2 that reacts with NO3, other RO2, HO2, or NO. RO2-NO reactions for NO3-initiated chemistry are relatively unimportant due to rapid reaction of NO with NO3 at night (Perring et al., 2009), but they are included in models. Unimolecular rearrangements of the NO3-initiated RO2 radical are not currently considered in models (Crounse et al., 2011). The products of the initial NO3-BVOC reaction may retain the nitrate group, thus forming an organic nitrate or releasing nitrogen as NO2. The branching between organic nitrate formation and N recycling is parameterized in models. Table 4 summarizes the gas-phase organic nitrate yields for isoprene and monoterpene oxidation by NO3 in a number of currently available chemical mechanisms. The yields represent the first-generation yields since products may react to form further organic nitrates or release NO2. The organic nitrate yield values span from 0 (e.g., SAPRC07 isoprene) to 100 % (e.g., MCM isoprene). Although GEOS-Chem v10-01 does not consider gas-phase monoterpene chemistry, the model has recently been updated to consider a 10–50 % yield of organic nitrates from the monoterpene-NO3 reaction independent of the nitrate-RO2 fate but dependent on monoterpene identity (Fisher et al., 2016). Differences in the organic nitrate yield from NO3 oxidation result from a number of causes including treatment of RO2 fate, assumptions about decomposition versus retention, and prioritization of functional group identity.
Table 4.
Gas-phase organic nitrate yields (in percent) from BVOC + NO3 systems in current chemical mechanisms. Gas-phase organic nitrate yields depend on RO2 fate as indicated in the ternary diagrams; clockwise from the top: RO2 reacts with NO3, RO2, and HO2.
| Chemical mechanism | Gas-phase yield of organic nitrates from isoprene+NO3 | Gas-phase yield of organic nitrates from monoterpenes+NO3 | References |
|---|---|---|---|
|
| |||
| CB05 |
|
|
(Yarwood et al., 2005) |
| CB6r2 |
|
same as CB05 | (Perring et al., 2009; Hildebrandt Ruiz and Yarwood, 2013) |
| GECKO-A | up to 100% (see supporting information) | same as isoprene | (Aumont et al., 2005) |
| GEOS-Chem v10-01 |
|
NA (monoterpene oxidation is offline) | (Mao et al., 2013) |
| GFDL AM3 |
|
|
|
| MCM v3.3.1 |
|
|
(Jenkin et al., 1997; Saunders et al., 2003; Jenkin et al., 2015) |
| MOZART |
|
|
(Emmons et al., 2010) with updates on organic nitrates |
| SAPRC07 |
|
|
(Carter, 2010b; Carter, 2010a) Plots of RO2+RO2 based on RO2C |
| SAPRC07tic |
|
|
(Rollins et al., 2009; Xie et al., 2013) α-pinene (same as SAPRC07) Other monoterpenes: (Pye et al., 2015) |
Some models parameterize the yield of organic nitrates as a function of RO2 fate while others, such as the carbon bond-based (CB) mechanisms, treat all RO2 fates the same. The MCM v3.3.1 also considers the yield of isoprene organic nitrates to be independent of RO2 fate, but monoterpene organic nitrate yields are variable between 0 and 100 % depending on RO2 fate. Differences in organic nitrate formation, due to treating the organic nitrate yield as a function of RO2 fate, may vary with atmospheric conditions. Reactions with both HO2 and RO2 are significant at night (Xie et al., 2013; Pye et al., 2015). RO2-NO3 may be important in urban areas or locations where BVOC concentrations are not so high as to deplete NO3 (Rollins et al., 2012).
Mechanisms differ in their assumptions about whether or not the organic nitrates from NO3-initiated chemistry release NO2 or retain the nitrate group. An example of this difference in treatment of organic nitrates can been seen in the reactions of nitrated peroxy radicals with different radicals (NO, HO2, RO2) predicted by SAPRC07 and MCM. MCM predicts greater loss of the nitrate group, while SAPRC tends to retain it, leading to either < 5 % (MCM) or > 50 % (SAPRC) organic nitrate yields.
In order to predict accurately the fates of RO2 and yield of organic nitrates, models must also include information on RO2 reaction rate constants. Some mechanisms use the same set of RO2 rate constants for all hydrocarbons. However, the MCM (Jenkin et al., 1997; Saunders et al., 2003) indicates that the RO2-HO2 rate constant should vary with carbon number (n) and predict k = 2.91 × 10−13 exp(1300/T) [1 − exp(−0.245n)] molec−1 cm3 s−1. The MCM RO2-RO2 rate constant varies between 2 × 10−12 cm3 molec−1 s−1 (based on C1-C3 primary RO2 with adjacent O or Cl) and 6.7 × 10−15 cm3 molec−1 s−1 for tertiary alkyl RO2 (based on t-C4H9O2). RO2-NO3 and RO2-NO rate constants are estimated as 2.3 × 10−13 and 9.0 × 10−12 cm3 molec−1 s−1 at 298 K.
AQMs and chemistry–climate models typically cannot handle the complexity associated with tracking each individual VOC and all its possible reaction products. As a result, surrogate species are often used to represent classes of compounds (e.g., CB05, which uses the designation NTR to indicate organic nitrates). This mapping can cause yields of organic nitrates to be falsely low in a mechanism if other functional groups are prioritized over nitrate in the mapping of predicted products to mechanism species. Compared to the other mechanisms in Table 4, SAPRC07 monoterpenes tend to have very low organic nitrate yields as a result of prioritization of peroxide and non-nitrate functional groups. If nitrate groups were prioritized, SAPRC07 would more closely resemble the “other monoterpene” yields from SAPRC07tic. In addition, the diversity across mechanisms in the RO2-HO2 monoterpene organic nitrate yields would be reduced such that they would all indicate > 50 % organic nitrate yields and all but the CB mechanisms would predict a 100 % yield of organic nitrates from RO2-HO2. The RO2-HO2 pathway is relatively unstudied in laboratory conditions due to difficulties in maintaining sufficient concentrations of both NO3 and HO2 radicals (Boyd et al., 2015; Schwantes et al., 2015).
2.6.2 Influence on organic aerosol
Nitrate radical oxidation can lead to significant amounts of SOA on global and regional scales. Due to a lack of information on the identity and volatility of later-generation BVOC + NO3 products, most models parameterize SOA formation separately from gas-phase chemistry using either the Odum two-product (Odum et al., 1996) fit, volatility basis set (VBS) (Donahue et al., 2006) fit, or fixed yield (Table 5). Based on the understanding of SOA pathways at the time, Hoyle et al. (2007) found that up to 21 % of the global average SOA burden may be due to NO3 oxidation, and Pye et al. (2010) predicted ~ 10 % of global SOA production was due to NO3. Regional contributions to SOA concentrations can be much higher (Hoyle et al., 2007; Pye et al., 2010). Nitrate radical reactions themselves are estimated to account for less than 3 % of isoprene oxidation and less than 2 % of sesquiterpene oxidation globally; however, they account for 26 % of bicyclic monoterpene oxidation (Pye et al., 2010). Representations of monoterpene-NO3 SOA are more widespread in chemistry–climate models than other BVOC-NO3 SOA parameterizations due to the relatively early recognition of its high yields (e.g., Griffin et al., 1999) and relative importance for SOA. Inclusion of SOA from isoprene-NO3 is more variable as reflected in Table 5.
Table 5.
Treatment of SOA formation from BVOC-NO3 systems in current 3-D models.
| Model | Gas-phase chemistry | Isoprene + NO3 parameterization | Monoterpene + NO3 parameterization |
|---|---|---|---|
| CAMx v6.20 with SOAP | CB05, CB6, or SAPRC99 | No SOA from this path | NO3 SOA yields same as photooxidation (OH + ozone) yields1 |
| CAMx v6.20 with 1.5-D VBS | CB05, CB6, or SAPRC99 | NO3 SOA yields same as photooxidation (OH + ozone) yields2 | NO3 SOA yields same as photooxidation (OH + ozone) yields2 |
| CMAQ v5.1 cb05e51-AERO6 | CB05 with additional modification3 | Odum two-product approach based on Kroll et al. (2006) photooxidation (OH) yields3 | Odum two-product approach based on Griffin et al. (1999a) photooxidation (OH + ozone) yields4 |
| CMAQ v5.1 SAPRC07tc-AERO6 | SAPRC075 with two monoter- penes: α-pinene (APIN) and other monoterpenes (TERP) | Odum two-product approach based on Kroll et al. (2006) photooxidation (OH) yields3 | Odum two-product approach based on Griffin et al. (1999a) photooxidation (OH + ozone) yields4 |
| CMAQ v5.1 SAPRC07tic-AERO6i | SAPRC07tic6,7 | based on semivolatile organic nitrate from isoprene dinitrate8 | no SOA from α-pinene + NO3; SOA from other monoterpenes based on semivolatile organic nitrates8 |
| EURAD-IM | RACM | Odum two-product approach 9 | Odum two-product approach 10 with T dependence11,12 |
| GEOS-Chem v10-01 | GEOS-Chem v10-01 with speciated isoprene nitrates6,7 | VBS fit9,13 | VBS fit to β-pinene + NO3 experiment10,13 |
| GFDL AM3 | GFDL AM3 | no SOA from this pathway | Odum two-product approach based on β-pinene + NO310,14 |
| GISS-GCM II | NA (offline oxidants) | no SOA from this pathway | Odum two-product approach based on β-pinene + NO310,14 |
| GLOMAP/ UKESM-1 | VOC + NO3 | Based on Kroll et al. experiments (2006), set to 3 %15 | Based on Tunved et al. (2004), set to 13 %15 |
| STOCHEM-CRI | MCM | CRI species fit to MCMv3.1 simulations16,17,18 | CRI species fit to MCMv3.1 simulations 16,17,18 |
| WRF-Chem v3.6.1 | MOZART-MOSAIC | no SOA from this pathway | VBS fit to β-pinene + NO3 experiment10,19 |
Appel et al. (2016).
Utembe et al. (2009).
Johnson et al. (2006).
SOA from BVOC-NO3 reactions traditionally has been parameterized on the initial hydrocarbon reaction assuming semivolatile products and an Odum two-product approach (e.g., Chung and Seinfeld, 2002). This treatment is often implemented in parallel to the gas-phase chemistry, meaning that later-generation products leading to SOA are not identified. Information is still emerging on the fate of organic nitrates, and that information is just beginning to be included in models. Hydrolysis of particle-phase organic nitrates is one such process more recently considered with impacts for both O3 and PM in models (Hildebrandt Ruiz and Yarwood, 2013; Browne et al., 2014; Pye et al., 2015; Fisher et al., 2016).
2.6.3 Influence on reactive nitrogen and ozone
The influence of BVOC nighttime oxidation on the nitrogen budget remains unclear. Current modeling efforts have mainly focused on the nighttime oxidation of isoprene, which is dominated by isoprene-NO3 reaction. This pathway is initialized via addition of NO3 to one of the double bonds, as discussed in Sect. 2.1.2. Due to the additional stabilization from alkoxy radical and nitrate functional groups (Paulson and Seinfeld, 1992), the yield of first-generation organic nitrates is relatively high (62–78 %; Table 2); they may react with NO3 again to produce secondary dinitrates (Perring et al., 2009; Rollins et al., 2009, 2012). Assuming little NOx is recycled from these organic nitrates, most models suggest that nighttime oxidation of isoprene by NO3 contributes significantly to the budget of organic nitrates (von Kuhlmann et al., 2004; Horowitz et al., 2007; Mao et al., 2013; Xie et al., 2013). Two recent studies (Suarez-Bertoa et al., 2012; Müller et al., 2014), however, suggest fast photolysis of carbonyl nitrates with high efficiency of NOx recycling, which could lead to release of NOx in the next day. Further modeling is required to investigate the importance of nighttime isoprene oxidation on the nitrogen budget.
Very little modeling effort has been dedicated to the influence of nighttime terpene oxidation on the nitrogen budget, mainly due to the lack of laboratory data on oxidation products and their fate. In contrast to isoprene, terpene emissions are temperature sensitive but not light sensitive (Guenther et al., 1995), leading to a significant portion of terpene emissions being released at night. The high yield of organic nitrates and SOA from the terpene-NO3 reaction (Fry et al., 2009, 2011, 2014; Boyd et al., 2015) provides an important sink for NOx at night, likely larger than for isoprene-NO3 over the eastern US (Warneke et al., 2004). Recent laboratory experiments suggest that aerosol organic nitrates can be either a permanent or temporary NOx sink depending on their monoterpene precursors (and hence nature of the resulting RO2) as well as ambient RH (Boyd et al., 2015; Nah et al., 2016b). In order to understand the impact of terpenes on nighttime chemistry, a fully coupled model of terpene-NOx chemistry will be required, as monoterpenes can be the dominant loss process for NO3 and N2O5 at night (Ayres et al., 2015).
While a significant portion of nitrogen is emitted at night (Boersma et al., 2008), the impact of nighttime chemistry on the initiation of the following daytime chemistry has received little attention in regional and global models. Different treatments of NO3 chemistry can result in 20 % change in the following daytime O3 concentration, as shown by a 1-D model study (Wong and Stutz, 2010) and box model simulations (Millet et al., 2016). This impact can be further complicated by uncertainty in emissions of BVOC and model resolutions. For example, a recent study by Millet et al. (2016) shows that in a city downwind of an isoprene-rich forest, daytime O3 can be largely modulated by the chemical removal of isoprene throughout the night. Such local-scale events may only be captured by a very high-resolution model with detailed characterization of emission sources. It is important to assess this impact on a global scale using 3-D chemistry models, owing to the profound coupling of boundary layer dynamics and chemistry. Quantifying the impact of BVOC-NO3 chemistry on NOx fate is important given the long-standing problem in current global and regional AQMs of a large overestimate of O3 over the eastern US in summer (Fiore et al., 2009).
2.6.4 Comparison of field data with air quality models
Recent field campaigns (SOAS, SEAC4RS, EUCAARI, EMEP) have allowed for the attribution of SOA to NO3 oxidation to provide model constraints not previously available. Pye et al. (2015) and Fisher et al. (2016) implemented updated BVOC + NO3 chemistry in CMAQ and GEOS-Chem, respectively, to interpret data in the SE US during the summer of 2013 (SOAS and SEAC4RS). Model predictions of gas-phase monoterpene nitrates (primarily NO3 derived) were higher than the sum of C10H17NO4 and C10H17NO5 (Nguyen et al., 2015) by a factor of 2–3 (Fisher et al., 2016) and 7 (Pye et al., 2015), consistent with a significant fraction of the monoterpene nitrates being highly functionalized (Lee et al., 2016). The studies identified particle-phase hydrolysis as an important modulator of particulate organic-nitrate concentrations and organic nitrate lifetime. The GEOS-Chem simulation reproduced the particle-phase organic nitrate diurnal cycles (SOAS), boundary layer concentrations, and gas–particle partitioning reasonably well; however, it underestimated concentrations in the free troposphere, possibly due to measurement limitations and/or the implementation of rapid uptake followed by hydrolysis of all gas-phase organic nitrates in the model, which may not be valid for non-tertiary organic nitrates (Fisher et al., 2016).
3 Perspectives and outlook
Section 3 outlines perspectives on the implications of NO3-BVOC atmospheric chemistry with respect to (1) aerosol optical and physical properties; (2) health effects; (3) trends in NOx emissions and organic aerosols and their implications for control strategies related to particulate matter; (4) critical needs for analytical methods; (5) critical needs for models; (6) field studies in the developing world and under-studied areas; and (7) critical issues to address in future field and laboratory measurements in light of current understanding of this chemistry and trends in emissions.
3.1 Aerosol optical and physical properties
The climatic effects of atmospheric aerosols depend on their various physical and chemical properties. Hygroscopicity, cloud condensation nuclei (CCN) activity, optical properties (namely light absorption and scattering), and ability to act as CCN and ice nuclei (IN) are the key aerosol properties that would determine their ability to affect climate. Additional properties such as aerosol number size distribution, chemical composition, mixing state, and morphology will determine whether the aerosols will be optically important or whether they would affect cloud properties. These aerosol properties depend on the sources, aging processes, and removal pathways that aerosols experience in the atmosphere (Boucher, 2013).
Absorption by aerosol may affect the cloud lifetime and altitude due to heating of the atmosphere (Mishra et al., 2014). They can also change the atmospheric lapse rate, which in turn can result in modification in aerosol microphysics in mixed-phase, ice, and convective clouds (Boucher, 2013). In addition to direct emissions of known absorbing particles (black carbon, mineral dust, biomass burning aerosols), SOA may also have absorption properties. The absorbing component of organic carbon (OC), namely “brown carbon” (BrC), is associated with OC found in both primary and secondary OC and has a spectral-dependent absorption that smoothly increases from short visible to UV wavelengths (Bond and Bergstrom, 2006). It has been suggested that BrC is a component of SOA that is composed of high molecular weight and multifunctional species such as humic-like substances, organic nitrates, and organosulfate species (Andreae and Gelencser, 2006; Bond and Bergstrom, 2006; Ramanathan et al., 2007b; Laskin et al., 2015; Moise et al., 2015). Many modeling studies often assume that BC and mineral dust are the only two significant types of light-absorbing aerosols on the global scale. Therefore, they treat SOA as a purely scattering component that leads to climate cooling (Stier et al., 2007; Bond et al., 2011; Ma et al., 2012). However, observations suggest that BrC is widespread mostly around and downwind urban centers (Jacobson, 1999). In such places, BrC may have significant contribution, and in some cases it may dominate the total aerosol absorption at specific (short) wavelengths (Ramanathan et al., 2007a; Bahadur et al., 2012; Chung et al., 2012; Feng et al., 2013).
Based on observations, Chung et al. (2012) recently suggested that the direct radiative forcing of carbonaceous aerosols is +0.65 (0.5 to about 0.8) Wm−2, comparable to that of methane, the second most important greenhouse gas. This study emphasizes the important role of BrC and calls for better measurements of the absorption properties of BrC, specifically at short wavelengths where the absorption is most significant. Many previous studies have concentrated on primary particulate matter, mostly from biomass burning. However, these studies often neglected contributions to absorption due to BrC in SOA. There is ample laboratory and field evidence for the formation of such absorbing material in SOA (Chung et al., 2012; Lack et al., 2012). This absorbing component is the least characterized component of the atmospheric absorbing aerosols and constitutes a major knowledge gap, calling for an urgent need to identify the optical properties of the organic (BrC) component in SOA, and the chemical pathways leading to its formation and losses (Laskin et al., 2015; Lin et al., 2015; Moise et al., 2015).
Recently, Washenfelder et al. (2015) measured aerosol optical extinction and absorption in rural Alabama during the SOAS campaign. While they found that the majority of BrC aerosol mass was associated with biomass burning, a smaller (but not negligible) contribution was attributed to biogenically derived SOA. This fraction reached a daily maximum at night and correlated with particle-phase organic nitrates and is associated with nighttime reactions between monoterpenes and the NO3 radical (Xu et al., 2015a). Based on the above, it is concluded that SOA produced from reactions of NO3 with BVOC can be a major source of SOA during the night that may affect daytime aerosol loading. This important fraction of NO3-derived SOA can contribute to the direct radiative effect of SOA through scattering and absorption of incoming solar radiation.
Nitration of aromatic compounds (oxidation via NO2, NO3, N2O5) has a potential to form chromophores that can absorb solar radiation. Theoretical and experimental studies have shown that nitration of PAHs leads to nitro PAHs and their derivatives such as nitrophenols (Jacobson, 1999; Harrison et al., 2005; Lu et al., 2011). The nitro substituents on the aromatic ring in compounds enhance and shift the absorption to longer wavelengths (> 350 nm). Field studies report that nitrogen-containing mono- and polyaromatic SOA constituents absorb light at short (near-UV and visible) wave-lengths. The reaction products between NO3 and BVOC have the potential to form effective chromophores. Multifunctional organic nitrates and organosulfate compounds formed during the nighttime suggest that the SOA produced from NO3 reactions leads to formation of BrC that can absorb solar radiation (Iinuma et al., 2007).
Only a few studies have investigated optical properties of SOA partially composed of organic nitrates (Moise et al., 2015). Most existing literature on optical properties of organic nitrates in SOA has been focused on oxidation of anthropogenic precursor compounds (Jacobson, 1999; Nakayama et al., 2010; Lu et al., 2011; Liu et al., 2012b), while a few partially contradictory studies have examined SOA formed from NO3 reaction with biogenic precursors (Song et al., 2013; Varma et al., 2013). The typically high mass absorption coefficient (MAC) that was observed for anthropogenic high-NOx SOA can be partially attributed to the presence of nitroaromatic groups, for example, via the nitration of PAHs (Jacobson, 1999; Lu et al., 2011). Song et al. (2013) examined optical properties of SOA formed by NO3+ O3+ α-pinene. With neutral seed aerosol, organic nitrates were present but observed to be non-absorbing; however, with acidic seed aerosol, SOA were strongly light absorbing, which the authors attributed to nitrooxy organosulfates formed via aldol condensation. Varma et al. (2013) measured absorption of NO3 +β-pinene SOA and found a higher refractive index than when oxidation was via OH or O3, and attributed to the difference to the low HC / NOx ratio and presence of organic nitrates in the particle phase.
Laboratory and field studies suggest that SOA formed by nighttime chemistry can have profound regional and possible global climatic effects via their absorbing properties. However, the optical properties of NO3-containing SOA are not well known. Varma et al. (2013) measured a high value for the refractive index real part value of 1.61 (±0.03) at λ = 655– 687 nm following reactions of NO3 with β-pinene. This value is significantly higher than values observed following OH- and ozone-initiated terpene oxidation (Fig. 6) (Moise et al., 2015). This has been attributed to the high content (up to 45 %) of organic nitrates in the particle phase (Varma et al., 2013).
Figure 6.

The real part of refractive index (RI) (mr) for biogenic SOA compiled from several chamber studies. The legend specifies the precursor type and oxidation pathway as well as the reference. The figure is reprinted with permission from Moise et al. (2015).
Key physical parameters of aerosols include particle size and number, volatility, viscosity, hygroscopicity, and CCN activity. While it is clear that atmospheric particle size increases through condensation of BVOC + NO3 oxidation products, the effect of NO3 oxidation on particle number is not usually studied in laboratory experiments. Very little is known about the volatility of SOA from NO3, with field studies from Hyytiälä indicating that organic nitrates may have low volatility (Häkkinen et al., 2012). Viscosity is not known. Few studies report the hygroscopicity and CCN activity of SOA from NO3 oxidation of BVOC. A study by Suda et al. (2014) showed that organic compounds with nitrate functionality (compared to other functional groups such as hydroxyl, carbonyl, hydroperoxide) have the lowest hygroscopicity and CCN efficiency. Recently, Cerully et al. (2015) reported that the hygroscopicity of less-oxidized OOA (LO-OOA, mostly from BVOC + NO3) is lower than other OA subtypes (MO-OOA and isoprene-OA) resolved by PMF analysis of AMS data from the SOAS campaign. As monoterpenes + NO3 reactions can contribute ~ 50 % of nighttime OA production (Xu et al., 2015a), results from Cerully et al. (2015) suggested that it is possible that SOA formed from NO3 oxidation of BVOC is less hygroscopic than OA formed from other oxidation pathways.
3.2 Health effects
Nitrated organic compounds also pose adverse health effects (Franze et al., 2003, 2005; Pöschl, 2005; Gruijthuijsen et al., 2006; Pöschl and Shiraiwa, 2015). In particular, several studies have reported that biological particles such as birch pollen protein can be nitrated by O3 and NO2 in polluted urban air (Franze et al., 2005; Reinmuth-Selzle et al., 2014). The mechanism of protein nitration involves the formation of long-lived reactive oxygen intermediates, which are most likely tyrosyl radicals (phenoxy radical derivatives of tyrosine) (Shiraiwa et al., 2011). The resulting organic nitrates were found to enhance the immune response and the allergenicity of proteins and biomedical data suggest strong links between protein nitration and various diseases (Gruijthuijsen et al., 2006). Inhalation and deposition of organic nitrates into lung lining fluid in the human respiratory tract may lead to hydrolysis of organic nitrates forming HNO3, which may reduce pulmonary functions (Koenig et al., 1989). Consequently, inhalation of aerosols partially composed of nitrated proteins or nitrating reagents might promote (i) immune reactions, (ii) the genesis of allergies, (iii) the intensity of allergic diseases, and (iv) airway inflammation. Toxicity of nitrated SOA compounds is still unclear. In the light of these observations and remaining uncertainties, the effect of organic nitrates present in biogenic SOA on human health should be a focus of future studies.
Formaldehyde is an important source of atmospheric radicals as well as a major hazardous air pollutant (HAP). It is a degradation product of almost every VOC in the atmosphere, and BVOC are known to contribute substantially to ambient concentrations of formaldehyde (Luecken et al., 2012). The overall yield of formaldehyde from BVOC-NO3 reactions is lower than from corresponding OH reactions, indicating that any changes in the relative distribution of oxidation routes will have a corresponding change in formaldehyde (and thus oxidant regeneration and HAP exposure).
3.3 Trends in NOx emissions and organic aerosols – implications for air quality control strategies
Nitrogen oxide emissions are converted to NO3 and thus affect nitrate-derived SOA. In the United States, where NOx emissions are dominated by fuel combustion, regulatory actions have resulted in decreasing NOx levels after increases from 1940 to 1970 (Nizich et al., 2000) and relatively stable levels between ~ 1970 and ~ 2000 (Richter et al., 2005). NOx emissions in the US are estimated to have decreased by roughly 30–40 % in the recent past (between 2005 and 2011/2012), as reflected in satellite-observed NO2, ground-based measurements, and the Environmental Protection Agency (EPA) National Emission Inventory (NEI) (Russell et al., 2012; Xing et al., 2013, 2015; Hidy et al., 2014; Tong et al., 2015). Recent decreases in NOx have been attributed to the mobile sector, and power plant controls including the EPA NOx State Implementation Plan Call implemented between 2003 and 2004 (Kim et al., 2006; Russell et al., 2012; Hidy et al., 2014; Foley et al., 2015; Lu et al., 2015). In the United States, NOx emissions are expected to continue to decrease and reach 72 and 61 % of their 2011 levels in 2018 and 2025, respectively (Eyth et al., 2014). Furthermore, recent work indicates that NOx emissions may be overestimated in models for the United States (Travis et al., 2016) particularly for on-road gasoline vehicles (McDonald et al., 2012).
Globally, the Representative Concentration Pathway trajectories indicate that NOx emissions will decrease below year 2000 levels by the middle of the 21st century (Lamarque et al., 2011). Europe has experienced declines in NOx with NO2 concentrations decreasing by 20 % over western Europe between 1996 and 2002 (Richter et al., 2005) and decreasing by an additional ~ 20 % in the more recent past (2004–2010) (Castellanos and Boersma, 2012). In contrast, NOx emissions in China have increased by large amounts since 1996 (Richter et al., 2005; Stavrakou et al., 2008; Verstraeten et al., 2015) with a more recent leveling out or decrease of NO2 concentrations (Krotkov et al., 2016). NO2 concentrations in India have continued to increase (Krotkov et al., 2016; Duncan et al., 2016).
These large past and expected future changes in anthropogenic NOx emissions indicate that analysis of historical data could reveal how NOx emissions affect organic aerosol formation and more specifically SOA from NO3-initiated chemistry. Long-term monitoring networks often measure NOx and OC, which could allow for correlation analysis. In addition, air quality trends in organic aerosol from traditionally less-sampled locations (e.g., Streets et al., 2008) and emissions for locations such as China have been characterized and could be used for analysis.
In addition to examining measurement data for relationships between NO3-derived SOA and NOx, chemical transport modeling with emission sensitivity simulations can be used to provide estimates of how various SOA pathways respond to changes in NOx emissions. For example, Carlton et al. (2010b) used the CMAQ model to determine that controllable NOx emissions were responsible for just over 20 % of total SOA in the United States based on the NO3-BVOC mechanism available at the time. Pye et al. (2015) predicted nitrate-derived SOA concentrations would decrease by 25 % due to a 25 % reduction in NOx emissions, but the overall change including all organic aerosol components would be only 9 % as a result of other less sensitive (or increasing) components. Other modeling studies (Lane et al., 2008; Zheng et al., 2015; Fisher et al., 2016) have shown that total organic aerosol or particle-phase organic nitrates may not respond strongly to decreased NOx emissions, but significant spatial and composition changes can occur.
3.4 Organic nitrate standards
The CIMS technique allows for highly time-resolved, chemically speciated measurements of multifunctional organic nitrates (Beaver et al., 2012; Paulot et al., 2012; Lee et al., 2014a; Xiong et al., 2015). Synthesis, purification, and independent quantification of an individual, isomerically specific organic nitrate is, however, required for calibration because standards are not commercially available, except for a few monofunctional alkyl nitrates.
The synthesis of monofunctional alkyl nitrates can be performed via several methods (Boschan et al., 1955), including nitration of alkyl halides with silver nitrate, direct nitration of alcohols or alkanes with nitric acid (Luxenhofer et al., 1996; Woidich et al., 1999), or treatment of alcohols with dinitrogen pentoxide (Kames et al., 1993). Techniques for the synthesis of multifunctional nitrates (in particular, hydroxynitrates) have been described in previous reports (Muthuramu et al., 1993; Kastler and Ballschmiter, 1998; Werner et al., 1999; Treves et al., 2000). Carbonyl nitrates have also been synthesized using the same protocol, i.e., nitration of hydroxy ketones with dinitrogen pentoxide (Kames et al., 1993; Suarez-Bertoa et al., 2012).
Most recently, three isomers of isoprene hydroxynitrates were synthesized (Lockwood et al., 2010; Lee et al., 2014b). As the precursor ingredient is an organic epoxide on which hydroxy and nitrate functional groups are attached, the same protocol (Nichols et al., 1953; Cavdar and Saracoglu, 2008) can be applied to synthesize hydroxynitrates of various VOC backbones assuming availability of precursor compounds. Oxidation of a single-parent compound can yield numerous isomerically unique byproducts possessing various functional groups, including one or more nitrates. As such, synthesis of and calibration for each nitrate rapidly become prohibitive. Given that multifunctional organic nitrates possessing more than four oxygen atoms, for which synthesis protocols currently do not exist, dominate the particulate nitrate mass of submicron particles (Lee et al., 2016), a more comprehensive calibration technique is needed. Three broad approaches are currently utilized. One is to cryogenically collect a suite of oxidation byproducts (present in the atmosphere, formed in a simulation chamber or flow tube, etc.) on a GC column. The desorbing eluent, separated in time by volatility/polarity as it is thermally desorbed, is measured simultaneously by CIMS and a quantitative instrument such as the TD-LIF (Day et al., 2002; Lee et al., 2014b). The corresponding eluting peaks detected by both instruments allow for calibration of each surviving, isobarically unique (at least for unit mass resolution spectrometers) organic nitrate (Bates et al., 2014; Schwantes et al., 2015; Teng et al., 2015). The second approach employed for the iodide adduct ionization technique is to deduce the instrument response from a comparison of the binding energies of the numerous iodide organic nitrate clusters to those of compounds with known sensitivities by applying variable voltages in the ion molecule reaction region to break up charged clusters systematically. The rate at which the signal of an organic nitrate cluster decays with voltage is a function of its binding energy, which governs its transmission efficiency through the electric fields and thus its sensitivity (Lopez-Hilfiker et al., 2016). Lastly, quantum chemical calculations of specific compounds allow the determination of the sensitivity of their iodide adduct (Iyer et al., 2016) and CF3O− (Kwan et al., 2012; Paulot et al., 2012) ionizations.
3.5 Critical needs for models
3.5.1 Robust and efficient representation of gas-phase chemistry
Previous sections have detailed the reactions of BVOC with NO3 and the need to include this chemistry to represent more accurately processes that control O3 and SOA formation. But applying that information in a way that can be used for air quality studies presents a serious challenge. As highlighted in Sect. 2.6.1, the chemical mechanisms currently being used in AQMs are limited in their representation of NO3-BVOC chemistry, largely lumping all monoterpenes together, and with no agreement on yields. The lack of detail in current mechanisms is reflected in the variety of methods by which SOA formation from BVOC-NO3 chemistry is estimated (Sect. 2.6.2).
Typically, the NO3-BVOC chemistry is implemented in AQMs into the existing system of organic and inorganic chemical reactions that occur in the atmosphere. Because there may be hundreds or thousands of different chemical reactions occurring simultaneously and the computational efforts required to solve those on a 3-D grid are onerous, the chemical mechanisms used in AQMs are typically condensed to a certain extent. The greatest challenges in modeling the reactions initiated by NO3 and BVOC in AQMs are (1) deciding how much detail must be included to accurately represent the chemistry; (2) estimating intermediate reactions and/or products when direct experimental observations are not available; (3) integrating the new reactions into existing chemical mechanisms; and (4) validating the complete schemes against observational data.
Including all of the attack pathways and isomers that are formed in the reactions of NO3 and BVOC and their subsequent products rapidly becomes an intractable problem, as the number of species and reactions produced from a VOC grows exponentially with the number of carbons in the compound (Aumont et al., 2005), resulting in an estimate of almost 400 million products from a single C10 hydrocarbon. Even restricting the chemistry solely to the RO2 formed from α-pinene, β-pinene, and limonene via addition of NO3 to the double bond results in 861 unique product species and 2646 reactions as estimated from the MCM (http://mcm.leeds.ac.uk/MCM-devel/home.htt; Saunders et al., 2003). In comparison, the chemical mechanisms used in AQMs typically consider a total of 100–200 species and less than 400 reactions to model the entire gas-phase chemistry occurring in the troposphere. One challenge is to find a balance between complexity and computational efficiency that involves both deriving complete mechanisms as well as condensing them to the extent possible.
The second major challenge is that many of the chemical pathways must be estimated given the limited experimental measurements of intermediate reaction rate constants and products. Structure–activity predictions have been used heavily in the past, but these have been formulated for a limited number of compounds. Their predictions become less accurate as the complexity of the molecule increases (Calvert et al., 2015). When heterogeneous reactions play a significant role in the transport and fate of reaction products, as they do in monoterpene chemistry, the challenge becomes even greater. With recent research, new product structures that contribute to SOA have been identified (Boyd et al., 2015). However, these are not covered by existing predictive theory, and these new pathways must be characterized, including reaction rate constants, co-reactants, and products. Physical parameters of all of these new species, such as solubility, radiative properties, emission rates, and deposition velocities also are required, but data are often unavailable for these or even comparable species.
The last challenge is integrating the chemistry within the rest of the chemistry occurring in the atmosphere. The major chemical mechanisms used in AQMs today were developed primarily to address episodes of elevated O3 under conditions of high NOx and have been evaluated for this purpose. Thus, the mechanisms often do not lend themselves well to predicting the chemistry of complex VOC or other air quality endpoints (Kaduwela et al., 2015). Minor pathways with respect to O3 formation have been removed from the mechanisms to reduce the computational burden, but these pathways may be important for formation of SOA. In addition, the detailed chemistry of multistep alkoxy and peroxy radical chemistry is condensed into a single step in some mechanisms, but identifying whether these radicals react with NOx or HOx or isomerize is critical for predicting the types of organic molecules that are formed. As described in Sect. 2.6, existing mechanisms include the capability for a limited number of nitrates, and in many cases the links to facilitate expansion to more detailed representations are missing.
Significant work must be done to allow modelers to implement this new information in AQMs and thus use this updated knowledge to develop improved predictions of future air quality. One approach is to focus on key chemicals of interest, derive mechanisms that are suitable for specialized applications, and append these on to existing frameworks (for example, Xie et al., 2013). The longer-term view requires a more comprehensive approach that draws on the development of community archives that can better accommodate rapidly changing information and better represent the interactions of biogenic with anthropogenic chemistry. Here, we put forward our recommendations for future work in the following areas:
Development of tools for the semi-automated production of the reaction pathways and products of later-generation products resulting from alternate pathways of radical reactions with BVOC. These tools should be able to incorporate experimental data when available. In conjunction with the automated development, we require advanced methods for condensing these large mechanisms into computationally feasible reaction schemes.
Improvements in estimation techniques for uncertain pathways, including reaction rate constants for multifunctional stable compounds and radicals for which measurements are not available, and the quantification of the errors associated with these estimation methods.
Development of theory and techniques for integrating gas-phase products with SOA production, in this case, describing the transformation of gas-phase organic nitrates to their SOA products.
Development of more versatile base mechanisms that have the flexibility to accept increased detail in VOC description and the continuing validation of the complete tropospheric chemical mechanisms against observational data.
3.5.2 Improved techniques and protocols for evaluation of complex and reduced gas-phase mechanisms
Generally speaking, once detailed mechanisms are developed, they are evaluated through some form of benchmarking. Systematic strategies for mechanism evaluation include validation of highly detailed mechanisms unable to be run in 3-D models against benchmark data from well-characterized simulation chamber experiments (Jenkin et al., 1997; Aumont et al., 2005) and the incorporation of these mechanisms into box or 1-D models to validate radical and short-lived species against field campaign observations. Less-detailed air quality (AQ) mechanisms can then be compared to these reference mechanisms by way of sensitivity experiments in idealized modeling studies – often aimed at assessing the sensitivity in O3 to changing NOx and VOC emissions (Archibald et al., 2010; Squire et al., 2015). AQ mechanisms are often also then re-evaluated against chamber and/or field experiment data before they are implemented into 3-D models and then undergo evaluation against extensive measurements in the residual layer.
One of the greatest challenges in the BVOC-NO3 system is that current nighttime measurements are mainly collected from surface sites, which are confined to a shallow surface layer at night and not representative of the whole nighttime boundary layer. The impact of nighttime chemistry on daytime ozone and nitrogen/aerosol budget would require careful investigation of nighttime chemistry in the residual layer, which contains > 80 % of air masses at night.
Moreover, the benchmarking activities mentioned above and the development process discussed in Sect. 3.5.1 are not well aligned. A more unified approach that identifies some key mechanistic problems and identifies strategies to evaluate them is required in order to make improved progress on simulating the changing composition of the atmosphere.
3.5.3 Reduce uncertainties in sub-grid-scale processes
Uncertainties in AQM predictions also arise from the representation of physical sub-grid-scale processes. The ones particularly relevant for the NO3-BVOC chemistry include, but are not limited to, the following.
Nighttime boundary layer mixing
The spatial distribution of BVOC and NOx precursors is highly variable, but the current AQMs neglect these heterogeneities and assume perfect mixing within grid cells of typically 3–10 km in the horizontal. At those resolutions, models are unable to resolve the localized surface emission sources and the microscale structure of boundary layer turbulence, and therefore cannot resolve spatial heterogeneities in chemistry, partitioning, and mixing of chemicals, which are essential for predicting the concentrations of secondary pollutants.
Typically, the freshly emitted monoterpene species have a tendency to accumulate in the shallow nighttime boundary layer (typically < 200 m), and can react with NO3 if available. However, often NO3 is located in the residual layer that is decoupled from the nighttime boundary layer (NBL), and the BVOC + NO3 reactions would depend on the model’s ability to mix the two layers. Thus, mixing within and out of the boundary layer provides a key challenge for modeling the impacts of BVOC-NO3 chemistry, as the measured gradients of NO3 and BVOC are very strong in the vertical (e.g., Brown et al., 2007b; Fuentes et al., 2007).
A large focus on model evaluation has been on the impacts of higher horizontal resolution (Jang et al., 1995). It has been shown in several cases that owing to the complex interplay of chemical families, the sensitivity of the chemical system is not captured at lower resolution (e.g., Cohan et al., 2006). However, very little work has focused on the role of improvements in vertical resolution, despite the fact that inter-model differences in properties like the height of the boundary layer vary by over a factor of 2 in some cases (e.g., Hu et al., 2010). Moreover, the NBL is not well mixed, so evaluation of nocturnal physics requires more than just evaluating the NBL height.
Plume parameterizations
Typically, parameterizations have been applied to anthropogenic emission sources (e.g., aircraft plumes, urban plumes) and not to biogenic sources. Partly, this is a result of the differences in the source terms, anthropogenic emissions often being well represented as point sources in space, whereas biogenic emissions are often large area sources. However, as the emissions of BVOC are often very species specific, and observations highlight large spatial variability over small areas (e.g., Niinemets et al., 2010), the adoption of the anthropogenic plume parameterizations to BVOC emissions could lead to improvements in model performance.
One approach is the plume-in-grid (PiG) parameterization (Karamchandani et al., 2002). This aims to solve the problem of sub-grid-scale chemical processes by implementing ensembles of Gaussian puffs within the AQM (e.g., Vijayaraghavan et al., 2006). Other approaches include hybrid Eulerian–Lagrangian models (Alessandrini and Ferrero, 2009). These differ from the PiG models by simulating large numbers of stochastic trajectories that can make use of variable reactive volumes to simulate their diffusion into background air masses simulated on Eulerian grids.
Global models have generally used a different approach to the problem of plumes. Broadly, following one of the two paradigms (Paoli et al., 2011) to (i) modify the emissions of the reaction mix (using so-called effective emissions or applying emission conversion factors) and (ii) modify the rates of reaction (effective reaction rates).
3.6 Field studies in the developing world and under-studied areas
In light of the questions raised earlier in this review, assessing the role of NO3-BVOC chemistry will require field experiments over a wide range of ratios of isoprene to monoterpene emissions and of NO3 to BVOC. Future studies of NO3-BVOC chemistry are in the planning stages for North America. These studies will provide access to environments with different NOx levels and over a modest range of isoprene and monoterpene emission rates. A wider range of these parameters can be accessed in countries where NOx emission controls are not as completely implemented and where BVOC emissions are abundant. Bringing the state-of-the-art capabilities developed for study of NO3-BVOC chemistry to locations in China and India would allow insight not only into the role of that chemistry in those countries now but also into the role this chemistry played in Europe and the US prior to implementation of current emission standards. Experiments in the tropics potentially would allow observations of the confluence of BVOC and very low NOx to be explored, thus providing insight into BVOC-NO3 as a sink of NOx.
3.7 Future needs for chamber studies
Field studies, by definition, include the entire complexity of the real atmosphere, so that the identification of single processes and quantification of their impact is challenging. Specific experiments in chambers allow investigating processes without effects from meteorology, which largely impacts observations in the real atmosphere specifically during nighttime, when the lower troposphere is not as well mixed as it is during daytime. In chamber experiments, specific compounds of interest can be isolated and studied under well-controlled oxidation environments, allowing a more detailed and direct characterization of the composition, chemical, and physical properties of aerosols. Because such laboratory chamber data provide the basic understanding for predicting SOA formation, it is important that the design of such experiments mimics the oxidation environments in the atmosphere to the greatest extent possible. Several important needs for understanding NO3-BVOC chemistry in chambers include (1) elucidation of kinetic and mechanistic information for NO3-BVOC reactions; (2) characterization of wall losses for low-volatility products in the NO3-BVOC system; (3) understanding the fate of peroxy radicals in the nighttime atmosphere and its influence on this chemistry; (4) hydrolysis and photooxidation of BVOC-derived organic nitrates from specific BVOC plus specific oxidant pairs over a range of appropriate conditions; (5) optical properties of aerosol organic nitrate; and (6) intercomparison of instrumental methods for key species in the NO3-BVOC system.
Kinetic and mechanistic elucidation
The number of chamber studies investigating NO3 chemistry is small compared to the number of studies for photochemical oxidation and ozonolysis. In most of the studies, gas-phase oxidation products and SOA yields from the oxidation of BVOC have been measured. Studies include the investigation of SOA from monoterpenes (Wangberg et al., 1997; Griffin et al., 1999; Hallquist et al., 1999; Spittler et al., 2006; Fry et al., 2009, 2011; Boyd et al., 2015; Nah et al., 2016b), methyl butenol (Fantechi et al., 1998a, b), and isoprene (Rollins et al., 2009; Ng et al., 2010; Schwantes et al., 2015). A few more studies investigated gas-phase reaction kinetics, including the reactions of NO3 with aldehydes (Clifford et al., 2005; Bossmeyer et al., 2006), amines (Zhou and Wenger, 2013), or cresol (Olariu et al., 2013). As a consequence of the small number of studies, the oxidation mechanisms of organic compounds by NO3 and the yields of oxidation products in the gas phase and particle phase have larger uncertainties. The well-controlled oxidation environments in chamber experiments, coupled with complimentary gas-phase and particle-phase measurements (online and offline), allow for elucidating detailed oxidation mechanisms under varying reaction conditions (Ng et al., 2008; Boyd et al., 2015; Schwantes et al., 2015). Identification of gas- and particle-phase reaction products from NO3-BVOC chemistry within controlled chamber environments can also greatly aid in the interpretation of field data in which multiple oxidants and BVOC are present. Future chamber experiments will naturally take advantage of new advanced gas–aerosol instrumentation and aim to constrain the formation yields of gas-phase oxidation products and establish a fundamental under-standing of aerosol formation mechanisms from NO3-BVOC under a wide range of oxidation conditions.
Wall losses
Although chamber studies allow separating processes driven by chemistry and physics from transport processes that occur in the real atmosphere, careful characterization of the behavior of NO3 in chambers as well as the organic products of the NO3 oxidation remains a research priority. Yields of gas-phase oxidation products can be influenced by chamber-specific loss processes (surface loss on the chamber wall) and SOA yields can be impacted by both direct loss of particles and loss of species that can condense on particle or chamber wall surfaces (McMurry and Grosjean, 1985; Loza et al., 2010; Matsunaga and Ziemann, 2010; Yeh and Ziemann, 2014; Zhang et al., 2014a, 2015; Krechmer et al., 2016; La et al., 2016; Nah et al., 2016a; Ye et al., 2016). The extent to which vapor wall loss affects SOA yields appears to be dependent on the VOC system, from relatively small effects to as high as a factor of 4 (Zhang et al., 2014a; Nah et al., 2016a). Studies on the effects of vapor loss on SOA formation from BVOC + NO3 are limited. With minimal or no competing gas–particle partitioning processes, substantial vapor wall loss of organic nitrates has been observed in experiments not specific to NO3 oxidation (Yeh and Ziemann, 2014; Krechmer et al., 2016). However, the use of excess oxidant concentrations and rapid SOA formation in BVOC + NO3 experiments (hence, shorter experiments) could potentially mitigate the effects of vapor wall loss on SOA yields in chamber studies (Boyd et al., 2015; Nah et al., 2016a). In light of the developing understanding of this issue, an important consideration for the design of any future systematic chamber studies is the influence of vapor wall loss on SOA formation from nitrate radical oxidation under different reaction conditions, such as peroxy radical fates, relative humidity, seeds, oxidant level, chamber volume, etc.
Peroxy radical fate
As discussed above, the fate of peroxy radicals directly governs the product distribution in the NO3-BVOC system, including SOA yields and composition. Dark reactions of peroxy radicals differ significantly from their photochemical analogs, and are directly related to the development of mechanistic understanding in the NO3-BVOC system. There is a need to systematically investigate reaction products and SOA formation from NO3-BVOC reactions under different peroxy radical reaction regimes, but this aspect has only recently become a focus of chamber studies (Ng et al., 2008; Boyd et al., 2015; Schwantes et al., 2015). Rapid formation of highly oxygenated organic nitrates has been observed in laboratory studies of β-pinene + NO3 and α-pinene + NO3; these products could be formed by unimolecular isomerization of peroxy radicals or autoxidation (Nah et al., 2016b). The importance of this peroxy radical reaction channel in NO3-BVOC chemistry warrants further studies. Future chamber studies will need to be explicit in their specification of the peroxy radical chemistry regime that is investigated in a particular experiment, and will need to relate that regime to the conditions of ambient nighttime atmosphere.
Organic nitrate hydrolysis and photooxidation
Recent field studies have shown that organic nitrates formed from NO3-BVOC are important components of ambient OA. However, the reactivity in both gaseous and condensed phases of these biogenic nitrates, in particular of polyfunctional nitrates, has been subject to few studies and requires better characterization to evaluate the role of these compounds as reservoirs/sinks of NOx. Field results suggest that the fate of organic nitrates in both the gas and aerosol phase have variable lifetimes with respect to hydrolysis. The difference in the relative amount of primary/secondary/tertiary organic nitrates (which hydrolyze with different rates) from nitrate radical oxidation versus photochemical oxidation needs to be constrained. Most of the hydrolysis studies thus far are conducted in bulk, except for a few recent studies on monoterpene organic nitrates (e.g., Boyd et al., 2015; Rindelaub et al., 2015). The solubility of multifunctional organic nitrates in water and the extent to which hydrolysis occurs in aerosol water warrant future studies. The effect of particle acidity on hydrolysis might also be important for organic nitrates formed in different BVOC systems.
While there are extensive studies on photochemical aging of ozonolysis SOA, studies on photochemical aging of NO3-initiated SOA and organic nitrates are extremely limited. A recent study shows that the particle-phase organic nitrates from NO3+β-pinene and NO3+α-pinene reactions exhibit completely different behavior upon photochemical aging during the night-to-day transition, and act as permanent and temporary NOx sinks, respectively (Nah et al., 2016b). With the ~ 1-week lifetime of aerosols in the atmosphere and the majority of NO3-BVOC organic nitrates that are formed at night, the photochemical fates of these organic nitrates could impact next-day NOx cycling and ozone formation. Therefore, there is a critical need to understand the multigenerational chemistry and characterize the evolution of organic nitrates over its diurnal life cycle, including aging NO3-initiated SOA and organic nitrates by photolysis and/or OH radicals.
Aerosol optical properties
The optical properties, especially in the short wavelength region, of NO3-derived SOA may be most conveniently measured during coordinated chamber studies that also include detailed measurements of gas-phase oxidation chemistry and aerosol composition. Such studies could also serve to isolate the specific optical properties of NO3-BVOC-derived aerosol to obtain better optical closure in the interpretation of field data. Field studies that include aerosol optical properties measurements in conjunction with other instrumentation can help quantify the bulk organic nitrate abundance and identify organic nitrate molecular composition in the SOA.
Instrument intercomparisons
The discussion above shows that recent advances in analytical instrumentation are key to the developing science of NO3-BVOC chemistry. Chamber studies provide an excellent opportunity for the comparison and validation of such instrumentation. State-of-the-art and developing instruments for measurement of NO3 and N2O5 were compared approximately a decade ago (Fuchs et al., 2012; Dorn et al., 2013). These instruments have improved and proliferated since that time, and further validation studies are needed. Measurements of total and speciated gas and aerosol-phase organic nitrates, as well as other oxygenated compounds that result from NO3-BVOC reactions, have not been the subject of a specific intercomparison study. Their comparison and validation will be a priority in future coordinated chamber studies.
Utility of coordinated chamber studies
Because of the need for a better understanding of NO3 oxidation and because of the challenges of chamber studies, investigating NO3 chemistry, coordination between studies carried out in different chambers, and between chamber and field studies, can augment efforts of single or standalone chamber studies. Coordinated studies that would include several chambers could increase the accuracy and reliability of results and quantify realistic errors associated with product yield estimates. This can be achieved by determining the same quantities in similar experiments in different chambers. Studies could benefit from complementary capabilities and properties of chambers. Chambers that typically operate at higher concentration ranges, and therefore increased oxidation rates, are suitable to perform a larger number of experiments that are useful for screening experiments and a series of experiments with systematic variations of chemical conditions. Other chambers are suited to perform experiments at atmospheric reactant concentrations. Experiments in these chambers may take place on a longer timescale, for example, a scale characteristic of the duration of at least one night. Analytical instrumentation and capability also differs considerably among chambers, so that coordinated chamber studies can make use of the determination of complementary quantities such as product yields of different organic compounds and characterization of various properties of particles for the same chemical system. For instance, it would be invaluable to conduct coordinated studies where a variety of instrument techniques are used to measure total and speciated gas- and particle-phase organic nitrates, as well as aerosol physical and chemical properties in the same chamber.
Substantial insights into aerosol sources, formation, and processing can be gained from coordinated laboratory chamber and field studies. Laboratory chamber experiments provide the fundamental data to interpret field measurements. The analysis of field data in turn can provide important insights for constraining chamber experiment parameters so that the oxidation conditions in chambers can be as representative as possible of those in the atmosphere. Two recent sets of experiments serve as examples of this approach. Fundamental chamber studies on β-pinene+NO3 in the Georgia Tech Environmental Chamber (GTEC) facility under conditions relevant to the SE US provided constraints on the contribution of monoterpenes + NO3 to ambient OA during the 2013 SOAS campaign (Boyd et al., 2015; Xu et al., 2015a). The Focused Isoprene eXperiment at California Institute of Technology (FIXCIT) chamber study following SOAS advanced the understanding of isoprene oxidation chemistry relevant to the SE US (Nguyen et al., 2014). It is important not to consider fundamental laboratory studies as isolated efforts, but they should be an integrated part of field studies. Similarly, having the modeling community involved in early planning stages of laboratory and field studies will greatly aid in the identification of critically needed measurement data.
4 Impacts of NO3-BVOC chemistry on air quality
The previous sections have demonstrated that understanding how NO3 reacts with BVOC, including the ultimate fate of products, encompasses all aspects of atmospheric physics, chemistry, and transport. These sections have raised numerous complex and fascinating scientific questions and highlighted the critical need for much more basic science to fill in unknown aspects of this system. However, “getting this system right” is not just an interesting scientific problem because it has direct implications for policy decisions that governments across the world are taking to protect citizens and ecosystems from harmful effects of air pollutants. Addressing the uncertainties raised in the previous sections is critical for developing efficient, accurate, and cost-effective strategies to reduce the harmful effects of air pollution.
BVOC have long been predicted to be significant contributors to regional and global O3 (e.g., Pierce et al., 1998; Curci et al., 2009) and PM2.5 (Pandis et al., 1991), with NO3 reactions providing a major pathway for loss of ambient BVOC (Winer et al., 1984; Pye et al., 2010; Xie et al., 2013). If BVOC react with NO3 instead of OH, the O3 production of the BVOC can be reduced relative to reactions through OH, although in some instances they may slightly increase O3 by reducing next-day NOx. For example, measurements in St. Louis (Millet et al., 2016) demonstrate that nights with lower levels of NO3 resulted in higher isoprene concentrations the following morning, producing higher and earlier O3 peaks. Recent insights into the role of biogenic nitrates, which are produced in large quantities through the reactions of NO3 with primary emitted BVOC and subsequent reactions of their stable products, demonstrate that these compounds can substantially alter the availability of NOx (Perring et al., 2013). This highlights the importance of accurate treatment of fates of organic nitrates that form from nighttime chemistry in models, which will impact the next-day NOx and ozone levels. Organic nitrates from BVOC + NO3 also can contribute to nitrogen deposition (Nguyen et al., 2015), which adversely impacts ecosystems. The ways in which the patterns of deposition for biogenic nitrates affect inorganic nitrate deposition remain poorly understood.
Implications for spatial distribution of ozone and PM2.5
While it is clear that NO3-BVOC reactions affect oxidant availability and SOA, it remains unclear how large that role is in the ambient atmosphere relative to other VOC and other oxidants and where it occurs. The extent of O3 formation downwind of sources is influenced by the transport of NOy species, including organic nitrates, which can release NOx downwind, where O3 may be formed more efficiently. Biogenically derived nitrates are the dominant organic nitrates in many places (Pratt et al., 2012). A variety of different organic nitrates are formed from different BVOC, with some being short lived (releasing NO2 locally) and others being long lived (releasing NO2 downwind unless they are removed in the meantime). Errors in our attribution of the lifetime of individual biogenic nitrate compounds can cause errors in predicted NOx redistributions regionally and globally, and modify the spatial distributions of O3 (Perring et al., 2013). Updates to the chemistry of BVOC-NO3 also could alter calculations of the relative role of biogenic species versus anthropogenic pollutants to O3 and PM2.5 formation.
Implications for control strategy development
Air quality models are used not only to understand the production of air pollutants in the current atmosphere but also to guide the development of strategies to reduce the future pollution burden. Uncertainties in the chemistry and removal of BVOC can contribute to uncertainties in the sensitivity of O3 and PM to emission reduction strategies. This increases the risk of implementing expensive control strategies that are found later to be inefficient (more control specified than needed) or ineffective (do not meet the air quality goals for which they were developed). As noted by Millet et al. (2016), in urban areas downwind of high isoprene emissions, the loss of isoprene by NO3 at night can produce the opposite O3-NOx behavior that would normally be expected in urban areas, potentially causing a reassessment of optimum control strategies. In addition, the early O3 peaks noted on low NO3 nights expands the high ozone time window, resulting in higher 8 h O3 averages, on which regulatory compliance in the US is based.
The uncertainties in our understanding of NO3-BVOC chemistry propagate into chemical mechanisms, as described in Sect. 3. Past work has shown that vastly different chemical mechanisms may predict similar O3 in current atmospheres but show huge differences for intermediate species (e.g., Luecken et al., 2008) and different potential responses to precursor reductions, including different indicators of O3 sensitivity to VOC versus NOx reductions (Knote et al., 2015). The presence of large weekend effects in NOx makes identifying such errors more likely in current analyses.
Incorporating new information on biogenic chemistry within a chemical mechanism will impact the availability of NOx (e.g., Archibald et al., 2010; Xie et al., 2013) and modify the predicted effectiveness of anthropogenic NOx controls. Incorporating new chemical information into models can also impact PM2.5 sensitivities to NOx reductions. In one example, organic PM2.5 was almost twice as responsive to a NOx reduction than in older mechanisms (Pye et al., 2015). Because much of the NOx dependence of O3 and aerosols from NO3-BVOC reactions is inadequately accounted for in models, the few examples we have hint that current NOx control strategies might result in more significant improvements to air quality than currently assumed. Retrospective analyses should focus on elucidating the elements of this hypothesis that are represented in the historical record.
The role of climate change in modifying air quality is also a highly uncertain issue and may be particularly sensitive to the characterization of BVOC. Biogenic emissions may increase or decrease in the future, depending on many factors including increased temperatures, changes in water availability, occurrence of biotic and abiotic stress (e.g., Kleist et al., 2012; Wu et al., 2015), CO2 fertilization, CO2 inhibition, and land use changes (Chen et al., 2009; Squire et al., 2014). Uncertainties in biogenic reactions may be amplified as they become a larger share of the VOC burden in some places. The predicted response of O3 to future climate has been found to be especially sensitive to assumptions about the chemical pathways of BVOC reactions, in particular the treatment of nitrates. Mao et al. (2013) and several earlier researchers found that predictions of the O3 response to NOx reductions change from negative to positive depending solely on how the isoprene chemistry was represented. Similarly, a comparison of several widely used chemical mechanisms with varied descriptions of BVOC-derived nitrates (Squire et al., 2015) found that description of BVOC chemistry significantly alters not only the amount of oxidant change predicted under future scenarios but also the direction of the change. Direct measurements of the key steps in isoprene oxidation should eliminate the ambiguity in such model calculations. Nonetheless, the exquisite sensitivity of model predictions of ozone trends to the representation of isoprene and NOx indicates that ambient observations of those trends are an excellent strategy for evaluating the accuracy of mechanisms.
The relative distribution of emissions among different types of BVOC may also shift as climate and land use changes, emphasizing the need to understand differences among terpenes in their chemistry, transport, and fate (Pratt et al., 2012). While most of the research to date has been done on isoprene, with some on α-pinene and β-pinene, little has been done on products or reaction parameters of other terpenes. The previous sections have demonstrated that different terpenoid structures can have vastly different atmospheric chemistry and physical properties, so it is unclear whether assuming one “representative” species or distribution, as is done in most chemical mechanisms, will adequately account for future impacts of BVOC on O3 and PM.
Summary of impacts
This review has illustrated that accurate characterization of NO3-BVOC chemistry is critical to our understanding of both the air quality and climate impacts of NOx emissions. Our knowledge of the complexity of NO3-BVOC reaction pathways and multigenerational products has advanced rapidly, especially in the last decade. Despite the fact that much of that information is not yet in a form that can be included in current air quality models, we anticipate improved predictive capabilities in models in the coming years through sustained laboratory and field studies coupled to model development. While the current levels of uncertainty make it difficult to accurately quantify the impact of NO3-BVOC chemistry on air pollutant concentrations, we expect that developments in this field will improve the effectiveness of air pollution control strategies going forward. The limited studies available demonstrate that even small changes to BVOC chemistry modify the production of oxidants (NO3, OH, and O3) and change the transport of NOy. Therefore, NO3-BVOC oxidation modifies the chemical regime in which additional BVOC oxidation occurs. Of most importance will be the studies that indicate changes in the direction of predicted future pollutant concentrations as chemical mechanisms of BVOC are updated. Emission control strategies and attainment of air quality goals rely on the best possible chemical models. Current and future laboratory and field research is critical to the improvement of chemical mechanisms that account for biogenic chemical processes and products which will augment efforts to reduce harmful air pollutants.
Supplementary Material
Acknowledgments
The authors acknowledge support from the International Global Atmospheric Chemistry project (IGAC), the US National Science Foundation (NSF grants AGS-1541331 and AGS-1644979), and Georgia Tech College of Engineering and College of Sciences for support of the workshop on nitrate radicals and biogenic hydrocarbons that led to this review article. N. L. Ng acknowledges support from NSF CAREER AGS-1555034 and US Environmental Protection Agency STAR (Early Career) RD-83540301. S. S. Brown acknowledges support from the NOAA Atmospheric Chemistry, Carbon Cycle and Climate program. A. T. Archibald and B. Ouyang thank NERC for funding through NE/M00273X/1. E. Atlas acknowledges NSF grant AGS-0753200. R. C. Cohen acknowledges NSF grant AGS-1352972. J. N. Crowley acknowledges the Max Planck Society. J. L. Fry, D. A. Day, and J. L. Jimenez acknowledge support from the NOAA Climate Program Office’s AC4 program, award no. NA13OAR4310063 (Colorado)/NA13OAR4310070 (Reed). N. M. Donahue acknowledges NSF AGS-1447056. M. I. Guzman wishes to acknowledge support from NSF CAREER award (CHE-1255290). J. L. Jimenez and D. A. Day acknowledge support from NSF AGS-1360834 and EPA 83587701-0. R. McLaren acknowledges NSERC grant RGPIN/183982-2012. H. Herrmann, A. Tilgner, and A. Mutzel acknowledge the DARK KNIGHT project funded by DFG under HE 3086/25-1. B. Picquet-Varrault acknowledges support from the French National Agency for Research (project ONCEM-ANR-12-BS06-0017-01). R. H. Schwantes acknowledges NSF AGS-1240604. Y. Rudich and S. S. Brown acknowledge support from the USA-Israel Binational Science Foundation (BSF) grant no. 2012013. Y. Rudich acknowledges support from the Henri Gutwirth Foundation. J. Mao acknowledges support from the NOAA Climate Program Office grant no. NA13OAR4310071. J. A. Thornton acknowledges support from NSF AGS 1360745. B. H. Lee was supported by the NOAA Climate and Global Change Postdoctoral Fellowship. R. A. Zaveri acknowledges support from the US Department of Energy (DOE) Atmospheric System Research (ASR) program under contract DE-AC06-76RLO 1830 at Pacific Northwest National Laboratory. The US Environmental Protection Agency (EPA), through its Office of Research and Development (ORD), collaborated in the research described herein. It has been subjected to Agency administrative review and approved for publication, but may not necessarily reflect official Agency policy.
Appendix A
Glossary of abbreviations and common chemical formulas.
- ACSM
Aerosol chemical speciation monitor
- AM3
Atmospheric Model 3
- AMS
Aerosol mass spectrometry/spectrometer
- AQM
Air quality model
- AR
Absolute rate in simulation chamber
- BB-CEAS
Broadband cavity-enhanced absorption spectroscopy
- BB-CRDS
Broadband cavity ring-down spectroscopy
- BDE
Bond dissociation energy
- BEACHON-RoMBAS
Rocky Mountain Biogenic Aerosol Study
- BERLIOZ
Berliner Ozonexperiment
- BEWA
Regional Biogenic Emissions of Reactive Volatile Organic Compounds from Forests
- BC
Black carbon
- BrC
Brown carbon
- BVOC
Biogenic volatile organic compound(s)
- CalNex
California Research at the Nexus of Air Quality and Climate Change
- CAMx
Comprehensive Air Quality Model with extensions
- CARES
Carbonaceous Aerosol and Radiative Effects Study
- CAPRAM
Chemical aqueous-phase radical mechanism
- CAPS
Cavity-attenuated phase shift spectroscopy/spectrometer
- CB05
Carbon Bond 2005 chemical mechanism
- CCN
Cloud condensation nuclei
- CEAS
Cavity-enhanced absorption spectroscopy/spectrometer
- CE-DOAS
Cavity-enhanced differential optical absorption spectroscopy
- CIMS
Chemical ionization mass spectrometry/spectrometer
- CMAQ
Community Multiscale Air Quality
- CRDS
Cavity ring-down spectroscopy/spectrometer
- DF-A
Discharge flow – absorption
- DF-CEAS
Discharge flow – cavity-enhanced absorption spectroscopy
- DF-LIF
Discharge flow – laser-induced fluorescence
- DF-MS
Discharge flow – mass spectrometry
- DMS
Dimethyl sulfide
- DOAS
Differential optical absorption spectroscopy/spectrometer
- ELVOC
Extremely low volatility organic compounds
- EMEP
European Monitoring and Evaluation Program
- EPA
Environmental Protection Agency
- ESI
Electrospray ionization
- EUCAARI
European Integrated project on Aerosol, Cloud, Climate, and Air Quality Interactions
- EURAD-IM
EURopean Air pollution Dispersion – Inverse Model
- F-A
Flow system – absorption
- F-CIMS
Flow system – chemical ionization mass spectrometry
- F-LIF
Flow system – laser-induced fluorescence
- FIGAERO
Filter inlet for gases and aerosols
- FIXCIT
Focused Isoprene eXperiment at California Institute of Technology
- FTICR
Fourier transform ion cyclotron
- FTIR
Fourier transform infrared spectroscopy
- GC
Gas chromatography
- GC-MS
Gas chromatography mass spectrometry
- GCM
Global climate model
- GECKO-A
Generator of Explicit Chemistry and Kinetics of Organics in the Atmosphere
- GEOS-Chem
Goddard Earth Observing System – Chemistry
- GLOMAP
Global Model of Aerosol Processes
- GTEC
Georgia Tech Environmental Chamber
- HAP
Hazardous air pollutant(s)
- HOHPEX
HOHenpeissenberg Photochemistry Experiment
- HPLC
High-performance liquid chromatography
- HO2
Hydroperoxy radical
- HR-ToF
High-resolution time-of-flight
- IC
Ion chromatography
- ICARTT
International Consortium for Atmospheric Research on Transport and Transformation
- IGAC
International Global Atmospheric Chemistry
- IN
Ice nuclei
- IUPAC
International Union of Pure and Applied Chemistry
- LC
Liquid chromatography
- LED
Light-emitting diode
- LIF
Laser-induced fluorescence
- LO-OOA
Less-oxidized oxygenated organic aerosol
- LV-OOA
Low-volatility oxygenated organic aerosol
- MAC
Mass absorption coefficient
- MCM
Master chemical mechanism
- MBO
2-methyl-3-buten-2-ol
- MIESR
Matrix isolation electron spin resonance
- MO-OOA
More-oxidized oxygenated organic aerosol
- MOSAIC
Model for Simulating Aerosol Interactions and Chemistry
- MOZART
Model for OZone and Related chemical Tracers
- NARSTO
North American Research Strategy for Tropospheric Ozone
- NEAQS
New England Air Quality Study
- NBL
Nocturnal boundary layer
- NEI
National Emissions Inventory
- NO
Nitric oxide
- NO2
Nitrogen dioxide
- NO3
Nitrate radical
- N2O5
Dinitrogen pentoxide
- NOx
Nitrogen oxides, NO + NO2
- NOy
Total reactive nitrogen
- NOS
Nitrooxy organosulfate
- NSF
National Science Foundation
- O3
Ozone
- OA
Organic aerosol
- OC
Organic carbon
- OH
Hydroxyl radical
- OS
Organosulfate
- PACIFIC
Pacific Air Quality Study
- PAHs
Polyaromatic (or polycyclic aromatic) hydrocarbons
- PiLS
Particle-into-liquid sampler
- PiG
Plume-in-grid
- PM
Particulate matter
- PM1/2.5
Particulate matter smaller than 1/2.5 μ
- PMF
Positive matrix factorization
- PROPHET
Program for Research on Oxidants: Photochemistry, Emissions and Transport
- PTR-MS
Proton transfer reaction mass spectrometry
- PR-A
Pulse radiolysis – absorption
- RH
Relative humidity
- RI
Refractive index
- RL
Residual layer
- RONO2
Organic nitrate
- RO2
Organic peroxy radical
- RR
Relative rate
- SAPRC
Statewide Air Pollution Research Center
- SAR
Structure activity relationship
- SEAC4RS
Studies of Emissions and Atmospheric Composition, Clouds and Climate Coupling by Regional Surveys
- SEARCH
Southeastern Aerosol Research and Characterization
- SOA
Secondary organic aerosol
- SOAS
Southern Oxidant and Aerosol Study
- SOS
Southern Oxidant Study
- STOCHEM-CRI
STOchastic lagrangian CHEMistry model using Common Representative Intermediates
- SV-OOA
Semi-volatile oxygenated organic aerosol
- TD-CAPS
Thermal dissociation cavity-attenuated phase shift spectroscopy
- TD-CRDS
Thermal dissociation cavity ring-down spectroscopy
- TD-LIF
Thermal dissociation laser-induced fluorescence
- UKESM-1
UK Earth System Model 1
- VBS
Volatility basis set
- VOC
Volatile organic compound(s)
- WRF-Chem
Weather Research and Forecasting Model with Chemistry
Footnotes
The Supplement related to this article is available online at doi:10.5194/acp-17-2103-2017-supplement.
Competing interests. The authors declare that they have no conflict of interest.
References
- Abida O, Mielke LH, Osthoff HD. Observation of gas-phase peroxynitrous and peroxynitric acid during the photolysis of nitrate in acidified frozen solutions. Chem Phys Lett. 2011;511:187–192. doi: 10.1016/j.cplett.2011.06.055. [DOI] [Google Scholar]
- Alessandrini S, Ferrero E. A hybrid Lagrangian–Eulerian particle model for reacting pollutant dispersion in non-homogeneous non-isotropic turbulence. Physica A. 2009;388:1375–1387. doi: 10.1016/j.physa.2008.12.015. [DOI] [Google Scholar]
- Aliwell SR, Jones RL. Measurement of atmospheric NO3 2. Diurnal variation of stratospheric NO3 at midlatitude. Geophys Res Lett. 1996a;23:2589–2592. [Google Scholar]
- Aliwell SR, Jones RL. Measurement of atmospheric NO3 1. Improved removal of water vapour absorption features in the analysis for NO3. Geophys Res Lett. 1996b;23:2585–2588. [Google Scholar]
- Aliwell SR, Jones RL. Measurements of tropospheric NO3 at midlatitude. J Geophys Res. 1998;103:5719–5727. [Google Scholar]
- Allan BJ, Carslaw N, Coe H, Burgess RA, Plane JMC. Observations of the Nitrate Radical in the Marine Boundary Layer. J Atmos Chem. 1999;33:129–154. [Google Scholar]
- Allan BJ, McFiggans G, Plane JMC, Coe H, Mc-Fadyen GG. The nitrate radical in the remote marine boundary layer. J Geophys Res. 2000;105:24191–124204. [Google Scholar]
- Allan BJ, Plane JMC, Coe H, Shillito J. Observations of NO3 concentration profiles in the troposphere. J Geophys Res. 2002;107:4588. doi: 10.1029/2002JD002112. [DOI] [Google Scholar]
- Allan JD, Delia AE, Coe H, Bower KN, Alfarra MR, Jimenez JL, Middlebrook AM, Drewnick F, Onasch TB, Canagaratna MR, Jayne JT, Worsnop DR. A generalised method for the extraction of chemically resolved mass spectra from aerodyne aerosol mass spectrometer data. J Aerosol Sci. 2004;35:909–922. [Google Scholar]
- Allan JD, Alfarra MR, Bower KN, Coe H, Jayne JT, Worsnop DR, Aalto PP, Kulmala M, Hyötyläinen T, Cavalli F, Laaksonen A. Size and composition measurements of background aerosol and new particle growth in a Finnish forest during QUEST 2 using an Aerodyne Aerosol Mass Spectrometer. Atmos Chem Phys. 2006;6:315–327. doi: 10.5194/acp-6-315-2006. [DOI] [Google Scholar]
- Alvarado A, Arey J, Atkinson R. Kinetics of the Gas-Phase Reactions of OH and NO3 Radicals and O3 with the Monoterpene Reaction Products Pinonaldehyde, Caronaldehyde, and Sabinaketone. J Atmos Chem. 1998;31:281–297. [Google Scholar]
- Andreae MO, Gelencser A. Black carbon or brown carbon? The nature of light-absorbing carbonaceous aerosols. Atmos Chem Phys. 2006;6:3131–3148. doi: 10.5194/acp-6-3131-2006. [DOI] [Google Scholar]
- Angove D, Fookes C, Hynes R, Walters C, Azzi M. The characterisation of secondary organic aerosol formed during the photodecomposition of 1, 3-butadiene in air containing nitric oxide. Atmos Environ. 2006;40:4597–4607. [Google Scholar]
- Arangio AM, Slade JH, Berkemeier T, Pöschl U, Knopf DA, Shiraiwa M. Multiphase Chemical Kinetics of OH Radical Uptake by Molecular Organic Markers of Biomass Burning Aerosols: Humidity and Temperature Dependence, Surface Reaction, and Bulk Diffusion. J Phys Chem A. 2015;119:4533–4544. doi: 10.1021/jp510489z. [DOI] [PubMed] [Google Scholar]
- Archibald AT, Cooke MC, Utembe SR, Shallcross DE, Derwent RG, Jenkin ME. Impacts of mechanistic changes on HOx formation and recycling in the oxidation of isoprene. Atmos Chem Phys. 2010;10:8097–8118. doi: 10.5194/acp-10-8097-2010. [DOI] [Google Scholar]
- Asaf DDP, Matveev V, Peleg M, Kern C, Zingler J, Platt U, Luria M. Long-Term Measurements of NO3 Radical at a Semiarid Urban Site: 1. Extreme Concentration Events and Their Oxidation Capacity. Environ Sci Technol. 2009;43:9117–9123. doi: 10.1021/es900798b. [DOI] [PubMed] [Google Scholar]
- Atkinson R, Arey J. Atmospheric Degradation of Volatile Organic Compounds. Chem Rev. 2003;103:4605–4638. doi: 10.1021/cr0206420. [DOI] [PubMed] [Google Scholar]
- Atkinson R, Aschmann SM, Winer AM, Pitts JN. Kinetics of the Gas-Phase Reactions of NO3 Radicals with a Series of Dialkenes, Cycloalkenes, and Monoterpenes at 295 ± 1-K. Environ Sci Technol. 1984;18:370–375. doi: 10.1021/es00123a016. [DOI] [PubMed] [Google Scholar]
- Atkinson R, Aschmann SM, Winer AM, Pitts JN. Kinetics and atmospheric implications of the gas-phase reactions of NO3 radicals with a series of monoterpenes and related organics at 294 ± 2-K. Environ Sci Technol. 1985;19:159–163. doi: 10.1021/es00132a009. [DOI] [Google Scholar]
- Atkinson R, Aschmann SM, Pitts JN. Rate constants for the gas-phase reactions of the nitrate radical with a series of organic compounds at 296 ± 2 K. J Phys Chem A. 1988;92:3454–3457. [Google Scholar]
- Atkinson R, Aschmann SM, Arey J. Rate constants for the gas-phase reactions of OH and NO3 radicals and O3 with sabinene and camphene at 296 ± 2 K. Atmos Environ. 1990;24:2647–2654. [Google Scholar]
- Atkinson R, Arey J, Aschmann SM, Corchnoy SB, Shu Y. Rate constants for the gas-phase reactions of cis-3-Hexen-1-ol, cis-3-Hexenylacetate, trans-2-Hexenal, and Linalool with OH and NO3 radicals and O3 at 296 ± 2 K, and OH radical formation yields from the O3 reactions. Int J Chem Kinet. 1995;27:941–955. [Google Scholar]
- Atlas E. Evidence for Greater-Than-or-Equal-to-C-3 Alkyl Nitrates in Rural and Remote Atmospheres. Nature. 1988;331:426–428. [Google Scholar]
- Atlas E, Schauffler S. Analysis of alkyl nitrates and selected halocarbons in the ambient atmosphere using a charcoal preconcentration technique. Environ Sci Technol. 1991;25:61–67. [Google Scholar]
- Aumont B, Szopa S, Madronich S. Modelling the evolution of organic carbon during its gas-phase tropospheric oxidation: development of an explicit model based on a self generating approach. Atmos Chem Phys. 2005;5:2497–2517. doi: 10.5194/acp-5-2497-2005. [DOI] [Google Scholar]
- Ayers JD, Simpson WR. Measurements of N2O5 near Fairbanks, Alaska. J Geophys Res-Atmos. 2006;111:13377–13392. doi: 10.1029/2006JD007070. [DOI] [Google Scholar]
- Ayers JD, Apodaca RL, Simpson WR, Baer DS. Off-axis cavity ringdown spectroscopy: application to atmospheric nitrate radicaldetection. Appl Optics. 2005;44:7239–7242. doi: 10.1364/AO.44.007239. [DOI] [PubMed] [Google Scholar]
- Ayres BR, Allen HM, Draper DC, Brown SS, Wild RJ, Jimenez JL, Day DA, Campuzano-Jost P, Hu W, de Gouw J, Koss A, Cohen RC, Duffey KC, Romer P, Baumann K, Edgerton E, Takahama S, Thornton JA, Lee BH, Lopez-Hilfiker FD, Mohr C, Wennberg PO, Nguyen TB, Teng A, Goldstein AH, Olson K, Fry JL. Organic nitrate aerosol formation via biogenic volatile organic compounds in the southeastern United States. Atmos Chem Phys. 2015;15:13377–13392. doi: 10.5194/acp-15-13377-2015. [DOI] [Google Scholar]
- Bahadur R, Praveen PS, Xu Y, Ramanathan V. Solar absorption by elemental and brown carbon determined from spectral observations. P Natl Acad Sci USA. 2012;109:17366–17371. doi: 10.1073/pnas.1205910109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker J, Arey J, Atkinson R. Kinetics of the gas-phase reactions of OH radicals, NO3 radicals and O3 with three C7 carbonyls formed from the atmospheric reactions of myrcene, ocimene and terpinolene. J Atmos Chem. 2004;48:241–260. [Google Scholar]
- Ball SM, Povey IM, Norton EG, Jones RL. Broadband cavity ringdown spectroscopy of the NO3 radical. Chem Phys Lett. 2001;342:113–120. doi: 10.1016/S0009-2614(01)00573-5. [DOI] [Google Scholar]
- Ball SM, Langridge JM, Jones RL. Broadband cavity enhanced absorption spectroscopy using light emitting diodes. Chem Phys Lett. 2004;398:68–74. doi: 10.1016/j.cplett.2004.08.144. [DOI] [Google Scholar]
- Barnes I, Bastian V, Becker KH, Tong Z. Kinetics and Products of the Reactions of NO3 with Monoalkenes, Dialkenes, and Monoterpenes. J Phys Chem-Us. 1990;94:2413–2419. [Google Scholar]
- Bartenbach S, Williams J, Plass-Dülmer C, Berresheim H, Lelieveld J. In-situ measurement of reactive hydrocarbons at Hohenpeissenberg with comprehensive two-dimensional gas chromatography (GC × GC-FID): use in estimating HO and NO3. Atmos Chem Phys. 2007;7:1–14. doi: 10.5194/acp-7-1-2007. [DOI] [Google Scholar]
- Bates KH, Crounse JD, St Clair JM, Bennett NB, Nguyen TB, Seinfeld JH, Stoltz BM, Wennberg PO. Gas Phase Production and Loss of Isoprene Epoxydiols. J Phys Chem A. 2014;118:1237–1246. doi: 10.1021/jp4107958. [DOI] [PubMed] [Google Scholar]
- Beaver MR, Clair JMS, Paulot F, Spencer KM, Crounse JD, LaFranchi BW, Min KE, Pusede SE, Woolridge PJ, Schade GW, Park C, Cohen RC, Wennberg PO. Importance of biogenic precursors to the budget of organic nitrates during BEARPEX 2009: observations of multifunctional organic nitrates by CIMS and TD-LIF. Atmos Chem Phys. 2012;12:5773–5785. doi: 10.5194/acpd-12-319-2012. [DOI] [Google Scholar]
- Benter T, Schindler RN. Absolute rate coefficients for the reaction of NO3 radicals with simple dienes. Chem Phys Lett. 1988;145:67–70. doi: 10.1016/0009-2614(88)85134-0. [DOI] [Google Scholar]
- Benton AK, Langridge JM, Ball SM, Bloss WJ, Dall’Osto M, Nemitz E, Harrison RM, Jones RL. Night-time chemistry above London: measurements of NO3 and N2O5 from the BT Tower. Atmos Chem Phys. 2010;10:9781–9795. doi: 10.5194/acp-10-9781-2010. [DOI] [Google Scholar]
- Berkemeier T, Ammann M, Mentel TF, Pöschl U, Shiraiwa M. Organic Nitrate Contribution to New Particle Formation and Growth in Secondary Organic Aerosols from α-Pinene Ozonolysis. Environ Sci Technol. 2016;50:6334–6342. doi: 10.1021/acs.est.6b00961. [DOI] [PubMed] [Google Scholar]
- Berndt T, Boge O. Gas-phase reaction of NO3 radicals with isoprene: A kinetic and mechanistic study. Int J Chem Kinet. 1997a;29:755–765. [Google Scholar]
- Berndt T, Boge O. Products and mechanism of the gas-phase reaction of NO3 radicals with α-pinene. J Chem Soc Faraday Trans. 1997b;93:3021–3027. doi: 10.1039/a702364b. [DOI] [Google Scholar]
- Berndt T, Boge O, Kind I, Rolle W. Reaction of NO3 radicals with 1,3-cyclohexadiene alpha-terpinene, and alpha-phellandrene: Kinetics and products. Ber Bunsen-Ges Phys Chem Chem Phys. 1996;100:462–469. [Google Scholar]
- Biesenthal T, Bottenheim J, Shepson P, Li SM, Brickell P. The chemistry of biogenic hydrocarbons at a rural site in eastern Canada. J Geophys Res-Atmos. 1998;103:25487–25498. [Google Scholar]
- Bitter M, Ball SM, Povey IM, Jones RL. A broadband cavity ringdown spectrometer for in-situ measurements of atmospheric trace gases. Atmos Chem Phys. 2005;5:2547–2560. doi: 10.5194/acp-5-2547-2005. [DOI] [Google Scholar]
- Blake NJ, Blake DR, Wingenter OW, Sive BC, Kang CH, Thornton DC, Bandy AR, Atlas E, Flocke F, Harris JM, Rowland FS. Aircraft measurements of the latitudinal, vertical, and seasonal variations of NMHCs, methyl nitrate, methyl halides, and DMS during the First Aerosol Characterization Experiment (ACE 1) J Geophys Res-Atmos. 1999;104:21803–21817. [Google Scholar]
- Blake NJ, Blake DR, Sive BC, Katzenstein AS, Meinardi S, Wingenter OW, Atlas EL, Flocke F, Ridley BA, Rowland FS. The seasonal evolution of NMHCs and light alkyl nitrates at middle to high northern latitudes during TOPSE. J Geophys Res-Atmos. 2003a;108:8359. doi: 10.1029/2001jd001467. [DOI] [Google Scholar]
- Blake NJ, Blake DR, Swanson AL, Atlas E, Flocke F, Rowland FS. Latitudinal, vertical, and seasonal variations of C-1-C-4 alkyl nitrates in the troposphere over the Pacific Ocean during PEM-Tropics A and B: Oceanic and continental sources. J Geophys Res-Atmos. 2003b;108:8242. doi: 10.1029/2001jd001444. [DOI] [Google Scholar]
- Boersma KF, Jacob DJ, Eskes HJ, Pinder RW, Wang J, van der ARJ. Intercomparison of SCIAMACHY and OMI tropospheric NO2 columns: Observing the diurnal evolution of chemistry and emissions from space. J Geophys Res-Atmos. 2008;113:D16S26. doi: 10.1029/2007JD008816. [DOI] [Google Scholar]
- Bond TC, Bergstrom RW. Light absorption by carbonaceous particles: An investigative review. Aerosol Sci Tech. 2006;40:1–41. [Google Scholar]
- Bond TC, Zarzycki C, Flanner MG, Koch DM. Quantifying immediate radiative forcing by black carbon and organic matter with the Specific Forcing Pulse. Atmos Chem Phys. 2011;11:1505–1525. doi: 10.5194/acp-11-1505-2011. [DOI] [Google Scholar]
- Bonn B, Moorgat GK. New particle formation during a-and b-pinene oxidation by O3, OH and NO3, and the influence of water vapour: particle size distribution studies. Atmos Chem Phys. 2002;2:183–196. doi: 10.5194/acp-2-183-2002. [DOI] [Google Scholar]
- Boschan R, Merrow RT, Vandolah RW. The Chemistry of Nitrate Esters. Chem Rev. 1955;55:485–510. [Google Scholar]
- Bossmeyer J, Brauers T, Richter C, Rohrer F, Wegener R, Wahner A. Simulation chamber studies of the NO3 chemistry of atmospheric aldehydes. Geophys Res Lett. 2006;33:L18810. doi: 10.11029/12006GL026778. [DOI] [Google Scholar]
- Boucher O, Randall D, Artaxo P, Bretherton C, Feingold G, Forster P, Kerminen V-M, Kondo Y, Liao H, Lohmann U, Rasch P, Satheesh SK, Sherwood S, Stevens B, Zhang XY. Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change. Cambridge, United Kingdom and New York, NY, USA: 2013. Clouds and Aerosols. [Google Scholar]
- Boyd CM, Sanchez J, Xu L, Eugene AJ, Nah T, Tuet WY, Guzman MI, Ng NL. Secondary organic aerosol formation from the β-pinene + NO3 system: effect of humidity and peroxy radical fate. Atmos Chem Phys. 2015;15:7497–7522. doi: 10.5194/acp-15-7497-2015. [DOI] [Google Scholar]
- Bräuer P, Mouchel-Vallon C, Tilgner A, Mutzel A, Böge O, Rodigast M, Poulain L, van Pinxteren D, Aumont B, Herrmann H. Development of a protocol designed for the self-generation of explicit aqueous phase oxidation schemes of organic compounds. Atmos Chem Phys Discuss. 2017 in preparation. [Google Scholar]
- Brown SS, Stutz J. Nighttime Radical Observations and Chemistry. Chem Soc Rev. 2012;41:6405–6447. doi: 10.1039/c2cs35181a. [DOI] [PubMed] [Google Scholar]
- Brown SS, Stark H, Ciciora SJ, Ravishankara AR. In-situ measurement of atmospheric NO3 and N2O5 via cavity ring-down spectroscopy. Geophys Res Lett. 2001;28:3227–3230. [Google Scholar]
- Brown SS, Stark H, Ciciora SJ, McLaughlin RJ, Rav-ishankara AR. Simultaneous in-situ detection of atmospheric NO3 and N2O5 via cavity ring-down spectroscopy. Rev Sci Instr. 2002;73:3291–3301. [Google Scholar]
- Brown SS, Stark H, Ryerson TB, Williams EJ, Nicks DKJ, Trainer M, Fehsenfeld FC, Ravishankara AR. Nitrogen oxides in the nocturnal boundary layer: Simultaneous, in-situ detection of NO3, N2O5, NO, NO2 and O3. J Geophys Res. 2003;108:D94299. doi: 10.1029/2002JD002917. [DOI] [Google Scholar]
- Brown SS, Dubé WP, Osthoff HD, Stutz J, Ryerson TB, Wollny AG, Brock CA, Warneke C, de Gouw JA, Atlas E, Neuman JA, Holloway JS, Lerner BM, Williams EJ, Kuster WC, Goldan PD, Angevine WM, Trainer M, Fehsenfeld FC, Ravishankara AR. Vertical profiles in NO3 and N2O5 measured from an aircraft: Results from the NOAA P-3 and surface platforms during NEAQS 2004. J Geophys Res. 2007a;112:D22304. doi: 10.1029/2007JD008883. [DOI] [Google Scholar]
- Brown SS, Dubé WP, Osthoff HD, Wolfe DE, Angevine WM, Ravishankara AR. High resolution vertical distributions of NO3 and N2O5 through the nocturnal boundary layer. Atmos Chem Phys. 2007b;7:139–149. doi: 10.5194/acp-7-139-2007. [DOI] [Google Scholar]
- Brown SS, de Gouw JA, Warneke C, Ryerson TB, Dubé WP, Atlas E, Weber RJ, Peltier RE, Neuman JA, Roberts JM, Swanson A, Flocke F, McKeen SA, Brioude J, Sommariva R, Trainer M, Fehsenfeld FC, Ravishankara AR. Nocturnal isoprene oxidation over the Northeast United States in summer and its impact on reactive nitrogen partitioning and secondary organic aerosol. Atmos Chem Phys. 2009;9:3027–3042. doi: 10.5194/acp-9-3027-2009. [DOI] [Google Scholar]
- Brown SS, Dubé WP, Peischl J, Ryerson TB, Atlas E, Warneke C, de Gouw J, Te Lintel Hekkert S, Brock CA, Flocke F, Trainer M, Parrish DD, Fehsenfeld FC, Ravishankara AR. Budgets for nocturnal VOC oxidation by nitrate radicals aloft during the 2006 Texas Air Quality Study. J Geophys Res. 2011;116:D24305. doi: 10.1029/2011JD016544. [DOI] [Google Scholar]
- Brown SS, Dubé WP, Bahreini R, Middlebrook AM, Brock CA, Warneke C, de Gouw JA, Washenfelder RA, Atlas E, Peischl J, Ryerson TB, Holloway JS, Schwarz JP, Spackman R, Trainer M, Parrish DD, Fehsenfeld FC, Ravishankara AR. Biogenic VOC oxidation and organic aerosol formation in an urban noctural boundary layer: Aircraft vertical profiles in Houston, TX. Atmos Chem Phys. 2013;13:11317–11337. doi: 10.5194/acp-13-11317-2013. [DOI] [Google Scholar]
- Browne EC, Min KE, Wooldridge PJ, Apel E, Blake DR, Brune WH, Cantrell CA, Cubison MJ, Diskin GS, Jimenez JL, Weinheimer AJ, Wennberg PO, Wisthaler A, Cohen RC. Observations of total RONO2 over the boreal forest: NOx sinks and HNO3 sources. Atmos Chem Phys. 2013;13:4543–4562. doi: 10.5194/acp-13-4543-2013. [DOI] [Google Scholar]
- Browne EC, Wooldridge PJ, Min KE, Cohen RC. On the role of monoterpene chemistry in the remote continental boundary layer. Atmos Chem Phys. 2014;14:1225–1238. doi: 10.5194/acp-14-1225-2014. [DOI] [Google Scholar]
- Bruns EA, Perraud V, Zelenyuk A, Ezell MJ, Johnson SN, Yu Y, Imre D, Finlayson-Pitts BJ, Alexander ML. Comparison of FTIR and Particle Mass Spectrometry for the Measurement of Particulate Organic Nitrates. Environ Sci Technol. 2010;44:1056–1061. doi: 10.1021/es9029864. [DOI] [PubMed] [Google Scholar]
- Calvert JG, Orlando J, Stockwell WR, Wallington TJ. The Mechanisms of Reactions Influencing Atmospheric Ozone. Oxford University Press; New York, New York: 2015. [Google Scholar]
- Canosa-Mas CE, Carr S, King MD, Shallcross DE, Thompson KC, Wayne RP. A kinetic study of the reactions of NO3 with methyl vinyl ketone, methacrolein, acrolein, methyl acrylate and methyl methacrylate. Phys Chem Chem Phys. 1999a;1:4195–4202. [Google Scholar]
- Canosa-Mas CE, King MD, Scarr PJ, Thompson KC, Wayne RP. An experimental study of the gas-phase reactions of the NO3 radical with three sesquiterpenes: isolongifolene, alloisolongifolene, and neoclovene. Phys Chem Chem Phys. 1999b;1:2929–2933. [Google Scholar]
- Carlton AG, Bhave PV, Napelenok SL, Edney EO, Sarwar G, Pinder RW, Pouliot GA, Houyoux M. Model Representation of Secondary Organic Aerosol in CMAQv4.7. Environ Sci Technol. 2010a;44:8553–8560. doi: 10.1021/es100636q. [DOI] [PubMed] [Google Scholar]
- Carlton AG, Pinder RW, Bhave PV, Pouliot GA. To What Extent Can Biogenic SOA be Controlled? EnvironSci Technol. 2010b;44:3376–3380. doi: 10.1021/es903506b. [DOI] [PubMed] [Google Scholar]
- Carter WPL. Final report to the California Air Resources Board Contract No. 03-318, 27. Jan, 2010a. Development of the SAPRC-07 Chemical Mechanism and Updated Ozone Reactivity Scales. [Google Scholar]
- Carter WPL. Development of the SAPRC-07 chemical mechanism. Atmos Environ. 2010b;44:5324–5335. doi: 10.1016/j.atmosenv.2010.01.026. [DOI] [Google Scholar]
- Castellanos P, Boersma KF. Reductions in nitrogen oxides over Europe driven by environmental policy and economic recession. Sci Rep. 2012;2:265. doi: 10.1038/srep00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavdar H, Saracoglu N. Synthesis of new beta-hydroxy nitrate esters as potential glycomimetics or vasodilators. Eur J Org Chem. 2008:4615–4621. [Google Scholar]
- Cerully KM, Bougiatioti A, Hite JR, Jr, Guo H, Xu L, Ng NL, Weber R, Nenes A. On the link between hygroscopicity, volatility, and oxidation state of ambient and water-soluble aerosols in the southeastern United States. Atmos Chem Phys. 2015;15:8679–8694. doi: 10.5194/acp-15-8679-2015. [DOI] [Google Scholar]
- Chacon-Madrid HJ, Henry KM, Donahue NM. Photo-oxidation of pinonaldehyde at low NOx: from chemistry to organic aerosol formation. Atmos Chem Phys. 2013;13:3227–3236. doi: 10.5194/acp-13-3227-2013. [DOI] [Google Scholar]
- Chameides WL. Photo-chemical role of tropospheric nitrogen oxides. Geophys Res Lett. 1978;5:17–20. [Google Scholar]
- Chen J, Avise J, Guenther A, Wiedinmyer C, Salathe E, Jackson RB, Lamb B. Future land use and land cover influences on regional biogenic emissions and air quality in the United States. Atmos Environ. 2009;43:5771–5780. doi: 10.1016/j.atmosenv.2009.08.015. [DOI] [Google Scholar]
- Chew AA, Atkinson R, Aschmann SM. Kinetics of the gas-phase reactions of NO3 radicals with a series of alcohols, glycol ethers, ethers and chloroalkenes. J Chem Soc Faraday Trans. 1998;94:1083–1089. [Google Scholar]
- Chung CE, Ramanathan V, Decremer D. Observationally constrained estimates of carbonaceous aerosol radiative forcing. P Natl Acad Sci USA. 2012;109:11624–11629. doi: 10.1073/pnas.1203707109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung SH, Seinfeld JH. Global distribution and climate forcing of carbonaceous aerosols. J Geophys Res. 2002;107:4407. doi: 10.1029/2001JD001397. [DOI] [Google Scholar]
- Clifford GM, Thüner LP, Wenger JC, Shallcross DE. Kinetics of the gas-phase reactions of OH and NO3 radicals with aromatic aldehydes. J Photoch Photobio A. 2005;176:172–182. doi: 10.1016/j.jphotochem.2005.09.022. [DOI] [Google Scholar]
- Coe H, Allan BJ, Plane JMC. Retrieval of vertical profiles of NO3 from zenith sky measurements using an optimal estimation method. J Geophys Res. 2002;107:4587. doi: 10.1029/2002JD002111. [DOI] [Google Scholar]
- Cohan DS, Hu Y, Russell AG. Dependence of ozone sensitivity analysis on grid resolution. Atmos Environ. 2006;40:126–135. doi: 10.1016/j.atmosenv.2005.09.031. [DOI] [Google Scholar]
- Corchnoy SB, Atkinson R. Kinetics of the gas-phase reactions of hydroxyl and nitrogen oxide (NO3) radicals with 2-carene, 1,8-cineole, p-cymene, and terpinolene, Environ. Sci Technol. 1990;24:1497–1502. [Google Scholar]
- Cosman LM, Knopf DA, Bertram AK. N2O5 Reactive Uptake on Aqueous Sulfuric Acid Solutions Coated with Branched and Straight-Chain Insoluble Organic Surfactants. J Phys Chem A. 2008;112:2386–2396. doi: 10.1021/jp710685r. [DOI] [PubMed] [Google Scholar]
- Crippa M, Canonaco F, Lanz VA, Äijälä M, Allan JD, Carbone S, Capes G, Ceburnis D, Dall’Osto M, Day DA, De-Carlo PF, Ehn M, Eriksson A, Freney E, Hildebrandt Ruiz L, Hillamo R, Jimenez JL, Junninen H, Kiendler-Scharr A, Kortelainen A-M, Kulmala M, Laaksonen A, Mensah AA, Mohr C, Nemitz E, O’Dowd C, Ovadnevaite J, Pandis SN, Petäjä T, Poulain L, Saarikoski S, Sellegri K, Swietlicki E, Tiitta P, Worsnop DR, Baltensperger U, Prévôt ASH. Organic aerosol components derived from 25 AMS data sets across Europe using a consistent ME-2 based source apportionment approach. Atmos Chem Phys. 2014;14:6159–6176. doi: 10.5194/acp-14-6159-2014. [DOI] [Google Scholar]
- Crounse JD, Paulot F, Kjaergaard HG, Wennberg PO. Peroxy radical isomerization in the oxidation of isoprene. Phys Chem Chem Phys. 2011;13:13607–13613. doi: 10.1039/C1CP21330J. [DOI] [PubMed] [Google Scholar]
- Crowley JN, Schuster G, Pouvesle N, Parchatka U, Fischer H, Bonn B, Bingemer H, Lelieveld J. Nocturnal nitrogen oxides at a rural mountain-site in south-western Germany. Atmos Chem Phys. 2010;10:2795–2812. doi: 10.5194/acp-10-2795-2010. [DOI] [Google Scholar]
- Crowley JN, Thieser J, Tang MJ, Schuster G, Bozem H, Beygi ZH, Fischer H, Diesch JM, Drewnick F, Borrmann S, Song W, Yassaa N, Williams J, Pöhler D, Platt U, Lelieveld J. Variable lifetimes and loss mechanisms for NO3 and N2O5 during the DOMINO campaign: contrasts between marine, urban and continental air. Atmos Chem Phys. 2011;11:10853–10870. doi: 10.5194/acp-11-10853-2011. [DOI] [Google Scholar]
- Crutzen PJ. A Discussion of the Chemistry of Some Minor Constituents in the Stratosphere and Troposphere. Pure Appl Geophys. 1973;106:1385–1399. [Google Scholar]
- Curci G, Beekmann M, Vautard R, Smiatek G, Steinbrecher R, Theloke J, Friedrich R. Modelling study of the impact of isoprene and terpene biogenic emissions on European ozone levels. Atmos Environ. 2009;43:1444–1455. doi: 10.1016/j.atmosenv.2008.02.070. [DOI] [Google Scholar]
- Darer AI, Cole-Filipiak NC, O’Connor AE, Elrod MJ. Formation and Stability of Atmospherically Relevant Isoprene-Derived Organosulfates and Organonitrates. Environ Sci Technol. 2011;45:1895–1902. doi: 10.1021/es103797z. [DOI] [PubMed] [Google Scholar]
- Day D, Wooldridge P, Dillon M, Thornton J, Cohen R. A thermal dissociation laser-induced fluorescence instrument for in situ detection of NO2, peroxy nitrates, alkyl nitrates, and HNO3. J Geophys Res-Atmos. 2002;107:4046. doi: 10.1029/2001JD000779. [DOI] [Google Scholar]
- Day DA, Dillon MB, Wooldridge PJ, Thornton JA, Rosen RS, Wood EC, Cohen RC. On alkyl nitrates, O3, and the “missing NOy. J Geophys Res. 2003;108:4501. doi: 10.129/2003JD003685. [DOI] [Google Scholar]
- Day DA, Liu S, Russell LM, Ziemann PJ. Organonitrate group concentrations in submicron particles with high nitrate and organic fractions in coastal southern California. Atmos Environ. 2010;44:1970–1979. [Google Scholar]
- Dlugokencky EJ, Howard CJ. Studies of NO3 radical reactions with some atmospheric organic compounds at low pressures. J Phys Chem-Us. 1989;93:1091–1096. doi: 10.1021/j100340a015. [DOI] [Google Scholar]
- Docherty KS, Ziemann PJ. Reaction of Oleic Acid Particles with NO3 Radicals: Products, Mechanism, and Implications for Radical-Initiated Organic Aerosol Oxidation. J Phys Chem A. 2006;110:3567–3577. doi: 10.1021/jp0582383. [DOI] [PubMed] [Google Scholar]
- Donahue NM, Robinson AL, Stanier CO, Pandis SN. Coupled Partitioning, Dilution, and Chemical Aging of Semivolatile Organics. Environ Sci Technol. 2006;40:2635–2643. doi: 10.1021/es052297c. [DOI] [PubMed] [Google Scholar]
- Dorn HP, Apodaca RL, Ball SM, Brauers T, Brown SS, Crowley JN, Dubé WP, Fuchs H, Häseler R, Heitmann U, Jones RL, Kiendler-Scharr A, Labazan I, Langridge JM, Meinen J, Mentel TF, Platt U, Pöhler D, Rohrer F, Ruth AA, Schlosser E, Schuster G, Shillings AJL, Simpson WR, Thieser J, Tillmann R, Varma R, Venables DS, Wahner A. Intercomparison of NO3 radical detection instruments in the atmosphere simulation chamber SAPHIR. Atmos Meas Tech. 2013;6:1111–1140. doi: 10.5194/amt-6-1111-2013. [DOI] [Google Scholar]
- Draper DC, Farmer DK, Desyaterik Y, Fry JL. A qualitative comparison of secondary organic aerosol yields and composition from ozonolysis of monoterpenes at varying concentrations of NO2. Atmos Chem Phys. 2015;15:12267–12281. doi: 10.5194/acp-15-12267-2015. [DOI] [Google Scholar]
- Dubé WP, Brown SS, Osthoff HD, Nunley MR, Ciciora SJ, Paris MW, McLaughlin RJ, Ravishankara AR. Aircraft instrument for simultaneous, in situ measurement of NO3 and N2O5 via pulsed cavity ring-down spectroscopy. Rev Sci Instrum. 2006;77:034101. doi: 10.1063/1.2176058. [DOI] [Google Scholar]
- Duncan BN, Lamsal LN, Thompson AM, Yoshida Y, Lu Z, Streets DG, Hurwitz MM, Pickering KE. A space-based, high-resolution view of notable changes in urban NOx pollution around the world (2005–2014) J Geophys Res-Atmos. 2016;121:976–996. doi: 10.1002/2015JD024121. [DOI] [Google Scholar]
- Ehn M, Thornton JA, Kleist E, Sipila M, Junninen H, Pullinen I, Springer M, Rubach F, Tillmann R, Lee B, Lopez-Hilfiker F, Andres S, Acir I-H, Rissanen M, Jokinen T, Schobesberger S, Kangasluoma J, Kontkanen J, Nieminen T, Kurten T, Nielsen LB, Jorgensen S, Kjaergaard HG, Canagaratna M, Maso MD, Berndt T, Petaja T, Wahner A, Kerminen V-M, Kulmala M, Worsnop DR, Wildt J, Mentel TF. A large source of low-volatility secondary organic aerosol. Nature. 2014;506:476–479. doi: 10.1038/nature13032. [DOI] [PubMed] [Google Scholar]
- Ellermann T, Nielsen OJ, Skov H. Absolute rate constants for the reaction of NO3 radicals with a series of dienes at 295-K. Chem Phys Lett. 1992;200:224–229. doi: 10.1016/0009-2614(92)80002-s. [DOI] [Google Scholar]
- Emmons LK, Walters S, Hess PG, Lamarque JF, Pfister GG, Fillmore D, Granier C, Guenther A, Kinnison D, Laepple T, Orlando J, Tie X, Tyndall G, Wiedinmyer C, Baughcum SL, Kloster S. Description and evaluation of the Model for Ozone and Related chemical Tracers, version 4 (MOZART-4) Geosci Model Dev. 2010;3:43–67. doi: 10.5194/gmd-3-43-2010. [DOI] [Google Scholar]
- Eyth A, Zubrow A, Mason R. Preparation of Emissions Inventories for the Version 6.1, 2011 Emissions Modeling Platform Report. Technical Support Document (TSD) 2014 [Google Scholar]
- Fantechi G, Jensen NR, Hjorth J, Peeters J. Determination of the rate constants for the gas-phase reactions of methyl butenol with OH radicals, ozone, NO3 radicals, and Cl atoms. Int J Chem Kinet. 1998a;30:589–594. [Google Scholar]
- Fantechi G, Jensen NR, Hjorth J, Peeters J. Mechanistic studies of the atmospheric oxidation of methyl butenol by OH radicals, ozone and NO3 radicals. Atmos Environ. 1998b;32:3547–3556. doi: 10.1016/S1352-2310(98)00061-2. [DOI] [Google Scholar]
- Farmer DK, Matsunaga A, Docherty KS, Surratt JD, Seinfeld JH, Ziemann PJ, Jimenez JL. Response of the Aerosol Mass Spectrometer to organonitrates and organosulfates and implications for field studies. PNAS. 2010;107:6670–6675. doi: 10.1073/pnas.0912340107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Ramanathan V, Kotamarthi V. Brown carbon: a significant atmospheric absorber of solar radiation? Atmos Chem Phys. 2013;13:8607–8621. doi: 10.5194/acp-13-8607-2013. [DOI] [Google Scholar]
- Fiedler SE, Hoheisel G, Ruth AA, Hese A. Incoherent broad-band cavity-enhanced absorption spectroscopy of azulene in a supersonic jet. Chem Phys Lett. 2003;382:447–453. doi: 10.1016/j.cplett.2003.10.075. [DOI] [Google Scholar]
- Fiore AM, Dentener FJ, Wild O, Cuvelier C, Schultz MG, Hess P, Textor C, Schulz M, Doherty RM, Horowitz LW, MacKenzie IA, Sanderson MG, Shindell DT, Stevenson DS, Szopa S, Van Dingenen R, Zeng G, Atherton C, Bergmann D, Bey I, Carmichael G, Collins WJ, Duncan BN, Faluvegi G, Folberth G, Gauss M, Gong S, Hauglustaine D, Holloway T, Isaksen ISA, Jacob DJ, Jonson JE, Kaminski JW, Keating TJ, Lupu A, Marmer E, Montanaro V, Park RJ, Pitari G, Pringle KJ, Pyle JA, Schroeder S, Vivanco MG, Wind P, Wojcik G, Wu S, Zuber A. Multimodel estimates of intercontinental source-receptor relationships for ozone pollution. J Geophys Res. 2009;114:D04301. doi: 10.1029/2008JD010816. [DOI] [Google Scholar]
- Fischer RG, Kastler J, Ballschmiter K. Levels and pattern of alkyl nitrates, multifunctional alkyl nitrates, and halocarbons in the air over the Atlantic Ocean. J Geophys Res-Atmos. 2000;105:14473–14494. doi: 10.1029/1999jd900780. [DOI] [Google Scholar]
- Fisher JA, Jacob DJ, Travis KR, Kim PS, Marais EA, Chan Miller C, Yu K, Zhu L, Yantosca RM, Sulprizio MP, Mao J, Wennberg PO, Crounse JD, Teng AP, Nguyen TB, StClair JM, Cohen RC, Romer P, Nault BA, Wooldridge PJ, Jimenez JL, Campuzano-Jost P, Day DA, Hu W, Shepson PB, Xiong F, Blake DR, Goldstein AH, Misztal PK, Hanisco TF, Wolfe GM, Ryerson TB, Wisthaler A, Mikoviny T. Organic nitrate chemistry and its implications for nitrogen budgets in an isoprene- and monoterpene-rich atmosphere: constraints from aircraft (SEAC4RS) and ground-based (SOAS) observations in the Southeast US. Atmos Chem Phys. 2016;16:5969–5991. doi: 10.5194/acp-16-5969-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flocke F, Volzthomas A, Kley D. Measurements of Alkyl Nitrates in Rural and Polluted Air Masses. Atmos Environ A. 1991;25:1951–1960. [Google Scholar]
- Flocke F, Atlas E, Madronich S, Schauffler SM, Aikin K, Margitan JJ, Bui TP. Observations of methyl nitrate in the lower stratosphere during STRAT: Implications for its gas phase production mechanisms. Geophys Res Lett. 1998;25:1891–1894. [Google Scholar]
- Foley KM, Hogrefe C, Pouliot G, Possiel N, Roselle SJ, Simon H, Timin B. Dynamic evaluation of CMAQ part I: Separating the effects of changing emissions and changing meteorology on ozone levels between 2002 and 2005 in the eastern US. Atmos Environ. 2015;103:247–255. doi: 10.1016/j.atmosenv.2014.12.038. [DOI] [Google Scholar]
- Franze T, Weller MG, Niessner R, Pöschl U. Enzyme immunoassays for the investigation of protein nitration by air pollutants. Analyst. 2003;128:824–831. doi: 10.1039/b303132b. [DOI] [PubMed] [Google Scholar]
- Franze T, Weller MG, Niessner R, Pöschl U. Protein Nitration by Polluted Air. Environ Sci Technol. 2005;39:1673–1678. doi: 10.1021/es0488737. [DOI] [PubMed] [Google Scholar]
- Fry JL, Sackinger K. Model evaluation of NO3 secondary organic aerosol (SOA) source and heterogeneous organic aerosol (OA) sink in the Western United States. Atmos Chem Phys. 2012;12:8797–8811. doi: 10.5194/acpd-12-5189-2012. [DOI] [Google Scholar]
- Fry JL, Kiendler-Scharr A, Rollins AW, Wooldridge PJ, Brown SS, Fuchs H, Dubé W, Mensah A, dal Maso M, Tillmann R, Dorn H-P, Brauers T, Cohen RC. Organic nitrate and secondary organic aerosol yield from NO3 oxidation of β-pinene evaluated using a gas-phase kinetics/aerosol partitioning model. Atmos Chem Phys. 2009;9:1431–1449. doi: 10.5194/acp-9-1431-2009. [DOI] [Google Scholar]
- Fry JL, Kiendler-Scharr A, Rollins AW, Brauers T, Brown SS, Dorn H-P, Dubé WP, Fuchs H, Mensah A, Rohrer F, Tillmann R, Wahner A, Wooldridge PJ, Cohen RC. SOA from limonene: role of NO3 in its generation and degradation. Atmos Chem Phys. 2011;11:3879–3894. doi: 10.5194/acp-11-3879-2011. [DOI] [Google Scholar]
- Fry JL, Draper DC, Zarzana KJ, Campuzano-Jost P, Day DA, Jimenez JL, Brown SS, Cohen RC, Kaser L, Hansel A, Cappellin L, Karl T, Hodzic Roux A, Turnipseed A, Cantrell C, Lefer B, Grossberg N. Observations of gas- and aerosol-phase organic nitrates at BEACHON-RoMBAS 2011. Atmos Chem Phys. 2013;13:8585–8605. doi: 10.5194/acp-13-8585-2013. [DOI] [Google Scholar]
- Fry JL, Draper DC, Barsanti KC, Smith JN, Ortega J, Winkler PM, Lawler MJ, Brown SS, Edwards PM, Cohen RC, Lee L. Secondary Organic Aerosol Formation and Organic Nitrate Yield from NO3 Oxidation of Biogenic Hydrocarbons. Environ Sci Technol. 2014;48:11944–11953. doi: 10.1021/es502204x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs H, Dubé WP, Ciciora SJ, Brown SS. Determination of Inlet Transmission and Conversion Efficiencies for in Situ Measurements of the Nocturnal Nitrogen Oxides, NO3, N2O5 and NO2, via Pulsed Cavity Ring-Down Spectroscopy. Anal Chem. 2008;80:6010–6017. doi: 10.1021/ac8007253. [DOI] [PubMed] [Google Scholar]
- Fuchs H, Simpson WR, Apodaca RL, Brauers T, Cohen RC, Crowley JN, Dorn HP, Dubé WP, Fry JL, Häseler R, Kajii Y, Kiendler-Scharr A, Labazan I, Matsumoto J, Mentel TF, Nakashima Y, Rohrer F, Rollins AW, Schuster G, Tillmann R, Wahner A, Wooldridge PJ, Brown SS. Comparison of N2O5 mixing ratios during NO3Comp 2007 in SAPHIR. Atmos Meas Tech. 2012;5:2763–2777. doi: 10.5194/amtd-5-4927-2012. [DOI] [Google Scholar]
- Fuentes JD, Wang D, Bowling DR, Potosnak M, Monson RK, Goliff WS, Stockwell WR. Biogenic Hydrocarbon Chemistry within and Above a Mixed Deciduous Forest. J Atmos Chem. 2007;56:165–185. [Google Scholar]
- Gao S, Ng NL, Keywood M, Varutbangkul V, Bahreini R, Nenes A, He J, Yoo KY, Beauchamp JL, Hodyss RP, Flagan RC, Seinfeld JH. Particle Phase Acidity and Oligomer Formation in Secondary Organic Aerosol. Environ Sci Technol. 2004;38:6582–6589. doi: 10.1021/es049125k. [DOI] [PubMed] [Google Scholar]
- Gao S, Surratt JD, Knipping EM, Edgerton ES, Shahgholi M, Seinfeld JH. Characterization of polar organic components in fine aerosols in the southeastern United States: Identity, origin, and evolution. J Geophys Res-Atmos. 2006;111:D14314. doi: 10.1029/2005JD006601. [DOI] [Google Scholar]
- Garnes LA, Allen DT. Size Distributions of Organonitrates in Ambient Aerosol Collected in Houston, Texas. Aerosol Sci Tech. 2002;36:983–992. doi: 10.1080/02786820290092186. [DOI] [Google Scholar]
- Geyer A, Alicke B, Mihelcic D, Stutz J, Platt U. Comparison of tropospheric NO3 radical measurements by differential optical absorption spectroscopy and matrix isolation electron spin resonance. J Geophys Res. 1999;104:26097–26105. [Google Scholar]
- Geyer A, Ackermann R, Dubois R, Lohrmann B, Müller T, Platt U. Long-term observation of nitrate radicals in the continental boundary layer near Berlin. Atmos Environ. 2001a;35:3619–3631. doi: 10.1016/S1352-2310(00)00549-5. [DOI] [Google Scholar]
- Geyer A, Alicke B, Konrad S, Schmitz T, Stutz J, Platt U. Chemistry oxidation capacity of the nitrate radical in the continental boundary layer near Berlin. JGeophys Res-Atmos. 2001b;106:8013–8025. doi: 10.1029/2000JD900681. [DOI] [Google Scholar]
- Geyer A, Alicke B, Ackermann R, Martinez M, Harder H, Brune W, di Carlo P, Williams E, Jobson T, Hall S, Shetter R, Stutz J. Direct observations of daytime NO3: Implications for urban boundary layer chemistry. J Geophys Res. 2003;108:4368. doi: 10.1029/2002JD002967. [DOI] [Google Scholar]
- Glasius M, Calogirou A, Jensen NR, Hjorth J, Nielsen CJ. Kinetic study of gas-phase reactions of pinonaldehyde and structurally related compounds. Int J Chem Kinet. 1997;29:527–533. [Google Scholar]
- Goldan PD, Kuster WC, Fehsenfeld FC, Montzka SA. Hydrocarbon measurements in the southeastern United States: The rural oxidants in the southern environment (ROSE) program 1990. J Geophys Res-Atmos. 1995;100:25945–25963. [Google Scholar]
- Goldstein A, Galbally I. Known and Unexplored Organic Constituents in the Earth’s Atmosphere. Environ Sci Technol. 2007;41:1514–1521. doi: 10.1021/es072476p. [DOI] [PubMed] [Google Scholar]
- Gölz C, Senzig J, Platt U. NO3-initiated oxidation of biogenic hydrocarbons. Chemosphere. 2001;3:339–352. doi: 10.1016/S1465-9972(01)00015-0. [DOI] [Google Scholar]
- Gómez-González Y, Surratt JD, Cuyckens F, Szmigielski R, Vermeylen R, Jaoui M, Lewandowski M, Offenberg JH, Kleindienst TE, Edney EO, Blockhuys F, Van Alsenoy C, Maenhaut W, Claeys M. Characterization of organosulfates from the photooxidation of isoprene and unsaturated fatty acids in ambient aerosol using liquid chromatography/(−) electrospray ionization mass spectrometry. J Mass Spectrom. 2008;43:371–382. doi: 10.1002/jms.1329. [DOI] [PubMed] [Google Scholar]
- Griffin RJ, Cocker DR, III, Flagan RC, Seinfeld JH. Organic aerosol formation from the oxidation of biogenic hydrocarbons. J Geophys Res. 1999;104:3555–3567. [Google Scholar]
- Grifiths PT, Badger CL, Cox RA, Folkers M, Henk HH, Mentel TF. Reactive Uptake of N2O5 by Aerosols Containing Dicarboxylic Acids Effect of Particle Phase, Composition, and Nitrate Content. J Phys Chem. 2009;113:5082–5090. doi: 10.1021/jp8096814. [DOI] [PubMed] [Google Scholar]
- Gross S, Bertram AK. Reactive Uptake of NO3, N2O5, NO2, HNO3, and O3 on Three Types of Polycyclic Aromatic Hydrocarbon Surfaces. J Phys Chem A. 2008;112:3104–3113. doi: 10.1021/jp7107544. [DOI] [PubMed] [Google Scholar]
- Gross S, Bertram AK. Products and kinetics of the reaction of an alkane monolayer and a terminal alkene monolayer with NO3 radicals. J Geophys Res. 2009;114:D02307. doi: 10.1029/2008JD010987. [DOI] [Google Scholar]
- Gross S, Iannone RQ, Xiao S, Bertram AK. Reactive uptake studies of NO3 and N2O5 on alkenoic acid, alkanoate and polyalcohol substrates to probe nighttime aerosol chemistry. Phys Chem Chem Phys. 2009;11:7792–7803. doi: 10.1039/b904741g. [DOI] [PubMed] [Google Scholar]
- Grossenbacher JW, Couch T, Shepson PB, Thornberry T, Witmer-Rich M, Carroll MA, Faloona I, Tan D, Brune W, Ostling K, Bertman S. Measurements of isoprene nitrates above a forest canopy. J Geophys Res-Atmos. 2001;106:24429–24438. [Google Scholar]
- Gruijthuijsen YK, Grieshuber I, Stöcklinger A, Tischler U, Fehrenbach T, Weller MG, Vogel L, Vieths S, Pöschl U, Duschl A. Nitration Enhances the Allergenic Potential of Proteins. Int Arch Allerg Imm. 2006;141:265–275. doi: 10.1159/000095296. [DOI] [PubMed] [Google Scholar]
- Guenther A, Hewitt CN, Erickson D, Fall R, Geron C, Graedel TE, Harley P, Klinger L, Lerdau M, McKay WA, Pierce TE, Scholes B, Steinbrecher R, Tallamraju R, Taylor J, Zimmerman P. A global model of natural volatile organic compound emissions. J Geophys Res. 1995;100:8873–8892. [Google Scholar]
- Häkkinen SAK, Äijälä M, Lehtipalo K, Junninen H, Backman J, Virkkula A, Nieminen T, Vestenius M, Hakola H, Ehn M, Worsnop DR, Kulmala M, Petäjä T, Riipinen I. Long-term volatility measurements of submicron atmospheric aerosol in Hyytiälä, Finland. Atmos Chem Phys. 2012;12:10771–10786. doi: 10.5194/acp-12-10771-2012. [DOI] [Google Scholar]
- Hallquist M, Langer S, Ljungstrom E, Wangberg I. Rates of reaction between the nitrate radical and some unsaturated alcohols. Int J Chem Kinet. 1996;28:467–474. [Google Scholar]
- Hallquist M, Wangberg I, Ljungstrom E. Atmospheric Fate of Carbonyl Oxidation Products Originating from -Pinene and 3-Carene: Determination of Rate of Reaction with OH and NO3 Radicals, UV Absorption Cross Sections, and Vapor Pressures. Environ Sci Technol. 1997a;31:3166–3172. [Google Scholar]
- Hallquist M, Wängberg I, Ljungström E. Atmospheric Fate of Carbonyl Oxidation Products Originating from alpha-Pinene and delta3-Carene: Determination of Rate of Reaction with OH and NO3 Radicals, UV Absorption Cross Sections, and Vapor Pressures. Environ Sci Technol. 1997b;31:3166–3172. doi: 10.1021/es970151a. [DOI] [Google Scholar]
- Hallquist M, Wängberg I, Ljungstrom E, Barnes I, Becker KH. Aerosol and Product Yields from NO3 Radical-Initiated Oxidation of Selected Monoterpenes. Environ Sci Technol. 1999;33:553–559. [Google Scholar]
- Hallquist M, Wenger JC, Baltensperger U, Rudich Y, Simpson D, Claeys M, Dommen J, Donahue NM, George C, Goldstein AH, Hamilton JF, Herrmann H, Hoffmann T, Iinuma Y, Jang M, Jenkin ME, Jimenez JL, Kiendler-Scharr A, Maenhaut W, McFiggans G, Mentel TF, Monod A, Prévot ASH, Seinfeld JH, Surratt JD, Szmigielski R, Wildt J. The formation, properties and impact of secondary organic aerosol: current and emerging issues. Atmos Chem Phys. 2009;9:5155–5236. doi: 10.5194/acp-9-5155-2009. [DOI] [Google Scholar]
- Hao C, Shepson PB, Drummond JW, Muthuramu K. Gas chromatographic detector for selective and sensitive detection of atmospheric organic nitrates. Anal Chem. 1994;66:3737–3743. [Google Scholar]
- Hao LQ, Kortelainen A, Romakkaniemi S, Portin H, Jaatinen A, Leskinen A, Komppula M, Miettinen P, Sueper D, Pajunoja A, Smith JN, Lehtinen KEJ, Worsnop DR, Laaksonen A, Virtanen A. Atmospheric submicron aerosol composition and particulate organic nitrate formation in a boreal forestland–urban mixed region. Atmos Chem Phys. 2014;14:13483–13495. doi: 10.5194/acp-14-13483-2014. [DOI] [Google Scholar]
- Harrison MA, Barra S, Borghesi D, Vione D, Arsene C, Olariu RI. Nitrated phenols in the atmosphere: a review. Atmos Environ. 2005;39:231–248. [Google Scholar]
- Heintz F, Platt U, Flentje J, Dubois R. Long-term observation of nitrate radicals at the Tor Station, Kap Arkona (Rügen) J Geophys Res. 1996;101:22891–22910. [Google Scholar]
- Herrmann H. Kinetics of aqueous phase reactions relevant for atmospheric chemistry. Chem Rev. 2003;103:4691–4716. doi: 10.1021/cr020658q. [DOI] [PubMed] [Google Scholar]
- Herrmann H, Zellner R. Reactions of NO3-Radicals in Aqueous Solution. In: Alfassi ZB, editor. N-Centered Radicals. Wiley; Chichester: 1998. pp. 291–343. [Google Scholar]
- Herrmann H, Tilgner A, Barzaghi P, Majdik Z, Gligorovski S, Poulain L, Monod A. Towards a more detailed description of tropospheric aqueous phase organic chemistry: CAPRAM 3.0. Atmos Environ. 2005;39:4351–4363. [Google Scholar]
- Herrmann H, Hoffmann D, Schaefer T, Brauer P, Tilgner A. Tropospheric Aqueous-Phase Free-Radical Chemistry: Radical Sources, Spectra, Reaction Kinetics and Prediction Tools. Chem Phys Chem. 2010;11:3796–3822. doi: 10.1002/cphc.201000533. [DOI] [PubMed] [Google Scholar]
- Herrmann H, Schaefer T, Tilgner A, Styler SA, Weller C, Teich M, Otto T. Tropospheric Aqueous-Phase Chemistry: Kinetics, Mechanisms, and Its Coupling to a Changing Gas Phase. Chem Rev. 2015;115:4259–4334. doi: 10.1021/cr500447k. [DOI] [PubMed] [Google Scholar]
- Hidy GM, Blanchard CL, Baumann K, Edgerton E, Tanenbaum S, Shaw S, Knipping E, Tombach I, Jansen J, Walters J. Chemical climatology of the southeastern United States, 1999–2013. Atmos Chem Phys. 2014;14:11893–11914. doi: 10.5194/acp-14-11893-2014. [DOI] [Google Scholar]
- Hildebrandt Ruiz LH, Yarwood G. Prepared for the Texas AQRP (Project 12-012) University of Texas at Austin, and ENVIRON International Corporation; Novato, CA: 2013. Interactions between organic aerosol and NOy: Influence on oxidant production. [Google Scholar]
- Hodzic A, Jimenez JL, Prévôt ASH, Szidat S, Fast JD, Madronich S. Can 3-D models explain the observed fractions of fossil and non-fossil carbon in and near Mexico City? Atmos Chem Phys. 2010;10:10997–11016. doi: 10.5194/acp-10-10997-2010. [DOI] [Google Scholar]
- Hoffmann D, Weigert B, Barzaghi P, Herrmann H. Reactivity of poly-alcohols towards OH, NO3 and in aqueous solution. Phys Chem Chem Phys. 2009;11:9351–9363. doi: 10.1039/b908459b. [DOI] [PubMed] [Google Scholar]
- Hoffmann T, Odum JR, Bowman F, Collins D, Klockow D, Flagan RC, Seinfeld JH. Formation of organic aerosols from the oxidation of biogenic hydrocarbons. J Atmos Chem. 1997;26:189–222. [Google Scholar]
- Horowitz LW, Fiore AM, Milly GP, Cohen RC, Perrin A, Wooldridge PJ, Hess PG, Emmons LK, Lamarque JF. Observational constraints on the chemistry of isoprene nitrates over the eastern United States. J Geophys Res. 2007;112:D12S08. doi: 10.1029/2006JD007747. [DOI] [Google Scholar]
- Hou H, Wang B. A Systematic Computational Study on the Reactions of HO2 with RO2: The HO2 + CH3O2(CD3O2) and HO2 + CH2FO2 Reactions. J Phys Chem A. 2005;109:451–460. doi: 10.1021/jp046329e. [DOI] [PubMed] [Google Scholar]
- Hou H, Deng L, Li J, Wang B. A Systematic Computational Study of the Reactions of HO2 with RO2: The HO2 + CH2ClO2, CHCl2O2, and CCl3O2 Reactions. J Phys Chem A. 2005;109:9299–9309. doi: 10.1021/jp052718c. [DOI] [PubMed] [Google Scholar]
- Hoyle CR, Berntsen T, Myhre G, Isaksen ISA. Secondary organic aerosol in the global aerosol – chemical transport model Oslo CTM2. Atmos Chem Phys. 2007;7:5675–5694. doi: 10.5194/acp-7-5675-2007. [DOI] [Google Scholar]
- Hoyle CR, Boy M, Donahue NM, Fry JL, Glasius M, Guenther A, Hallar AG, Huff Hartz K, Petters MD, Petaja T, Rosenoern T, Sullivan AP. A review of the anthropogenic influence on biogenic secondary organic aerosol. Atmos Chem Phys. 2011;11:321–343. doi: 10.5194/acpd-10-19515-2010. [DOI] [Google Scholar]
- Hu KS, Darer AI, Elrod MJ. Thermodynamics and kinetics of the hydrolysis of atmospherically relevant organonitrates and organosulfates. Atmos Chem Phys. 2011;11:8307–8320. doi: 10.5194/acp-11-8307-2011. [DOI] [Google Scholar]
- Hu X-M, Nielsen-Gammon JW, Zhang F. Evaluation of Three Planetary Boundary Layer Schemes in the WRF Model. J Appl Meteorol Climatol. 2010;49:1831–1844. doi: 10.1175/2010JAMC2432.1. [DOI] [Google Scholar]
- Huey LG. Measurement of trace atmospheric species by chemical ionization mass spectrometry: Speciation of reactive nitrogen and future directions. Mass Spectrom Rev. 2007;26:166–184. doi: 10.1002/mas.20118. [DOI] [PubMed] [Google Scholar]
- Huie RE. Laboratory Studies of Atmospheric Heterogeneous Chemistry; Current Problems in Atmospheric Chemistry. In: Barker JR, editor. Advances in Physical Chemistry Series. World Scientific; Singapore: 1994. pp. 374–419. [Google Scholar]
- Hung H-M, Katrib Y, Martin ST. Products and Mechanisms of the Reaction of Oleic Acid with Ozone and Nitrate Radical. J Phys Chem A. 2005;109:4517–4530. doi: 10.1021/jp0500900. [DOI] [PubMed] [Google Scholar]
- Hurst JM, Barket DJ, Herrera-Gomez O, Couch TL, Shepson PB, Faloona I, Tan D, Brune W, Westberg H, Lamb B. Investigation of the nighttime decay of isoprene. J Geophys Res-Atmos. 2001;106:24335–24346. [Google Scholar]
- Hutzell WT, Luecken DJ, Appel KW, Carter WPL. Interpreting predictions from the SAPRC07 mechanism based on regional and continental simulations. Atmos Environ. 2012;46:417–429. doi: 10.1016/j.atmosenv.2011.09.030. [DOI] [Google Scholar]
- Iannone R, Xiao S, Bertram AK. Potentially important nighttime heterogeneous chemistry: NO3 with aldehydes and N2O5 with alcohols. Phys Chem Chem Phys. 2011;13:10214–10223. doi: 10.1039/C1CP20294D. [DOI] [PubMed] [Google Scholar]
- Iinuma Y, Müller C, Berndt T, Böge O, Claeys M, Herrmann H. Evidence for the Existence of Organosulfates from b-Pinene Ozonolysis in Ambient Secondary Organic Aerosol. Environ Sci Technol. 2007;41:6678–6683. doi: 10.1021/es070938t. [DOI] [PubMed] [Google Scholar]
- Iyer S, Lopez-Hilfiker F, Lee BH, Thornton JA, Kurten T. Modeling the Detection of Organic and Inorganic Compounds Using Iodide-Based Chemical Ionization. J Phys Chem A. 2016;120:576–587. doi: 10.1021/acs.jpca.5b09837. [DOI] [PubMed] [Google Scholar]
- Jacobs MI, Burke WJ, Elrod MJ. Kinetics of the reactions of isoprene-derived hydroxynitrates: gas phase epoxide formation and solution phase hydrolysis. Atmos Chem Phys. 2014;14:8933–8946. doi: 10.5194/acp-14-8933-2014. [DOI] [Google Scholar]
- Jacobson MZ. Isolating nitrated and aromatic aerosols and nitrated aromatic gases as sources of ultraviolet light absorption. J Geophys Res-Atmos. 1999;104:3527–3542. doi: 10.1029/1998jd100054. [DOI] [Google Scholar]
- Jang J-CC, Jeffries HE, Tonnesen S. Sensitivity of ozone to model grid resolution – II. Detailed process analysis for ozone chemistry. Atmos Environ. 1995;29:3101–3114. doi: 10.1016/1352-2310(95)00119-J. [DOI] [Google Scholar]
- Jaoui M, Kleindienst TE, Docherty KS, Lewandowski M, Offenberg JH. Secondary organic aerosol formation from the oxidation of a series of sesquiterpenes: α-cedrene, β-caryophyllene, α-humulene and α-farnesene with O3, OH and NO3 radicals. Environ Chem. 2013;10:178–193. [Google Scholar]
- Jayne JT, Leard DC, Zhang XF, Davidovits P, Smith KA, Kolb CE, Worsnop DR. Development of an aerosol mass spectrometer for size and composition analysis of submicron particles. Aerosol Sci Tech. 2000;33:49–70. [Google Scholar]
- Jenkin ME, Saunders SM, Pilling MJ. The tropospheric degradation of volatile organic compounds: a protocol for mechanism development. Atmos Environ. 1997;31:81–104. doi: 10.1016/S1352-2310(96)00105-7. [DOI] [Google Scholar]
- Jenkin ME, Young JC, Rickard AR. The MCM v3.3.1 degradation scheme for isoprene. Atmos Chem Phys. 2015;15:11433–11459. doi: 10.5194/acp-15-11433-2015. [DOI] [Google Scholar]
- Jimenez JL, Canagaratna MR, Donahue NM, Prevot ASH, Zhang Q, Kroll JH, DeCarlo PF, Allan JD, Coe H, Ng NL, Aiken AC, Docherty KS, Ulbrich IM, Grieshop AP, Robinson AL, Duplissy J, Smith JD, Wilson KR, Lanz VA, Hueglin C, Sun YL, Tian J, Laaksonen A, Raatikainen T, Rautiainen J, Vaattovaara P, Ehn M, Kulmala M, Tomlinson JM, Collins DR, Cubison MJ, Dunlea EJ, Huffman JA, Onasch TB, Alfarra MR, Williams PI, Bower K, Kondo Y, Schneider J, Drewnick F, Borrmann S, Weimer S, Demerjian K, Salcedo D, Cottrell L, Griffin R, Takami A, Miyoshi T, Hatakeyama S, Shimono A, Sun JY, Zhang YM, Dzepina K, Kimmel JR, Sueper D, Jayne JT, Herndon SC, Trimborn AM, Williams LR, Wood EC, Middlebrook AM, Kolb CE, Baltensperger U, Worsnop DR. Evolution of Organic Aerosols in the Atmosphere. Science. 2009;326:1525–1529. doi: 10.1126/science.1180353. [DOI] [PubMed] [Google Scholar]
- Johnston HS, Davis HF, Lee YT. NO3 Photolysis Product Channels: Quantum Yields from Observed Energy Thresholds. J Phys Chem. 1996;100:4713–4723. [Google Scholar]
- Jones BT, Ham JE. alpha-Terpineol reactions with the nitrate radical: Rate constant and gas-phase products. Atmos Environ. 2008;42:6689–6698. doi: 10.1016/j.atmosenv.2008.04.017. [DOI] [Google Scholar]
- Kaduwela A, Luecken D, Carter W, Derwent R. New directions: Atmospheric chemical mechanisms for the future. Atmos Environ. 2015;122:609–610. doi: 10.1016/j.atmosenv.2015.10.031. [DOI] [Google Scholar]
- Kaiser JC, Riemer N, Knopf DA. Detailed heterogeneous oxidation of soot surfaces in a particle-resolved aerosol model. Atmos Chem Phys. 2011;11:4505–4520. doi: 10.5194/acp-11-4505-2011. [DOI] [Google Scholar]
- Kames J, Schurath U, Flocke F, Volzthomas A. Preparation of Organic Nitrates from Alcohols and N2O5 for Species Identification in Atmospheric Samples. J Atmos Chem. 1993;16:349–359. [Google Scholar]
- Karamchandani P, Seigneur C, Vijayaraghavan K, Wu S-Y. Development and application of a state-of-the-science plume-in-grid model. J Geophys Res-Atmos. 2002;107:11–13. doi: 10.1029/2002JD002123. [DOI] [Google Scholar]
- Kastler J, Ballschmiter K. Bifunctional alkyl nitrates – trace constituents of the atmosphere. Fresen J Anal Chem. 1998;360:812–816. [Google Scholar]
- Kastler J, Jarman W, Ballschmiter K. Multifunctional organic nitrates as constituents in European and US urban photosmog. Fresen J Anal Chem. 2000;368:244–249. doi: 10.1007/s002160000550. [DOI] [PubMed] [Google Scholar]
- Kennedy O, Ouyang B, Langridge J, Daniels M, Bauguitte S, Freshwater R, McLeod M, Ironmonger C, Sendall J, Norris O. An aircraft based three channel broadband cavity enhanced absorption spectrometer for simultaneous measurements of NO3, N2O5 and NO2. Atmos Meas Tech. 2011;4:1759–1776. doi: 10.5194/amtd-4-3499-2011. [DOI] [Google Scholar]
- Kercher JP, Riedel TP, Thornton JA. Chlorine activation by N2O5: simultaneous, in situ detection of ClNO2 and N2O5 by chemical ionization mass spectrometry. Atmos Meas Tech. 2009;2:193–204. doi: 10.5194/amt-2-193-2009. [DOI] [Google Scholar]
- Kerdouci J, Picquet-Varrault B, Doussin JF. Prediction of Rate Constants for Gas-Phase Reactions of Nitrate Radical with Organic Compounds: A New Structure–Activity Relationship. Chem Phys Chem. 2010;11:3909–3920. doi: 10.1002/cphc.201000673. [DOI] [PubMed] [Google Scholar]
- Kerdouci J, Picquet-Varrault B, Durand-Jolibois R, Gaimoz C, Doussin J-F. An Experimental Study of the Gas-Phase Reactions of NO3 Radicals with a Series of Unsaturated Aldehydes: trans-2-Hexenal, trans-2-Heptenal, and trans-2-Octenal. J Phys Chem A. 2012;116:10135–10142. doi: 10.1021/jp3071234. [DOI] [PubMed] [Google Scholar]
- Kerdouci J, Picquet-Varrault B, Doussin J-F. Structure–activity relationship for the gas-phase reactions of NO3 radical with organic compounds: Update and extension to aldehydes. Atmos Environ. 2014;84:363–372. doi: 10.1016/j.atmosenv.2013.11.024. [DOI] [Google Scholar]
- Kern C, Trick S, Rippel B, Platt U. Applicability of light-emitting diodes as light sources for active differential optical absorption spectroscopy measurements. Appl Opt. 2006;45:2077–2088. doi: 10.1364/ao.45.002077. [DOI] [PubMed] [Google Scholar]
- Kesselmeier J, Staudt M. Biogenic Volatile Organic Compounds (VOC): An Overview on Emission, Physiology and Ecology. J Atmos Chem. 1999;33:23–88. doi: 10.1023/A:1006127516791. [DOI] [Google Scholar]
- Khan MAH, Cooke MC, Utembe SR, Archibald AT, Derwent RG, Xiao P, Percival CJ, Jenkin ME, Morris WC, Shallcross DE. Global modeling of the nitrate radical (NO3) for present and pre-industrial scenarios. Atmos Res. 2015;164/165:347–357. doi: 10.1016/j.atmosres.2015.06.006. [DOI] [Google Scholar]
- Kiendler-Scharr A, Mensah AA, Friese E, Topping D, Nemitz E, Prevot ASH, Äijälä M, Allan J, Canonaco F, Canagaratna M, Carbone S, Crippa M, Dall Osto M, Day DA, De Carlo P, Di Marco CF, Elbern H, Eriksson A, Freney E, Hao L, Herrmann H, Hildebrandt L, Hillamo R, Jimenez JL, Laaksonen A, McFiggans G, Mohr C, O’Dowd C, Otjes R, Ovadnevaite J, Pandis SN, Poulain L, Schlag P, Sellegri K, Swietlicki E, Tiitta P, Vermeulen A, Wahner A, Worsnop D, Wu HC. Organic nitrates from night-time chemistry are ubiquitous in the European submicron aerosol. Geophys Res Lett. 2016;43:7735–7744. doi: 10.1002/2016GL069239. [DOI] [Google Scholar]
- Kim PS, Jacob DJ, Fisher JA, Travis K, Yu K, Zhu L, Yantosca RM, Sulprizio MP, Jimenez JL, Campuzano-Jost P, Froyd KD, Liao J, Hair JW, Fenn MA, Butler CF, Wagner NL, Gordon TD, Welti A, Wennberg PO, Crounse JD, StClair JM, Teng AP, Millet DB, Schwarz JP, Markovic MZ, Perring AE. Sources, seasonality, and trends of southeast US aerosol: an integrated analysis of surface, aircraft, and satellite observations with the GEOS-Chem chemical transport model. Atmos Chem Phys. 2015;15:10411–10433. doi: 10.5194/acp-15-10411-2015. [DOI] [Google Scholar]
- Kim S-W, Heckel A, McKeen SA, Frost GJ, Hsie EY, Trainer MK, Richter A, Burrows JP, Peckham SE, Grell GA. Satellite-observed US power plant NOx emission reductions and their impact on air quality. Geophys Res Lett. 2006;33:L22812. doi: 10.1029/2006GL027749. [DOI] [Google Scholar]
- Kind I, Berndt T, Böge O. Gas-phase rate constants for the reaction of NO3 radicals with a series of cyclic alkenes, 2-ethyl-1-butene and 2,3-dimethyl-1,3-butadiene. Chem Phys Lett. 1998;288:111–118. [Google Scholar]
- King MD, Dick EM, Simpson WR. A new method for the atmospheric detection of the nitrate radical (NO3) Atmos Environ. 2000;34:685–688. doi: 10.1016/S1352-2310(99)00418-5. [DOI] [Google Scholar]
- Kleist E, Mentel TF, Andres S, Bohne A, Folkers A, Kiendler-Scharr A, Rudich Y, Springer M, Tillmann R, Wildt J. Irreversible impacts of heat on the emissions of monoterpenes, sesquiterpenes, phenolic BVOC and green leaf volatiles from several tree species. Biogeosciences. 2012;9:5111–5123. doi: 10.5194/bg-9-5111-2012. [DOI] [Google Scholar]
- Knopf DA, Mak J, Gross S, Bertram AK. Does atmospheric processing of saturated hydrocarbon surfaces by NO3 lead to volatilization. Geophys Res Lett. 2006;33:L17816. doi: 10.1029/2006GL026884. [DOI] [Google Scholar]
- Knopf DA, Forrester SM, Slade JH. Heterogeneous oxidation kinetics of organic biomass burning aerosol surrogates by O3, NO2, N2O5, and NO3. Phys Chem Chem Phys. 2011;13:21050–21062. doi: 10.1039/c1cp22478f. [DOI] [PubMed] [Google Scholar]
- Knote C, Hodzic A, Jimenez JL, Volkamer R, Orlando JJ, Baidar S, Brioude J, Fast J, Gentner DR, Goldstein AH, Hayes PL, Knighton WB, Oetjen H, Setyan A, Stark H, Thalman R, Tyndall G, Washenfelder R, Waxman E, Zhang Q. Simulation of semi-explicit mechanisms of SOA formation from glyoxal in aerosol in a 3-D model. Atmos Chem Phys. 2014;14:6213–6239. doi: 10.5194/acp-14-6213-2014. [DOI] [Google Scholar]
- Knote C, Tuccella P, Curci G, Emmons L, Orlando JJ, Madronich S, Baró R, Jiménez-Guerrero P, Luecken D, Hogrefe C, Forkel R, Werhahn J, Hirtl M, Pérez JL, San José R, Giordano L, Brunner D, Yahya K, Zhang Y. Influence of the choice of gas-phase mechanism on predictions of key gaseous pollutants during the AQMEII phase-2 intercomparison. Atmos Environ. 2015;115:553–568. doi: 10.1016/j.atmosenv.2014.11.066. [DOI] [Google Scholar]
- Koenig JQ, Covert DS, Pierson WE. Effects of Inhalation of Acidic Compounds on Pulmonary Function in Allergic Adolescent Subjects. Environ Health Persp. 1989;79:173–178. doi: 10.1289/ehp.8979173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo B, Knipping E, Yarwood G. 1.5-Dimensional volatility basis set approach for modeling organic aerosol in CAMx and CMAQ. Atmos Environ. 2014;95:158–164. doi: 10.1016/j.atmosenv.2014.06.031. [DOI] [Google Scholar]
- Koop T, Bookhold J, Shiraiwa M, Pöschl U. Glass transition and phase state of organic compounds: dependency on molecular properties and implications for secondary organic aerosols in the atmosphere. Phys Chem Chem Phys. 2011;13:19238–19255. doi: 10.1039/C1CP22617G. [DOI] [PubMed] [Google Scholar]
- Kotzias D, Hjorth JL, Skov H. A chemical mechanism for dry deposition – The role of biogenic hydrocarbon (terpene) emissions in the dry deposition of O3, SO2 and NOx in forest areas. Toxicol Environ Chem. 1989;20:95–99. doi: 10.1080/02772248909357364. [DOI] [Google Scholar]
- Krechmer JE, Pagonis D, Ziemann PJ, Jimenez JL. Quantification of Gas-Wall Partitioning in Teflon Environmental Chambers Using Rapid Bursts of Low-Volatility Oxidized Species Generated in Situ. Environ Sci Technol. 2016;50:5757–5765. doi: 10.1021/acs.est.6b00606. [DOI] [PubMed] [Google Scholar]
- Krotkov NA, McLinden CA, Li C, Lamsal LN, Celarier EA, Marchenko SV, Swartz WH, Bucsela EJ, Joiner J, Duncan BN, Boersma KF, Veefkind JP, Levelt PF, Fioletov VE, Dickerson RR, He H, Lu Z, Streets DG. Aura OMI observations of regional SO2 and NO2 pollution changes from 2005 to 2015. Atmos Chem Phys. 16:4605–4629. doi: 10.5194/acp-16-4605-2016. [DOI] [Google Scholar]
- Kulmala M, Asmi A, Lappalainen HK, Baltensperger U, Brenguier J-L, Facchini MC, Hansson H-C, Hov Ø, O’Dowd CD, Pöschl U, Wiedensohler A, Boers R, Boucher O, de Leeuw G, Denier van der Gon HAC, Feichter J, Krejci R, Laj P, Lihavainen H, Lohmann U, Mc-Figgans G, Mentel T, Pilinis C, Riipinen I, Schulz M, Stohl A, Swietlicki E, Vignati E, Alves C, Amann M, Ammann M, Arabas S, Artaxo P, Baars H, Beddows DCS, Bergström R, Beukes JP, Bilde M, Burkhart JF, Canonaco F, Clegg SL, Coe H, Crumeyrolle S, D’Anna B, Decesari S, Gilardoni S, Fischer M, Fjaeraa AM, Fountoukis C, George C, Gomes L, Halloran P, Hamburger T, Harrison RM, Herrmann H, Hoffmann T, Hoose C, Hu M, Hyvärinen A, Hõrrak U, Iinuma Y, Iversen T, Josipovic M, Kanakidou M, Kiendler-Scharr A, Kirkevåg A, Kiss G, Klimont Z, Kolmonen P, Komppula M, Kristjánsson J-E, Laakso L, Laaksonen A, Labonnote L, Lanz VA, Lehtinen KEJ, Rizzo LV, Makkonen R, Manninen HE, McMeeking G, Merikanto J, Minikin A, Mirme S, Morgan WT, Nemitz E, O’Donnell D, Panwar TS, Pawlowska H, Petzold A, Pienaar JJ, Pio C, Plass-Duelmer C, Prévôt ASH, Pryor S, Reddington CL, Roberts G, Rosenfeld D, Schwarz J, Seland Ø, Sellegri K, Shen XJ, Shiraiwa M, Siebert H, Sierau B, Simpson D, Sun JY, Topping D, Tunved P, Vaattovaara P, Vakkari V, Veefkind JP, Visschedijk A, Vuollekoski H, Vuolo R, Wehner B, Wildt J, Woodward S, Worsnop DR, van Zadelhoff G-J, Zardini AA, Zhang K, van Zyl PG, Kerminen V-M, Carslaw SK, Pandis SN. General overview: European Integrated project on Aerosol Cloud Climate and Air Quality interactions (EUCAARI) – integrating aerosol research from nano to global scales. Atmos Chem Phys. 2011;11:13061–13143. doi: 10.5194/acp-11-13061-2011. [DOI] [Google Scholar]
- Kwan AJ, Chan AWH, Ng NL, Kjaergaard HG, Seinfeld JH, Wennberg PO. Peroxy radical chemistry and OH radical production during the NO3-initiated oxidation of isoprene. Atmos Chem Phys. 2012;12:7499–7515. doi: 10.5194/acp-12-7499-2012. [DOI] [Google Scholar]
- Kwok ESC, Aschmann SM, Arey J, Atkinson R. Product formation from the reaction of the NO3 radical with isoprene and rate constants for the reactions of methacrolein and methyl vinyl ketone with the NO3 radical. Int J Chem Kinet. 1996;28:925–934. [Google Scholar]
- La YS, Camredon M, Ziemann PJ, Valorso R, Matsunaga A, Lannuque V, Lee-Taylor J, Hodzic A, Madronich S, Aumont B. Impact of chamber wall loss of gaseous organic compounds on secondary organic aerosol formation: explicit modeling of SOA formation from alkane and alkene oxidation. Atmos Chem Phys. 2016;16:1417–1431. doi: 10.5194/acp-16-1417-2016. [DOI] [Google Scholar]
- Lack D, Langridg NJM, Bahreni R, Cappa CD, Middlebrook AM, Schwarz JP. Brown carbon and internal mixing in biomass burning particles. P Natl Acad Sci USA. 2012;109:14802–14807. doi: 10.1073/pnas.1206575109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamarque J-F, Kyle GP, Meinshausen M, Riahi K, Smith SJ, Vuuren DP, Conley AJ, Vitt F. Global and regional evolution of short-lived radiatively-active gases and aerosols in the Representative Concentration Pathways. Climatic Change. 2011;109:191–212. doi: 10.1007/s10584-011-0155-0. [DOI] [Google Scholar]
- Lane TE, Donahue NM, Pandis SN. Effect of NOx on Secondary Organic Aerosol Concentrations. Environ Sci Technol. 2008;42:6022–6027. doi: 10.1021/es703225a. [DOI] [PubMed] [Google Scholar]
- Langridge JM, Ball SM, Shillings AJL, Jones RL. A broadband absorption spectrometer using light emitting diodes for ultrasensitive, in situ trace gas detection. Rev Sci Instrum. 2008;79:123110. doi: 10.1063/1.3046282. [DOI] [PubMed] [Google Scholar]
- Laskin A, Laskin J, Nizkorodov SA. Chemistry of Atmospheric Brown Carbon. Chem Rev. 2015;115:4335–4382. doi: 10.1021/cr5006167. [DOI] [PubMed] [Google Scholar]
- Le Breton M, Bacak A, Muller JBA, Bannan TJ, Kennedy O, Ouyang B, Xiao P, Bauguitte SJB, Shallcross DE, Jones RL, Daniels MJS, Ball SM, Percival CJ. The first airborne comparison of N2O5 measurements over the UK using a CIMS and BBCEAS during the RONOCO campaign. Analtyical Methods. 2014;6:9731–9743. doi: 10.1039/C4AY02273D. [DOI] [Google Scholar]
- Lee BH, Lopez-Hilfiker FD, Mohr C, Kurten T, Worsnop DR, Thornton JA. An Iodide-Adduct High-Resolution Time-of-Flight Chemical-Ionization Mass Spectrometer: Application to Atmospheric Inorganic and Organic Compounds. Environ Sci Technol. 2014a;48:6309–6317. doi: 10.1021/es500362a. [DOI] [PubMed] [Google Scholar]
- Lee BH, Mohr C, Lopez-Hilfiker FD, Lutz A, Hallquist M, Lee L, Romer P, Cohen RC, Iyer S, Kurten T, Hu W, Day DA, Campuzano-Jost P, Jimenez JL, Xu L, Ng NL, Guo H, Weber RJ, Wild RJ, Brown SS, Koss A, de Gouw J, Olson K, Goldstein AH, Seco R, Kim S, McAvery K, Shepson PB, Baumann K, Edgerton E, Liu J, Shilling JE, Miller DO, Brune WH, D’Ambro EL, Thornton JA. Highly functionalized organic nitrates in the southeast United States: Contribution to secondary organic aerosol and reactive nitrogen budgets. P Natl Acad Sci. 2016;113:1516–1521. doi: 10.1073/pnas.1508108113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee L, Wooldridge P, Nah T, Wilson K, Cohen R. Observation of rates and products in the reaction of NO3 with submicron squalane and squalene aerosol. Phys Chem Chem Phys. 2013;15:882–892. doi: 10.1039/C2CP42500A. [DOI] [PubMed] [Google Scholar]
- Lee L, Teng AP, Wennberg PO, Crounse JD, Cohen RC. On Rates and Mechanisms of OH and O3 Reactions with Isoprene-Derived Hydroxy Nitrates. J Phys Chem A. 2014b;118:1622–1637. doi: 10.1021/jp4107603. [DOI] [PubMed] [Google Scholar]
- Lee L, Wooldridge PJ, de Gouw J, Brown SS, Bates TS, Quinn PK, Cohen RC. Particulate organic nitrates observed in an oil and natural gas production region during winter-time. Atmos Chem Phys. 2015;15:9313–9325. doi: 10.5194/acp-15-9313-2015. [DOI] [Google Scholar]
- Li YP, Elbern H, Lu KD, Friese E, Kiendler-Scharr A, Mentel TF, Wang XS, Wahner A, Zhang YH. Updated aerosol module and its application to simulate secondary organic aerosols during IMPACT campaign May 2008. Atmos Chem Phys. 2013;13:6289–6304. doi: 10.5194/acp-13-6289-2013. [DOI] [Google Scholar]
- Liggio J, Mclaren R. An optimized method for the determination of volatile and semi-volatile aldehydes and ketones in ambient particulate matter. Int J Environ An Ch. 2003;83:819–835. [Google Scholar]
- Lin P, Liu J, Shilling JE, Kathmann SM, Laskin J, Laskin A. Molecular characterization of brown carbon (BrC) chromophores in secondary organic aerosol generated from photo-oxidation of toluene. Phys Chem Chem Phys. 2015;17:23312–23325. doi: 10.1039/C5CP02563J. [DOI] [PubMed] [Google Scholar]
- Liu S, Ahlm L, Day DA, Russell LM, Zhao Y, Gentner DR, Weber RJ, Goldstein AH, Jaoui M, Offenberg JH, Kleindienst TE, Rubitschun C, Surratt JD, Sheesley RJ, Scheller S. Secondary organic aerosol formation from fossil fuel sources contribute majority of summertime organic mass at Bakersfield. J Geophys Res-Atmos. 2012a;117:D00V26. doi: 10.1029/2012jd018170. [DOI] [Google Scholar]
- Liu S, Shilling JE, Song C, Hiranuma N, Zaveri RA, Russell LM. Hydrolysis of Organonitrate Functional Groups in Aerosol Particles. Aerosol Sci Tech. 2012b;46:1359–1369. doi: 10.1080/02786826.2012.716175. [DOI] [Google Scholar]
- Ljungström E, Hallquist M. Nitrate radical formation rates in Scandinavia. Atmos Environ. 1996;30:2925–2932. [Google Scholar]
- Lockwood AL, Shepson PB, Fiddler MN, Alaghmand M. Isoprene nitrates: preparation, separation, identification, yields, and atmospheric chemistry. Atmos Chem Phys. 2010;10:6169–6178. doi: 10.5194/acp-10-6169-2010. [DOI] [Google Scholar]
- Lopez-Hilfiker FD, Mohr C, Ehn M, Rubach F, Kleist E, Wildt J, Mentel ThF, Lutz A, Hallquist M, Worsnop D, Thornton JA. A novel method for online analysis of gas and particle composition: description and evaluation of a Filter Inlet for Gases and AEROsols (FIGAERO) Atmos Meas Tech. 2014;7:983–1001. doi: 10.5194/amt-7-983-2014. [DOI] [Google Scholar]
- Lopez-Hilfiker FD, Iyer S, Mohr C, Lee BH, D’Ambro EL, Kurtén T, Thornton JA. Constraining the sensitivity of iodide adduct chemical ionization mass spectrometry to multifunctional organic molecules using the collision limit and thermodynamic stability of iodide ion adducts. Atmos Meas Tech. 2016;9:1505–1512. doi: 10.5194/amt-9-1505-2016. [DOI] [Google Scholar]
- Loza CL, Chan AWH, Galloway MM, Keutsch FN, Flagan RC, Seinfeld JH. Characterization of Vapor Wall Loss in Laboratory Chambers. Environ Sci Technol. 2010;44:5074–5078. doi: 10.1021/es100727v. [DOI] [PubMed] [Google Scholar]
- Lu JW, Flores JM, Lavi A, Abo-Riziq A, Rudich Y. Changes in the optical properties of benzo[a]pyrene-coated aerosols upon heterogeneous reactions with NO2 and NO3. Phys Chem Chem Phys. 2011;13:6484–6492. doi: 10.1039/c0cp02114h. [DOI] [PubMed] [Google Scholar]
- Lu Z, Streets DG, de Foy B, Lamsal LN, Duncan BN, Xing J. Emissions of nitrogen oxides from US urban areas: estimation from Ozone Monitoring Instrument retrievals for 2005–2014. Atmos Chem Phys. 2015;15:10367–10383. doi: 10.5194/acp-15-10367-2015. [DOI] [Google Scholar]
- Luecken DJ, Phillips S, Sarwar G, Jang C. Effects of using the CB05 vsSAPRC99 vs CB4 chemical mechanism on model predictions: Ozone and gas-phase photochemical precursor concentrations. Atmos Environ. 2008;42:5805–5820. doi: 10.1016/j.atmosenv.2007.08.056. [DOI] [Google Scholar]
- Luecken DJ, Hutzell WT, Strum ML, Pouliot GA. Regional sources of atmospheric formaldehyde and acetaldehyde, and implications for atmospheric modeling. Atmos Environ. 2012;47:477–490. doi: 10.1016/j.atmosenv.2011.10.005. [DOI] [Google Scholar]
- Luxenhofer O, Schneider M, Dambach M, Ballschmiter K. Semivolatile long chain C6–C17 alkyl nitrates as trace compounds in air. Chemosphere. 1996;33:393–404. doi: 10.1016/0045-6535(96)00205-6. [DOI] [Google Scholar]
- Ma X, Yu F, Luo G. Aerosol direct radiative forcing based on GEOS-Chem-APM and uncertainties. Atmos Chem Phys. 2012;12:5563–5581. doi: 10.5194/acp-12-5563-2012. [DOI] [Google Scholar]
- Mao J, Paulot F, Jacob DJ, Cohen RC, Crounse JD, Wennberg PO, Keller CA, Hudman RC, Barkley MP, Horowitz LW. Ozone and organic nitrates over the eastern United States: Sensitivity to isoprene chemistry. J Geophys Res-Atmos. 2013;118:JD020231. doi: 10.1002/jgrd.50817. [DOI] [Google Scholar]
- Martinez E, Cabanas B, Aranda A, Martin P. Kinetics of the Reactions of NO3 Radical with Selected Monoterpenes: A Temperature Dependence Study. Environ Sci Technol. 1998;32:3730–3734. [Google Scholar]
- Martínez E, Cabañas B, Aranda A, Martín P, Salgado S. Absolute Rate Coefficients for the Gas-Phase Reactions of NO3 Radical with a Series of Monoterpenes at T = 298 to 433 K. J Atmos Chem. 1999;33:265–282. [Google Scholar]
- Martinez M, Perner D, Hackenthal EM, Külzer S, Schültz L. NO3 at Helgoland during the NORDEX campaign in October 1996. J Geophys Res. 2000;105:22685–22695. [Google Scholar]
- Matsumoto J, Imai H, Kosugi N, Kajii Y. In situ measurement of N2O5 in the urban atmosphere by thermal decomposition/laser-induced fluorescence technique. Atmos Environ. 2005a;39:6802–6811. doi: 10.1016/j.atmosenv.2005.07.055. [DOI] [Google Scholar]
- Matsumoto J, Kosugi N, Imai H, Kajii Y. Development of a measurement system for nitrate radical and dinitrogen pentoxide using a thermal conversion/laser-induced fluorescence technique. Rev Sci Instrum. 2005b;76:064101. doi: 10.1063/1.1927098. [DOI] [Google Scholar]
- Matsunaga A, Ziemann PJ. Gas-Wall Partitioning of Organic Compounds in a Teflon Film Chamber and Potential Effects on Reaction Product and Aerosol Yield Measurements. Aerosol Sci Technol. 2010;44:881–892. doi: 10.1080/02786826.2010.501044. [DOI] [Google Scholar]
- McDonald BC, Dallmann TR, Martin EW, Harley RA. Long-term trends in nitrogen oxide emissions from motor vehicles at national, state, and air basin scales. J Geophys Res. 2012;117:D00V18. doi: 10.1029/2012jd018304. [DOI] [Google Scholar]
- McLaren R, Salmon RA, Liggio J, Hayden KL, Anlauf KG, Leaitch WR. Nighttime chemistry at a rural site in the Lower Fraser Valley. Atmos Environ. 2004;38:5837–5848. [Google Scholar]
- McLaren R, Wojtal P, Majonis D, McCourt J, Halla JD, Brook J. NO3 radical measurements in a polluted marine environment: links to ozone formation. Atmos Chem Phys. 2010;10:4187–4206. doi: 10.5194/acp-10-4187-2010. [DOI] [Google Scholar]
- McMurry PH, Grosjean D. Gas and aerosol wall losses in Teflon film smog chambers. Environ Sci Technol. 1985;19:1176–1182. doi: 10.1021/es00142a006. [DOI] [PubMed] [Google Scholar]
- McNeill VF, Wolfe GM, Thornton JA. The Oxidation of Oleate in Submicron Aqueous Salt Aerosols?: Evidence of a Surface Process. J Phys Chem A. 2007;111:1073–1083. doi: 10.1021/jp066233f. [DOI] [PubMed] [Google Scholar]
- Meinen J, Thieser J, Platt U, Leisner T. Technical Note: Using a high finesse optical resonator to provide a long light path for differential optical absorption spectroscopy: CE-DOAS. Atmos Chem Phys. 2010;10:3901–3914. doi: 10.5194/acp-10-3901-2010. [DOI] [Google Scholar]
- Millet DB, Baasandorj M, Hu L, Mitroo D, Turner J, Williams BJ. Nighttime Chemistry and Morning Isoprene Can Drive Urban Ozone Downwind of a Major Deciduous Forest. Environ Sci Technol. 2016;50:4335–4342. doi: 10.1021/acs.est.5b06367. [DOI] [PubMed] [Google Scholar]
- Mishra AK, Klingmueller K, Fredj E, Lelieveld J, Rudich Y, Koren I. Radiative signature of absorbing aerosol over the eastern Mediterranean basin. Atmos Chem Phys. 2014;14:7213–7231. doi: 10.5194/acp-14-7213-2014. [DOI] [Google Scholar]
- Misztal PK, Hewitt CN, Wildt J, Blande JD, Eller ASD, Fares S, Gentner DR, Gilman JB, Graus M, Greenberg J, Guenther AB, Hansel A, Harley P, Huang M, Jardine K, Karl T, Kaser L, Keutsch FN, Kiendler-Scharr A, Kleist E, Lerner BM, Li T, Mak J, Nölscher AC, Schnitzhofer R, Sinha V, Thornton B, Warneke C, Wegener F, Werner C, Williams J, Worton DR, Yassaa N, Goldstein AH. Atmospheric benzenoid emissions from plants rival those from fossil fuels. Sci Rep. 2015;5:12064. doi: 10.1038/srep12064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moise T, Talukdar RK, Frost GJ, Fox RW, Rudich Y. Reactive uptake of NO3 by liquid and frozen organics. J Geophys Res. 2002;107:D24014. doi: 10.21029/22001JD000334. [DOI] [Google Scholar]
- Moise T, Flores JM, Rudich Y. Optical Properties of Secondary Organic Aerosols and Their Changes by Chemical Processes. Chem Rev. 2015;115:4400–4439. doi: 10.1021/cr5005259. [DOI] [PubMed] [Google Scholar]
- Moldanova J, Ljungstrom E. Modelling of particle formation from NO3 oxidation of selected monoterpenes. J Aerosol Sci. 2000;31:1317–1333. [Google Scholar]
- Müller JF, Peeters J, Stavrakou T. Fast photolysis of carbonyl nitrates from isoprene. Atmos Chem Phys. 2014;14:2497–2508. doi: 10.5194/acp-14-2497-2014. [DOI] [Google Scholar]
- Muthuramu K, Shepson PB, Obrien JM. Preparation, Analysis, and Atmospheric Production of Multifunctional Organic Nitrates. Environ Sci Technol. 1993;27:1117–1124. [Google Scholar]
- Mylonas DT, Allen DT, Ehrman SH, Pratsinis SE. The sources size distributions of organonitrates in Los Angeles aerosol Atmos. Environ, Part A, General Topics. 1991;25:2855–2861. doi: 10.1016/0960-1686(91)90211-O. [DOI] [Google Scholar]
- Nah T, McVay RC, Zhang X, Boyd CM, Seinfeld JH, Ng NL. Influence of seed aerosol surface area and oxidation rate on vapor wall deposition and SOA mass yields: a case study with α-pinene ozonolysis. Atmos Chem Phys. 2016a;16:9361–9379. doi: 10.5194/acp-16-9361-2016. [DOI] [Google Scholar]
- Nah T, Sanchez J, Boyd CM, Ng NL. Photochemical Aging of α-pinene and β-pinene Secondary Organic Aerosol formed from Nitrate Radical Oxidation. Environ Sci Technol. 2016b;50:222–231. doi: 10.1021/acs.est.5b04594. [DOI] [PubMed] [Google Scholar]
- Nakayama T, Ide T, Taketani F, Kawai M, Takahashi K, Matsumi Y. Nighttime measurements of ambient N2O5, NO2, NO and O3 in a sub-urban area, Toyokawa, Japan. Atmos Environ. 2008;42:1995–2006. doi: 10.1016/j.atmosenv.2007.12.001. [DOI] [Google Scholar]
- Nakayama T, Matsumi Y, Sato K, Imamura T, Yamazaki A, Uchiyama A. Laboratory studies on optical properties of secondary organic aerosols generated during the photooxidation of toluene and the ozonolysis of α-pinene. J Geophys Res-Atmos. 2010;115:D24204. doi: 10.1029/2010jd014387. [DOI] [Google Scholar]
- Naudet JP, Huguenin D, Rigaud P, Cariolle D. Stratospheric observations of NO3 and its experimental and theoretical distribution between 20 and 40 km. Planet Space Sci. 1981;29:707–712. [Google Scholar]
- Neta P, Huie RE, Ross AB. Rate Constants for Reactions of Inorganic Radicals in Aqueous Solution. J Phys Chem Ref Data. 1988;17:1027–1284. doi: 10.1063/1.555808. [DOI] [Google Scholar]
- Ng NL, Kwan AJ, Surratt JD, Chan AWH, Chhabra PS, Sorooshian A, Pye HOT, Crounse JD, Wennberg PO, Flagan RC, Seinfeld JH. Secondary organic aerosol (SOA) formation from reaction of isoprene with nitrate radicals (NO3) Atmos Chem Phys. 2008;8:4117–4140. doi: 10.5194/acp-8-4117-2008. [DOI] [Google Scholar]
- Ng NL, Canagaratna MR, Zhang Q, Jimenez JL, Tian J, Ulbrich IM, Kroll JH, Docherty KS, Chhabra PS, Bahreini R, Murphy SM, Seinfeld JH, Hildebrandt L, Donahue NM, DeCarlo PF, Lanz VA, Prévôt ASH, Dinar E, Rudich Y, Worsnop DR. Organic aerosol components observed in Northern Hemispheric datasets from Aerosol Mass Spectrometry. Atmos Chem Phys. 2010;10:4625–4641. doi: 10.5194/acp-10-4625-2010. [DOI] [Google Scholar]
- Ng NL, Herndon SC, Trimborn A, Canagaratna MR, Croteau PL, Onasch TB, Sueper D, Worsnop DR, Zhang Q, Sun YL, Jayne JT. An Aerosol Chemical Speciation Monitor (ACSM) for Routine Monitoring of the Composition and Mass Concentrations of Ambient Aerosol. Aerosol Sci Technol. 2011;45:780–794. doi: 10.1080/02786826.2011.560211. [DOI] [Google Scholar]
- Nguyen TB, Crounse JD, Schwantes RH, Teng AP, Bates KH, Zhang X, StClair JM, Brune WH, Tyndall GS, Keutsch FN, Seinfeld JH, Wennberg PO. Overview of the Focused Isoprene eXperiment at the California Institute of Technology (FIXCIT): mechanistic chamber studies on the oxidation of biogenic compounds. Atmos Chem Phys. 2014;14:13531–13549. doi: 10.5194/acp-14-13531-2014. [DOI] [Google Scholar]
- Nguyen TB, Crounse JD, Teng AP, Clair JMS, Paulot F, Wolfe GM, Wennberg PO. Rapid deposition of oxidized biogenic compounds to a temperate forest. P Natl Acad Sci USA. 2015;112:E392–E401. doi: 10.1073/pnas.1418702112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols PL, Magnusson AB, Ingham JD. Synthesis of Nitric Esters by the Addition of Nitric Acid to the Ethylene Oxide Ring. J Am Chem Soc. 1953;75:4255–4258. [Google Scholar]
- Niinemets Ü, Copolovici L, Hüve K. High within-canopy variation in isoprene emission potentials in temperate trees: Implications for predicting canopy-scale isoprene fluxes. J Geo-phys Res-Biogeo. 2010;115:G04029. doi: 10.1029/2010JG001436. [DOI] [Google Scholar]
- Nizich SV, Pope AA, Driver LM, Group P-A. National Air Pollutant Emission Trends Report. 2000:1900–1998. [Google Scholar]
- Noda J, Nyman G, Langer S. Kinetics of the gas-phase reaction of some unsaturated alcohols with the nitrate radical. J Phys Chem A. 2002;106:945–951. [Google Scholar]
- Noxon JF, Norton RB, Henderson WR. Observation of Atmospheric NO3. Geophys Res Lett. 1978;5:675–678. [Google Scholar]
- Noxon JF, Norton RB, Marovich E. NO3 in the troposphere. Geophys Res Lett. 1980;7:125–128. [Google Scholar]
- Noxon JF. NO3 and NO2 in the Mid-Pacific Troposphere. J Geophys Res. 1983;88:11017–11021. [Google Scholar]
- O’Brien RE, Laskin A, Laskin J, Rubitschun CL, Surratt JD, Goldstein AH. Molecular characterization of S- and N-containing organic constituents in ambient aerosols by negative ion mode high-resolution Nanospray Desorption Electrospray Ionization Mass Spectrometry: CalNex 2010 field study. J Geophys Res-Atmos. 2014;119:12706–712720. doi: 10.1002/2014JD021955. [DOI] [Google Scholar]
- O’Keefe A. Integrated cavity output analysis of ultra-weak absorption. Chem Phys Lett. 1998;293:331–336. doi: 10.1016/S0009-2614(98)00785-4. [DOI] [Google Scholar]
- O’Keefe A, Deacon DA. Cavity ring-down optical spectrometer for absorption measurements using pulsed laser sources. Rev Sci Instrum. 1988;59:2544–2551. [Google Scholar]
- O’Keefe A, Scherer JJ, Paul JB. cw Integrated cavity output spectroscopy. Chem Phys Lett. 1999;307:343–349. doi: 10.1016/S0009-2614(99)00547-3. [DOI] [Google Scholar]
- Odame-Ankrah CA, Osthoff HD. A compact diode laser cavity ring-down spectrometer for atmospheric measurements of NO3 and N2O5 with automated zeroing and calibration. Appl Spectrosc. 2011;65:1260–1268. doi: 10.1366/11-06384. [DOI] [PubMed] [Google Scholar]
- Odum JR, Hoffman T, Bowman F, Collins D, Flagan RC, Seinfeld JH. Gas/Particle Partitioning and Secondary Organic Aerosol Yields. Environ Sci Technol. 1996;30:2580–2585. [Google Scholar]
- Olariu RI, Barnes I, Bejan I, Arsene C, Vione D, Klotz B, Becker KH. FT-IR Product Study of the Reactions of NO3 Radicals With ortho-, meta-, and para-Cresol. Environ Sci Technol. 2013;47:7729–7738. doi: 10.1021/es401096w. [DOI] [PubMed] [Google Scholar]
- Osthoff HD, Pilling MJ, Ravishankara AR, Brown SS. Temperature dependence of the NO3 absorption cross section above 298 K and determination of the equilibrium constant for NO3+ NO2-N2O5 at atmospherically relevant conditions. Phys Chem Chem Phys. 2007;9:5785–5793. doi: 10.1039/b709193a. [DOI] [PubMed] [Google Scholar]
- Osthoff HD, Bates TS, Johnson JE, Kuster WC, Goldan PD, Sommariva R, Williams EJ, Lerner BM, Warneke C, de Gouw JA, Pettersson A, Baynard T, Meagher JF, Fehsenfeld FC, Ravishankara AR, Brown SS. Regional variation of dimethyl sulfide oxidation mechanism in the summertime marine boundary layer in the Gulf of Maine. J Geophys Res. 2009;114:D07301. doi: 10.1029/2008JD010990. [DOI] [Google Scholar]
- Pandis SN, Paulson SE, Seinfeld JH, Flagan RC. Aerosol formation in the photooxidation of isoprene and β-pinene. Atmos Environ Pt A. 1991;25:997–1008. doi: 10.1016/0960-1686(91)90141-S. [DOI] [Google Scholar]
- Paoli R, Cariolle D, Sausen R. Review of effective emissions modeling and computation. Geosci Model Dev. 2011;4:643–667. doi: 10.5194/gmd-4-643-2011. [DOI] [Google Scholar]
- Parrish DD, Buhr MP, Trainer M, Norton RB, Shimshock JP, Fehsenfeld FC, Anlauf KG, Bottenheim JW, Tang YZ, Wiebe HA, Roberts JM, Tanner RL, Newman L, Bowersox VC, Olszyna KJ, Bailey EM, Rodgers MO, Wang T, Berresheim H, Roychowdhury UK, Demerjian KL. The Total Reactive Oxidized Nitrogen Levels and the Partitioning between the Individual-Species at 6 Rural Sites in Eastern North-America. J Geophys Res-Atmos. 1993;98:2927–2939. [Google Scholar]
- Paul D, Furgeson A, Osthoff HD. Measurements of total peroxy and alkyl nitrate abundances in laboratory-generated gas samples by thermal dissociation cavity ring-down spectroscopy. Rev Sci Instr. 2009;80:114101. doi: 10.1063/1.3258204. [DOI] [PubMed] [Google Scholar]
- Paulot F, Henze DK, Wennberg PO. Impact of the isoprene photochemical cascade on tropical ozone. Atmos Chem Phys. 2012;12:1307–1325. doi: 10.5194/acp-12-1307-2012. [DOI] [Google Scholar]
- Paulson SE, Seinfeld JH. Development and Evaluation of a Photooxidation Mechnaism for Isoprene. J Geophys Res. 1992;97:20703–20715. [Google Scholar]
- Peeters J, Nguyen TL, Vereecken L. HOx radical regeneration in the oxidation of isoprene. Phys Chem Chem Phys. 2009;11:5935–5939. doi: 10.1039/B908511D. [DOI] [PubMed] [Google Scholar]
- Perraud V, Bruns EA, Ezell MJ, Johnson SN, Greaves J, Finlayson-Pitts BJ. Identification of organic nitrates in the NO3 radical initiated oxidation of α-pinene by atmospheric pressure chemical ionization mass spectrometry. Environ Sci Technol. 2010;44:5887–5893. doi: 10.1021/es1005658. [DOI] [PubMed] [Google Scholar]
- Perring AE, Wisthaler A, Graus M, Wooldridge PJ, Lockwood AL, Mielke LH, Shepson PB, Hansel A, Cohen RC. A product study of the isoprene + NO3 reaction. Atmos Chem Phys. 2009;9:4945–4956. doi: 10.5194/acp-9-4945-2009. [DOI] [Google Scholar]
- Perring AE, Pusede SE, Cohen RC. An Observational Perspective on the Atmospheric Impacts of Alkyl and Multifunctional Nitrates on Ozone and Secondary Organic Aerosol. Chem Rev. 2013;113:5848–5870. doi: 10.1021/cr300520x. [DOI] [PubMed] [Google Scholar]
- Pfrang C, Martin RS, Canosa-Mas CE, Wayne RP. Gas-phase reactions of NO3 and N2O5 with (Z)-hex-4-en-1-ol, (Z)-hex-3-en-1-ol (“leaf alcohol”), (E)-hex-3-en-1-ol, (Z)-hex-2-en-1-ol, (E)-hex-2-en-1-ol. Phys Chem Chem Phys. 2006;8:354–363. doi: 10.1039/b510835g. [DOI] [PubMed] [Google Scholar]
- Pierce T, Geron C, Bender L, Dennis R, Tonnesen G, Guenther A. Influence of increased isoprene emissions on regional ozone modeling. J Geophys Res-Atmos. 1998;103:25611–25629. doi: 10.1029/98JD01804. [DOI] [Google Scholar]
- Pitts JN, Jr, Bierman HW, Atkinson R, Winer AM. Atmospheric implications of simultaneous measurements of NO3 radicals and HONO. Geophys Res Lett. 1984;11:557–560. [Google Scholar]
- Platt U, Perner D, Winer AM, Harris GW, Pitts JNJ. Detection of NO3 in the polluted troposphere by differential optical absorption. Geophys Res Lett. 1980;7:89–92. [Google Scholar]
- Platt U, Perner D, Schröder J, Kessler C, Toennissen A. The Diurnal Variation of NO3. J Geophys Res. 1981;86:11965–11970. [Google Scholar]
- Platt U, Stutz J. Differential optical absorption spectroscopy: principles and applications. Springer; Berlin: 2008. [Google Scholar]
- Platt U, LeBras G, Poulet G, Burrows JP, Moortgat G. Peroxy radicals from night-time reactions of NO3 with organic compounds. Nature. 1990;348:147–149. [Google Scholar]
- Platt U, Meinen J, Pöhler D, Leisner T. Broadband Cavity Enhanced Differential Optical Absorption Spectroscopy (CE-DOAS) – applicability and corrections. Atmos Meas Tech. 2009;2:713–723. doi: 10.5194/amt-2-713-2009. [DOI] [Google Scholar]
- Platt UF, Winer AM, Bierman HW, Atkinson R, Pitts JN., Jr Measurement of Nitrate Radical Concentrations in Continental Air. Environ Sci Technol. 1984;18:365–369. doi: 10.1021/es00123a015. [DOI] [PubMed] [Google Scholar]
- Pöschl U. Atmospheric Aerosols: Composition, Transformation, Climate and Health Effects. Angewandte Chemie International Edition. 2005;44:7520–7540. doi: 10.1002/anie.200501122. [DOI] [PubMed] [Google Scholar]
- Pöschl U, Shiraiwa M. Multiphase Chemistry at the Atmosphere–Biosphere Interface Influencing Climate and Public Health in the Anthropocene. Chem Rev. 2015;115:4440–4475. doi: 10.1021/cr500487s. [DOI] [PubMed] [Google Scholar]
- Praske E, Crounse JD, Bates KH, Kurtén T, Kjaergaard HG, Wennberg PO. Atmospheric Fate of Methyl Vinyl Ketone: Peroxy Radical Reactions with NO and HO2. J Phys Chem A. 2015;119:4562–4572. doi: 10.1021/jp5107058. [DOI] [PubMed] [Google Scholar]
- Pratt KA, Mielke LH, Shepson PB, Bryan AM, Steiner AL, Ortega J, Daly R, Helmig D, Vogel CS, Griffith S, Dusanter S, Stevens PS, Alaghmand M. Contributions of individual reactive biogenic volatile organic compounds to organic nitrates above a mixed forest. Atmos Chem Phys. 2012;12:10125–10143. doi: 10.5194/acp-12-10125-2012. [DOI] [Google Scholar]
- Pye HOT, Chan AWH, Barkley MP, Seinfeld JH. Global modeling of organic aerosol: the importance of reactive nitrogen (NOx and NO3) Atmos Chem Phys. 2010;10:11261–11276. doi: 10.5194/acp-10-11261-2010. [DOI] [Google Scholar]
- Pye HOT, Luecken DJ, Xu L, Boyd CM, Ng NL, Baker KR, Ayres BR, Bash JO, Baumann K, Carter WPL, Edgerton E, Fry JL, Hutzell WT, Schwede DB, Shepson PB. Modeling the Current and Future Roles of Particulate Organic Nitrates in the Southeastern United States. Environ Sci Technol. 2015;49:14195–14203. doi: 10.1021/acs.est.5b03738. [DOI] [PubMed] [Google Scholar]
- Ramanathan V, Li F, Ramana M, Praveen P, Kim D, Corrigan C, Nguyen H, Stone EA, Schauer JJ, Carmichael G. Atmospheric brown clouds: Hemispherical and regional variations in long-range transport, absorption, and radiative forcing. J Geophys Res-Atmos. 2007a;112:D22S21. doi: 10.1029/2006JD008124. [DOI] [Google Scholar]
- Ramanathan V, Ramana MV, Roberts G, Kim D, Corrigan C, Chung C, Winker D. Warming trends in Asia amplified by brown cloud solar absorption. Nature. 2007b;448:575. doi: 10.1038/nature06019. [DOI] [PubMed] [Google Scholar]
- Reemtsma T, These A, Venkatachari P, Xia X, Hopke PK, Springer A, Linscheid M. Identification of Fulvic Acids and Sulfated and Nitrated Analogues in Atmospheric Aerosol by Electrospray Ionization Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal Chem. 2006;78:8299–8304. doi: 10.1021/ac061320p. [DOI] [PubMed] [Google Scholar]
- Reinmuth-Selzle K, Ackaert C, Kampf CJ, Samonig M, Shiraiwa M, Kofler S, Yang H, Gadermaier G, Brandstetter H, Huber CG, Duschl A, Oostingh GJ, Pöschl U. Nitration of the birch pollen allergen Bet v 1.0101: Efficiency and site-selectivity of liquid and gaseous nitrating agents. J Proteome Res. 2014;13:1570–1577. doi: 10.1021/pr401078h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renbaum-Wolff L, Grayson JW, Bateman AP, Kuwata M, Sellier M, Murray BJ, Shilling JE, Martin ST, Bertram AK. Viscosity of α-pinene secondary organic material and implications for particle growth and reactivity. P Natl Acad Sci USA. 2013;110:8014–8019. doi: 10.1073/pnas.1219548110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter A, Burrows JP, Nusz H, Granier C, Niemeier U. Increase in tropospheric nitrogen dioxide over China observed from space. Nature. 2005;437:129–132. doi: 10.1038/nature04092. [DOI] [PubMed] [Google Scholar]
- Rickard A. [last access: 5 Mai 2015];The Master Chemical Mechanism Version MCM v3.2. available at: http://mcm.leeds.ac.uk/MCMv3.2/
- Rindelaub JD, McAvey KM, Shepson PB. The photochemical production of organic nitrates from α-pinene and loss via acid-dependent particle phase hydrolysis. Atmos Environ. 2015;100:193–201. doi: 10.1016/j.atmosenv.2014.11.010. [DOI] [Google Scholar]
- Robinson AL, Donahue NM, Rogge WF. Photochemical oxidation and changes in molecular composition of organic aerosol in the regional context. J Geophys Res-Atmos. 2006;111:D03302. doi: 10.1029/2005JD006265. [DOI] [Google Scholar]
- Rollins AW, Kiendler-Scharr A, Fry JL, Brauers T, Brown SS, Dorn H-P, Dubé WP, Fuchs H, Mensah A, Mentel TF, Rohrer F, Tilmann R, Wegener R, Wooldridge PJ, Cohen RC. Isoprene oxidation by nitrate radical: alkyl nitrate and secondary organic aerosol yields. tmos Chem Phys. 2009;9:6685–6703. doi: 10.5194/acp-9-6685-2009. [DOI] [Google Scholar]
- Rollins AW, Smith JD, Wilson KR, Cohen RC. Real Time In Situ Detection of Organic Nitrates in Atmospheric Aerosols. Environ Sci Technol. 2010;44:5540–5545. doi: 10.1021/es100926x. [DOI] [PubMed] [Google Scholar]
- Rollins AW, Browne EC, Min KE, Pusede SE, Wooldridge PJ, Gentner DR, Goldstein AH, Liu S, Day DA, Russell LM, Cohen RC. Evidence for NOx Control over Nighttime SOA Formation. Science. 2012;337:1210–1212. doi: 10.1126/science.1221520. [DOI] [PubMed] [Google Scholar]
- Rollins AW, Pusede S, Wooldridge P, Min KE, Gentner DR, Goldstein AH, Liu S, Day DA, Russell LM, Rubitschun CL, Surratt JD, Cohen RC. Gas/particle partitioning of total alkyl nitrates observed with TD-LIF in Bakersfield. J Geophys Res-Atmos. 2013;118:6651–6662. doi: 10.1002/jgrd.50522. [DOI] [Google Scholar]
- Ross AB, Bielski BHJ, Buxton GV, Cabelli DE, Helman WP, Huie RE, Grodkovski J, Neta P, Mulazzani QG, Wilkinson F. NIST standard reference database 40: NDRL/NIST solution kinetics database version 3.0. 1998. [Google Scholar]
- Rudich Y, Talukdar RK, Fox RW, Ravinshankara AR. Rate coefficients for reactions of NO3 with a few olefins and oxygenated olefins. J Phys Chem A. 1996;100:5374–5381. [Google Scholar]
- Rudich Y, Talukdar RK, Ravishankara AR. Multiphase chemistry of NO3 in the remote troposphere. J Geophys Res-Atmos. 1998;103:16133–16143. doi: 10.1029/98jd01280. [DOI] [Google Scholar]
- Rudich Y. Laboratory Perspectives on the Chemical Transformations of Organic Matter in Atmospheric Particles. Chem Rev. 2003;103:5097–5124. doi: 10.1021/cr020508f. [DOI] [PubMed] [Google Scholar]
- Russell AR, Valin LC, Cohen RC. Trends in OMI NO2 observations over the United States: effects of emission control technology and the economic recession. Atmos Chem Phys. 2012;12:12197–12209. doi: 10.5194/acp-12-12197-2012. [DOI] [Google Scholar]
- Sadanaga Y, Takaji R, Ishiyama A, Nakajima K, Matsuki A, Bandow H. Thermal dissociation cavity attenuated phase shift spectroscopy for continuous measurement of total peroxy and organic nitrates in the clean atmosphere. Rev Sci Instrum. 2016;87:074102. doi: 10.1063/1.4958167. [DOI] [PubMed] [Google Scholar]
- Sanders RW, Solomon S, Mount GH, Bates MW, Schmeltekopf AL. Visible Spectroscopy at McMurdo Station, Antarctica 3, Observations of NO3. J Geophys Res. 1987;92:8339–8342. [Google Scholar]
- Saunders SM, Jenkin ME, Derwent RG, Pilling MJ. Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds. Atmos Chem Phys. 2003;3:161–180. doi: 10.5194/acp-3-161-2003. [DOI] [Google Scholar]
- Schichtel B, Malm WC, Bench G, Fallon S, McDade CE, Chow JC, Watson JG. Fossil and contemporary fine particulate carbon fractions at 12 rural and urban sites in the United States. J Geophys Res. 2008;113:D02311. doi: 10.1029/2007JD008605. [DOI] [Google Scholar]
- Schlag P, Kiendler-Scharr A, Blom MJ, Canonaco F, Henzing JS, Moerman M, Prévôt ASH, Holzinger R. Aerosol source apportionment from 1-year measurements at the CESAR tower in Cabauw, the Netherlands. Atmos Chem Phys. 2016;16:8831–8847. doi: 10.5194/acp-16-8831-2016. [DOI] [Google Scholar]
- Schneider M, Ballschmiter K. Alkyl nitrates as achiral and chiral solute probes in gas chromatography – Novel properties of a beta-cyclodextrin derivative and characterization of its enantioselective forces. J Chromatogr A. 1999;852:525–534. doi: 10.1016/s0021-9673(99)00635-4. [DOI] [PubMed] [Google Scholar]
- Schuster G, Labazan I, Crowley JN. A cavity ring down/cavity enhanced absorption device for measurement of ambient NO3 and N2O5. Atmos Meas Tech. 2009;2:1–13. doi: 10.5194/amt-2-1-2009. [DOI] [Google Scholar]
- Schwantes RH, Teng AP, Nguyen TB, Coggon MM, Crounse JD, StClair JM, Zhang X, Schilling KA, Seinfeld JH, Wennberg PO. Isoprene NO3 Oxidation Products from the RO2+ HO2 Pathway. J Phys Chem A. 2015;119:10158–10171. doi: 10.1021/acs.jpca.5b06355. [DOI] [PubMed] [Google Scholar]
- Scott CE, Rap A, Spracklen DV, Forster PM, Carslaw KS, Mann GW, Pringle KJ, Kivekäs N, Kulmala M, Lihavainen H, Tunved P. The direct and indirect radiative effects of biogenic secondary organic aerosol. Atmos Chem Phys. 2014;14:447–470. doi: 10.5194/acp-14-447-2014. [DOI] [Google Scholar]
- Setyan A, Zhang Q, Merkel M, Knighton WB, Sun Y, Song C, Shilling JE, Onasch TB, Herndon SC, Worsnop DR, Fast JD, Zaveri RA, Berg LK, Wiedensohler A, Flowers BA, Dubey MK, Subramanian R. Characterization of submicron particles influenced by mixed biogenic and anthropogenic emissions using high-resolution aerosol mass spectrometry: results from CARES. Atmos Chem Phys. 2012;12:8131–8156. doi: 10.5194/acp-12-8131-2012. [DOI] [Google Scholar]
- Shiraiwa M, Garland RM, Pöschl U. Kinetic double-layer model of aerosol surface chemistry and gas-particle interactions (K2-SURF): Degradation of polycyclic aromatic hydrocarbons exposed to O3, NO2, H2O, OH and NO3. Atmos Chem Phys. 2009;9:9571–9586. doi: 10.5194/acp-9-9571-2009. [DOI] [Google Scholar]
- Shiraiwa M, Sosedova Y, Rouviere A, Yang H, Zhang Y, Abbatt JPD, Ammann M, Pöschl U. The role of long-lived reactive oxygen intermediates in the reaction of ozone with aerosol particles. Nat Chem. 2011;4:291–295. doi: 10.1038/nchem.988. [DOI] [PubMed] [Google Scholar]
- Shiraiwa M, Pöschl U, Knopf DA. Multiphase Chemical Kinetics of NO3 Radicals Reacting with Organic Aerosol Components from Biomass Burning. Environ Sci Technol. 2012;46:6630–6636. doi: 10.1021/es300677a. [DOI] [PubMed] [Google Scholar]
- Shorees B, Atkinson R, Arey J. Kinetics of the gas-phase reactions of beta-phellandrene with OH and NO3 radicals and O3 at 297-K ± 2-K. Int J Chem Kinet. 1991;23:897–906. doi: 10.1002/kin.550231005. [DOI] [Google Scholar]
- Simpson WR. Continuous wave cavity ring-down spectroscopy applied to in situ detection of dinitrogen pentoxide (N2O5) Rev Sci Instrum. 2003;74:3442–3452. doi: 10.1063/1.1578705. [DOI] [Google Scholar]
- Slusher DL, Huey LG, Tanner DJ, Flocke FM, Roberts JM. A thermal dissociation–chemical ionization mass spectrometry (TD-CIMS) technique for the simultaneous measurement of peroxyacyl nitrates and dinitrogen pentoxide. J Geophys Res-Atmos. 2004;109:D19315. doi: 10.1029/2004JD004670. [DOI] [Google Scholar]
- Smith JP, Solomon S. Atmospheric NO3, 3 Sunrise Disappearance and the Stratospheric Profile. J Geophys Res. 1990;95:13819–13827. [Google Scholar]
- Smith JP, Solomon S, Sanders RW, Miller HL, Perliski LM, Keys JG, Schmeltekopf AL. Atmospheric NO3, 4 Vertical Profiles at Middle and Polar Latitudes at Sunrise. J Geophys Res. 1993;98:8983–8989. [Google Scholar]
- Sobanski N, Tang MJ, Thieser J, Schuster G, Pöhler D, Fischer H, Song W, Sauvage C, Williams J, Fachinger J, Berkes F, Hoor P, Platt U, Lelieveld J, Crowley JN. Chemical and meteorological influences on the lifetime of NO3 at a semi-rural mountain site during PARADE. Atmos Chem Phys. 2016;16:4867–4883. doi: 10.5194/acp-16-4867-2016. [DOI] [Google Scholar]
- Solomon S, Sanders RW, Mount GH, Carroll MA, Jakoubek RO, Schmeltekopf AL. Atmospheric NO3 2 Observations in Polar Regions. J Geophys Res. 1989;94:16423–16427. [Google Scholar]
- Solomon S, Smith JP, Sanders RW, Perliski L, Miller HL, Mount GH, Keys JG, Schmeltekopf AL. Visible and Near-Ultraviolet Spectroscopy at McMurdo Station, Antarctica, 8. Observations of Nighttime NO2 and NO3 from April to October 1991. J Geophys Res. 1993;98:993–1000. [Google Scholar]
- Song C, Gyawali M, Zaveri RA, Shilling JE, Arnott WP. Light absorption by secondary organic aerosol from α-pinene: Effects of oxidants, seed aerosol acidity, and relative humidity. J Geophys Res-Atmos. 2013;118:11741–711749. doi: 10.1002/jgrd.50767. [DOI] [Google Scholar]
- Spittler M, Barnes I, Bejan I, Brockmann KJ, Benter T, Wirtz K. Reactions of NO3 radicals with limonene and alpha-pinene: Product and SOA formation. Atmos Environ. 2006;40:S116–S127. [Google Scholar]
- Spracklen DV, Jimenez JL, Carslaw KS, Worsnop DR, Evans MJ, Mann GW, Zhang Q, Canagaratna MR, Allan J, Coe H, McFiggans G, Rap A, Forster P. Aerosol mass spectrometer constraint on the global secondary organic aerosol budget. Atmos Chem Phys. 2011;11:12109–12136. doi: 10.5194/acp-11-12109-2011. [DOI] [Google Scholar]
- Squire OJ, Archibald AT, Abraham NL, Beerling DJ, Hewitt CN, Lathière J, Pike RC, Telford PJ, Pyle JA. Influence of future climate and cropland expansion on isoprene emissions and tropospheric ozone. Atmos Chem Phys. 2014;14:1011–1024. doi: 10.5194/acp-14-1011-2014. [DOI] [Google Scholar]
- Squire OJ, Archibald AT, Griffiths PT, Jenkin ME, Smith D, Pyle JA. Influence of isoprene chemical mechanism on modelled changes in tropospheric ozone due to climate and land use over the 21st century. Atmos Chem Phys. 2015;15:5123–5143. doi: 10.5194/acp-15-5123-2015. [DOI] [Google Scholar]
- Stark H, Brown SS, Goldan PD, Aldener M, Kuster WC, Jakoubek R, Fehsenfeld FC, Meagher J, Bates TS, Ravishankara AR. Influence of the nitrate radical on the oxidation of dimethyl sulfide in a polluted marine environment. J Geophys Res. 2007;112:D10S10. doi: 10.1029/2006JD007669. [DOI] [Google Scholar]
- Starn T, Shepson P, Bertman S, Riemer D, Zika R, Olszyna K. Nighttime isoprene chemistry at an urban-impacted forest site. J Geophys Res-Atmos. 1998;103:22437–22447. [Google Scholar]
- Stavrakou T, Müller J-F, Boersma KF, De Smedt I, van der ARJ. Assessing the distribution and growth rates of NOx emission sources by inverting a 10-year record of NO2 satellite columns. Geophys Res Lett. 2008;35:L10801. doi: 10.1029/2008GL033521. [DOI] [Google Scholar]
- Steinbacher M, Dommen J, Ordonez C, Reimann S, Grüebler FC, Staehelin J, Andreani-Aksoyoglu S, Prevot ASH. Volatile Organic Compounds in the Po Basin, Part B: Biogenic VOCs. J Atmos Chem. 2005;51:293–315. doi: 10.1007/s10874-005-3577-0. [DOI] [Google Scholar]
- Stewart DJ, Almabrok SH, Lockhart JP, Mohamed OM, Nutt DR, Pfrang C, Marston G. The kinetics of the gas-phase reactions of selected monoterpenes and cycloalkenes with ozone and the NO3 radical. Atmos Environ. 2013;70:227–235. doi: 10.1016/j.atmosenv.2013.01.036. [DOI] [Google Scholar]
- Stier P, Seinfeld JH, Kinne S, Boucher O. Aerosol absorption and radiative forcing. Atmos Chem Phys. 2007;7:5237–5261. doi: 10.5194/acp-7-5237-2007. [DOI] [Google Scholar]
- Strader R, Lurmann F, Pandis SN. Evaluation of secondary organic aerosol formation in winter. Atmos Environ. 1999;33:4849–4863. doi: 10.1016/S1352-2310(99)00310-6. [DOI] [Google Scholar]
- Streets DG, Yu C, Wu Y, Chin M, Zhao Z, Hayasaka T, Shi G. Aerosol trends over China, 1980–2000. Atmos Res. 2008;88:174–182. doi: 10.1016/j.atmosres.2007.10.016. [DOI] [Google Scholar]
- Stroud C, Roberts J, Williams E, Hereid D, Angevine W, Fehsenfeld F, Wisthaler A, Hansel A, Martinez-Harder M, Harder H. Nighttime isoprene trends at an urban forested site during the 1999 Southern Oxidant Study. J Geophys Res-Atmos. 2002;107:4291. doi: 10.1029/2001JD000959. [DOI] [Google Scholar]
- Stutz J, Alicke B, Neftel A. Nitrous acid formation in the urban atmosphere: Gradient measurements of NO2 and HONO over grass in Milan, Italy. J Geophys Res-Atmos. 2002;107:8192. doi: 10.1029/2001JD000390. [DOI] [Google Scholar]
- Stutz J, Alicke B, Ackermann R, Geyer A, White AB, Williams E. Vertical profiles of NO3, N2O5, O3 and NOx in the nocturnal boundary layer: 1. Observations during the Texas Air Quality Study 2000. J Geophys Res. 2004;109:D12306. doi: 10.1029/2003JD004209. [DOI] [Google Scholar]
- Stutz J, Wong KW, Lawrence L, Ziemba L, Flynn JH, Rappenglück B, Lefer B. Nocturnal NO3 radical chemistry in Houston, TX. Atmos Environ. 2010;44:4099–4106. [Google Scholar]
- Suarez-Bertoa R, Picquet-Varrault B, Tamas W, Pangui E, Doussin JF. Atmospheric Fate of a Series of Carbonyl Nitrates: Photolysis Frequencies and OH-Oxidation Rate Constants. Environ Sci Technol. 2012;46:12502–12509. doi: 10.1021/es302613x. [DOI] [PubMed] [Google Scholar]
- Suda SR, Petters MD, Yeh GK, Strollo C, Matsunaga A, Faulhaber A, Ziemann PJ, Prenni AJ, Carrico CM, Sullivan RC, Kreidenweis SM. Influence of Functional Groups on Organic Aerosol Cloud Condensation Nucleus Activity. Environ Sci Technol. 2014;48:10182–10190. doi: 10.1021/es502147y. [DOI] [PubMed] [Google Scholar]
- Suh I, Lei WF, Zhang RY. Experimental and theoretical studies of isoprene reaction with NO3. J Phys Chem A. 2001;105:6471–6478. doi: 10.1021/jp0105950. [DOI] [Google Scholar]
- Sun YL, Zhang Q, Schwab JJ, Yang T, Ng NL, Demerjian KL. Factor analysis of combined organic and inorganic aerosol mass spectra from high resolution aerosol mass spectrometer measurements. Atmos Chem Phys. 2012;12:8537–8551. doi: 10.5194/acp-12-8537-2012. [DOI] [Google Scholar]
- Surratt JD, Kroll JH, Kleindienst TE, Edney EO, Claeys M, Sorooshian A, Ng NL, Offenberg JH, Lewandowski M, Jaoui M, Flagan RC, Seinfeld JH. Evidence for Organosulfates in Secondary Organic Aerosol. Environ Sci Technol. 2007;41:517–527. doi: 10.1021/es062081q. [DOI] [PubMed] [Google Scholar]
- Surratt JD, Gómez-González Y, Chan AWH, Vermeylen R, Shahgholi M, Kleindienst TE, Edney EO, Offenberg JH, Lewandowski M, Jaoui M, Maenhaut W, Claeys M, Flagan RC, Seinfeld JH. Organosulfate Formation in Biogenic Secondary Organic Aerosol. J Phys Chem A. 2008;112:8345–8378. doi: 10.1021/jp802310p. [DOI] [PubMed] [Google Scholar]
- Szmigielski R, Vermeylen R, Dommen J, Metzger A, Maenhaut W, Baltensperger U, Claeys M. The acid effect in the formation of 2-methyltetrols from the photooxidation of isoprene in the presence of NOx. Atmos Res. 2010;98:183–189. doi: 10.1016/j.atmosres.2010.02.012. [DOI] [Google Scholar]
- Takagi H, Washida N, Akimoto H, Okuda M. Analysis of Nitrate and Nitrite Esters by Gas-Chromatography Photoionization Mass-Spectrometry. Anal Chem. 1981;53:175–179. doi: 10.1021/ac00225a011. [DOI] [Google Scholar]
- Teng AP, Crounse JD, Lee L, StClair JM, Cohen RC, Wennberg PO. Hydroxy nitrate production in the OH-initiated oxidation of alkenes. Atmos Chem Phys. 2015;15:4297–4316. doi: 10.5194/acp-15-4297-2015. [DOI] [Google Scholar]
- Thieser J, Schuster G, Schuladen J, Phillips GJ, Reiffs A, Parchatka U, Pöhler D, Lelieveld J, Crowley JN. A two-channel thermal dissociation cavity ring-down spectrometer for the detection of ambient NO2, RO2NO2 and RONO2. Atmos Meas Tech. 2016;9:553–576. doi: 10.5194/amt-9-553-2016. [DOI] [Google Scholar]
- Tilgner A, Bräuer P, Wolke R, Herrmann H. Modelling multiphase chemistry in deliquescent aerosols and clouds using CAPRAM3.0i. J Atmos Chem. 2013;70:221–256. doi: 10.1007/s10874-013-9267-4. [DOI] [Google Scholar]
- Tolocka MP, Jang M, Ginter JM, Cox FJ, Kamens RM, Johnston MV. Formation of Oligomers in Secondary Organic Aerosol. Environ Sci Technol. 2004;38:1428–1434. doi: 10.1021/es035030r. [DOI] [PubMed] [Google Scholar]
- Tong DQ, Lamsal L, Pan L, Ding C, Kim H, Lee P, Chai T, Pickering KE, Stajner I. Long-term NOx trends over large cities in the United States during the great recession: Comparison of satellite retrievals, ground observations, and emission inventories. Atmos Environ. 2015;107:70–84. doi: 10.1016/j.atmosenv.2015.01.035. [DOI] [Google Scholar]
- Travis KR, Jacob DJ, Fisher JA, Kim PS, Marais EA, Zhu L, Yu K, Miller CC, Yantosca RM, Sulprizio MP, Thompson AM, Wennberg PO, Crounse JD, StClair JM, Cohen RC, Laughner JL, Dibb JE, Hall SR, Ullmann K, Wolfe GM, Pollack IB, Peischl J, Neuman JA, Zhou X. Why do models overestimate surface ozone in the Southeast United States? Atmos Chem Phys. 2016;16:13561–13577. doi: 10.5194/acp-16-13561-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treves K, Shragina L, Rudich Y. Henry’s law constants of some beta-, gamma-, and delta-hydroxy alkyl nitrates of atmospheric interest. Environ Sci Technol. 2000;34:1197–1203. [Google Scholar]
- Tröstl J, Chuang WK, Gordon H, Heinritzi M, Yan C, Molteni U, Ahlm L, Frege C, Bianchi F, Wagner R, Simon M, Lehtipalo K, Williamson C, Craven JS, Duplissy J, Adamov A, Almeida J, Bernhammer A-K, Breitenlechner M, Brilke S, Dias A, Ehrhart S, Flagan RC, Franchin A, Fuchs C, Guida R, Gysel M, Hansel A, Hoyle CR, Jokinen T, Junninen H, Kangasluoma J, Keskinen H, Kim J, Krapf M, Kürten A, Laaksonen A, Lawler M, Leiminger M, Mathot S, Möhler O, Nieminen T, Onnela A, Petäjä T, Piel FM, Miettinen P, Rissanen MP, Rondo L, Sarnela N, Schobesberger S, Sengupta K, Sipilä M, Smith JN, Steiner G, Tomè A, Virtanen A, Wagner AC, Weingartner E, Wimmer D, Winkler PM, Ye P, Carslaw KS, Curtius J, Dommen J, Kirkby J, Kulmala M, Riipinen I, Worsnop DR, Donahue NM, Baltensperger U. The role of low-volatility organic compounds in initial particle growth in the atmosphere. Nature. 2016;533:527–531. doi: 10.1038/nature18271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utembe SR, Cooke MC, Archibald AT, Shallcross DE, Derwent RG, Jenkin ME. Simulating secondary organic aerosol in a 3-D Lagrangian chemistry transport model using the reduced Common Representative Intermediates mechanism (CRI v2-R5) Atmos Environ. 2011;45:1604–1614. doi: 10.1016/j.atmosenv.2010.11.046. [DOI] [Google Scholar]
- Vaattovaara P, Petaja T, Joutsensaari J, Miettinen P, Zaprudin B, Kortelainen A, Heijari J, Yli-Pirila P, Aalto P, Worsnop DR, Laaksonen A. The evolution of nucleation- and Aitken-mode particle compsitions in a boreal forest environment during clean and pollution-affected new-particle formation events. Boreal Environ Res. 2009;14:662–682. [Google Scholar]
- Varma RM, Ball SM, Brauers T, Dorn HP, Heitmann U, Jones RL, Platt U, Pöhler D, Ruth AA, Shillings AJL, Thieser J, Wahner A, Venables DS. Light extinction by secondary organic aerosol: an intercomparison of three broadband cavity spectrometers. Atmos Meas Tech. 2013;6:3115–3130. doi: 10.5194/amt-6-3115-2013. [DOI] [Google Scholar]
- Venables DS, Gherman T, Orphal J, Wenger JC, Ruth AA. High Sensitivity in Situ Monitoring of NO3 in an Atmospheric Simulation Chamber Using Incoherent Broadband Cavity-Enhanced Absorption Spectroscopy. Environ Sci Technol. 2006;40:6758–6763. doi: 10.1021/es061076j. [DOI] [PubMed] [Google Scholar]
- Verstraeten WW, Neu JL, Williams JE, Bowman KW, Worden JR, Boersma KF. Rapid increases in tropospheric ozone production and export from China. Nat Geosci. 2015;8:690–695. doi: 10.1038/ngeo2493. [DOI] [Google Scholar]
- Vijayaraghavan K, Karamchandani P, Seigneur C. Plume-in-grid modeling of summer air pollution in Central California. Atmos Environ. 2006;40:5097–5109. doi: 10.1016/j.atmosenv.2005.12.050. [DOI] [Google Scholar]
- Virtanen A, Joutsensaari J, Koop T, Kannosto J, Yli-Pirila P, Leskinen J, Makela JM, Holopainen JK, Pöschl U, Kulmala M, Worsnop DR, Laaksonen A. An amorphous solid state of biogenic secondary organic aerosol particles. Nature. 2010;467:824–827. doi: 10.1038/nature09455. [DOI] [PubMed] [Google Scholar]
- von Friedeburg C, Wagner T, Geyer A, Kaiser N, Vogel B, Vogel H, Platt U. Derivation of tropospheric NO3 profiles using off-axis differential optical absorption spectroscopy measurements during sunrise and comparison with simulations. J Geophys Res-Atmos. 2002;107:4168. doi: 10.1029/2001JD000481. [DOI] [Google Scholar]
- von Kuhlmann R, Lawrence MG, Pöschl U, Crutzen PJ. Sensitivities in global scale modeling of isoprene. Atmos Chem Phys. 2004;4:1–17. doi: 10.5194/acp-4-1-2004. [DOI] [Google Scholar]
- Vrekoussis M, Kanakidou M, Mihalopoulos N, Crutzen PJ, Lelieveld J, Perner D, Berresheim H, Baboukas E. Role of the NO3 radicals in oxidation processes in the eastern Mediterranean troposphere during the MINOS campaign. Atmos Chem Phys. 2004;4:169–182. doi: 10.5194/acp-4-169-2004. [DOI] [Google Scholar]
- Wagner NL, Dubé WP, Washenfelder RA, Young CJ, Pollack IB, Ryerson TB, Brown SS. Diode laser-based cavity ring-down instrument for NO3, N2O5, NO, NO2 and O3 from aircraft. Atmos Meas Tech. 2011;4:1227–1240. doi: 10.5194/amt-4-1227-2011. [DOI] [Google Scholar]
- Wagner T, Otten C, Pfeilsticker K, Pundt I, Platt U. DOAS moonlight observation of atmospheric NO3 in the Arctic winter. Geophys Res Lett. 2000;27:3441–3444. doi: 10.1029/1999gl011153. [DOI] [Google Scholar]
- Wang X, Wang T, Yan C, Tham YJ, Xue L, Xu Z, Zha Q. Large daytime signals of N2O5 and NO3 inferred at 62 amu in a TD-CIMS: chemical interference or a real atmospheric phenomenon? Atmos Meas Tech. 2014;7:1–12. doi: 10.5194/amt-7-1-2014. [DOI] [Google Scholar]
- Wangberg I, Barnes I, Becker KH. Product and mechanistic study of the reaction of NO3 radicals with alpha-pinene. Environ Sci Technol. 1997;31:2130–2135. [Google Scholar]
- Warneke C, de Gouw JA, Goldan PD, Kuster WC, Williams EJ, Lerner BM, Brown SS, Stark H, Aldener M, Ravishankara AR, Roberts JM, Marchewka M, Bertman S, Sueper DT, McKeen SA, Meagher JF, Fehsenfeld FC. Comparison of day and nighttime oxidation of biogenic and anthropogenic VOCs along the New England coast in summer during New Enlgand Air Quality Study 2002. J Geophys Res. 2004;109:D10309. doi: 10.1029/2003JD004424. [DOI] [Google Scholar]
- Washenfelder R, Attwood A, Brock C, Guo H, Xu L, Weber R, Ng N, Allen H, Ayres B, Baumann K. Biomass burning dominates brown carbon absorption in the rural southeastern United States. Geophys Res Lett. 2015;42:653–664. [Google Scholar]
- Wayne RP, Barnes I, Biggs P, Burrows JP, Canosa-Mas CE, Hjorth J, LeBras G, Moortgat GK, Perner D, Poulet G, Restelli G, Sidebottom H. The Nitrate Radical: Physics, Chemistry, and the Atmosphere. Atmos Environ Pt A. 1991;25:1–203. [Google Scholar]
- Weaver A, Solomon S, Sanders RW, Arpag K, Miller HLJ. Atmospheric NO3 5, Off-axis measurements at sunrise: Estimates of tropospheric NO3 at 40° N. J Geophys Res. 1996;101:18605–18612. [Google Scholar]
- Weber RJ, Sullivan AP, Peltier R, Russell A, Yan B, Zheng M, de Gouw JA, Warneke C, Brock CA, Holloway JS, Atlas EL, Edgerton E. A study of secondary organic aerosol formation in the anthropogenic-influenced southeastern United States. J Geophys Res. 2007;112:D13302. doi: 10.1029/2007JD008408. [DOI] [Google Scholar]
- Werner G, Kastler J, Looser R, Ballschmiter K. Organic nitrates of isoprene as atmospheric trace compounds. Angew Chem Int Edit. 1999;38:1634–1637. doi: 10.1002/(SICI)1521-3773(19990601)38:11<1634::AID-ANIE1634>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Wille U, Becker E, Schindler RN, Lancar IT, Poulet G, Lebras G. A Discharge flow mass-spectrometric study of the reaction between the NO3 radical and isoprene. J Atmos Chem. 1991;13:183–193. doi: 10.1007/bf00115972. [DOI] [Google Scholar]
- Winer AM, Atkinson R, Pitts JNJ. Gaseous Nitrate Radical: Possible Nighttime Atmospheric Sink for Biogenic Organic Compounds. Science. 1984;224:156–158. doi: 10.1126/science.224.4645.156. [DOI] [PubMed] [Google Scholar]
- Woidich S, Froescheis O, Luxenhofer O, Ballschmiter K. El- and NCl-mass spectrometry of arylalkyl nitrates and their occurrence in urban air. Fresen J Anal Chem. 1999;364:91–99. [Google Scholar]
- Wolke R, Sehili AM, Simmel M, Knoth O, Tilgner A, Herrmann H. SPACCIM: A parcel model with detailed microphysics and complex multiphase chemistry. Atmos Environ. 2005;39:4375–4388. doi: 10.1016/J.Atmosenv.2005.02.038. [DOI] [Google Scholar]
- Wong KW, Stutz J. Influence of nocturnal vertical stability on daytime chemistry: A one-dimensional model study. Atmos Environ. 2010;44:3753–3760. [Google Scholar]
- Wood E, Bertram T, Wooldridge P, Cohen R. Measurements of N2O5, NO2, and O3 east of the San Francisco Bay. Atmos Chem Phys. 2005;5:483–491. doi: 10.5194/acp-5-483-2005. [DOI] [Google Scholar]
- Wood EC, Wooldridge PJ, Freese JH, Albrecht T, Cohen RC. Prototype for In Situ Detection of Atmospheric NO3 and N2O5 via Laser-Induced Fluorescence. Environ Sci Technol. 2003;37:5732–5738. doi: 10.1021/es034507w. [DOI] [PubMed] [Google Scholar]
- Worton DR, Mills GP, Oram DE, Sturges WT. Gas chromatography negative ion chemical ionization mass spectrometry: Application to the detection of alkyl nitrates and halocarbons in the atmosphere. J Chromatogr A. 2008;1201:112–119. doi: 10.1016/j.chroma.2008.06.019. [DOI] [PubMed] [Google Scholar]
- Wu C, Pullinen I, Andres S, Carriero G, Fares S, Goldbach H, Hacker L, Kasal T, Kiendler-Scharr A, Kleist E, Paoletti E, Wahner A, Wildt J, Mentel TF. Impacts of soil moisture on de novo monoterpene emissions from European beech, Holm oak, Scots pine, and Norway spruce. Biogeosciences. 2015;12:177–191. doi: 10.5194/bg-12-177-2015. [DOI] [Google Scholar]
- Wu T, Coeur-Tourneur C, Dhont G, Cassez A, Fertein E, He X, Chen W. Simultaneous monitoring of temporal profiles of NO3, NO2 and O3 by incoherent broadband cavity enhanced absorption spectroscopy for atmospheric applications. J Quant Spectrosc R. 2014;133:199–205. [Google Scholar]
- Xiao S, Bertram AK. Reactive uptake kinetics of NO3 on multicomponent and multiphase organic mixtures containing unsaturated and saturated organics. Phys Chem Chem Phys. 2011;13:6628–6636. doi: 10.1039/c0cp02682d. [DOI] [PubMed] [Google Scholar]
- Xie Y, Paulot F, Carter WPL, Nolte CG, Luecken DJ, Hutzell WT, Wennberg PO, Cohen RC, Pinder RW. Understanding the impact of recent advances in isoprene photooxidation on simulations of regional air quality. Atmos Chem Phys. 2013;13:8439–8455. doi: 10.5194/acp-13-8439-2013. [DOI] [Google Scholar]
- Xing J, Pleim J, Mathur R, Pouliot G, Hogrefe C, Gan CM, Wei C. Historical gaseous and primary aerosol emissions in the United States from 1990 to 2010. Atmos Chem Phys. 2013;13:7531–7549. doi: 10.5194/acp-13-7531-2013. [DOI] [Google Scholar]
- Xing J, Mathur R, Pleim J, Hogrefe C, Gan CM, Wong DC, Wei C, Gilliam R, Pouliot G. Observations and modeling of air quality trends over 1990–2010 across the Northern Hemisphere: China, the United States and Europe. Atmos Chem Phys. 2015;15:2723–2747. doi: 10.5194/acp-15-2723-2015. [DOI] [Google Scholar]
- Xiong F, McAvey KM, Pratt KA, Groff CJ, Hostetler MA, Lipton MA, Starn TK, Seeley JV, Bertman SB, Teng AP, Crounse JD, Nguyen TB, Wennberg PO, Misztal PK, Goldstein AH, Guenther AB, Koss AR, Olson KF, de Gouw JA, Baumann K, Edgerton ES, Feiner PA, Zhang L, Miller DO, Brune WH, Shepson PB. Observation of isoprene hydroxynitrates in the southeastern United States and implications for the fate of NOx. Atmos Chem Phys. 2015;15:11257–11272. doi: 10.5194/acp-15-11257-2015. [DOI] [Google Scholar]
- Xiong F, Borca CH, Slipchenko LV, Shepson PB. Photochemical degradation of isoprene-derived 4, 1-nitrooxy enal. Atmos Chem Phys. 2016;16:5595–5610. doi: 10.5194/acp-16-5595-2016. [DOI] [Google Scholar]
- Xu L, Guo H, Boyd CM, Klein M, Bougiatioti A, Cerully KM, Hite JR, Isaacman-VanWertz G, Kreisberg NM, Knote C, Olson K, Koss A, Goldstein AH, Hering SV, de Gouw J, Baumann K, Lee SH, Nenes A, Weber RJ, Ng NL. Effects of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the southeastern United States. P Natl Acad Sci USA. 2015a;112:37–42. doi: 10.1073/pnas.1417609112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Suresh S, Guo H, Weber RJ, Ng NL. Aerosol characterization over the southeastern United States using high-resolution aerosol mass spectrometry: patial, seasonal variation of aerosol compositionsources with a focus on organic nitrates. AtmosChem Phys. 2015b;15:7307–7336. doi: 10.5194/acp-15-7307-2015. [DOI] [Google Scholar]
- Yarwood G, Rao S, Yocke M, Whitten GZ. Updates to the Carbon Bond chemical mechanism: CB05 Final Report prepared for US EPA. 2005. [Google Scholar]
- Ye P, Ding X, Hakala J, Hofbauer V, Robinson ES, Donahue NM. Vapor wall loss of semi-volatile organic compounds in a Teflon chamber. Aerosol Sci Technol. 2016;50:822–834. doi: 10.1080/02786826.2016.1195905. [DOI] [Google Scholar]
- Yeh GK, Ziemann PJ. Alkyl Nitrate Formation from the Reactions of C8–C14 n-Alkanes with OH Radicals in the Presence of NOx: Measured Yields with Essential Corrections for Gas–Wall Partitioning. J Phys Chem A. 2014;118:8147–8157. doi: 10.1021/jp500631v. [DOI] [PubMed] [Google Scholar]
- Yokelson RJ, Burkholder JB, Fox RW, Talukdar RK, Ravishankara AR. Temperature Dependence of the NO3 Absorption Cross Section. J Phys Chem. 1994;98:13144–13150. [Google Scholar]
- Yvon SA, Plane JMC, Nien CF, Cooper DJ, Saltzman ES. Interaction between nitrogen and sulfur cycles in the polluted marine boundary layer. J Geophys Res. 1996;101:1379–1386. [Google Scholar]
- Zaveri RA, Berkowitz CM, Brechtel FJ, Gilles MK, Hubbe JM, Jayne JT, Kleinman LI, Laskin A, Madronich S, Onasch TB, Pekour MS, Springston SR, Thornton JA, Tivanski AV, Worsnop DR. Nighttime chemical evolution of aerosol and trace gases in a power plant plume: Implications for secondary organic nitrate and organosulfate aerosol formation, NO3 radical chemistry, and N2O5 heterogeneous hydrolysis. J Geophys Res. 2010;115:D12304. doi: 10.1029/2009jd013250. [DOI] [Google Scholar]
- Zhang Q, Jimenez JL, Canagaratna MR, Allan JD, Coe H, Ulbrich I, Alfarra MR, Takami A, Middlebrook AM, Sun YL, Dzepina K, Dunlea E, Docherty KS, De-Carlo PF, Salcedo D, Onasch TB, Jayne JT, Miyoshi T, Shimono A, Hatakeyama S, Takegawa N, Kondo Y, Scheider J, Drewnick F, Borrman S, Weimer S, Demerjian KL, Williams PI, Bower K, Bahreini R, Cottrell L, Griffin RJ, Rautiainen J, Sun JY, Zhang YM, Worsnop DR. Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically-influenced Northern Hemisphere midlatitudes. Geophys Res Lett. 2007;34:L13801. doi: 10.1029/2007GL029979. [DOI] [Google Scholar]
- Zhang X, Cappa CD, Jathar SH, McVay RC, Ensberg JJ, Kleeman MJ, Seinfeld JH. Influence of vapor wall loss in laboratory chambers on yields of secondary organic aerosol. P Natl Acad Sci USA. 2014a;111:5802–5807. doi: 10.1073/pnas.1404727111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Schwantes RH, McVay RC, Lignell H, Coggon MM, Flagan RC, Seinfeld JH. Vapor wall deposition in Teflon chambers. Atmos Chem Phys. 2015;15:4197–4214. doi: 10.5194/acp-15-4197-2015. [DOI] [Google Scholar]
- Zhang Y, Chapleski RC, Lu JW, Rockhold TH, Troya D, Morris JR. Gas-surface reactions of nitrate radicals with vinyl-terminated self-assembled monolayers. PhysChemChem Phys. 2014b;16:16659–16670. doi: 10.1039/C4CP01982B. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Morris JR. Hydrogen Abstraction Probability in Reactions of Gas-Phase NO3 with an OH-Functionalized Organic Surface. J Phys Chem C. 2015;119:14742–14747. doi: 10.1021/acs.jpcc.5b00562. [DOI] [Google Scholar]
- Zhao Z, Husainy S, Stoudemayer CT, Smith GD. Reactive uptake of NO3 radicals by unsaturated fatty acid particles. Phys Chem Chem Phys. 2011a;13:17809–17817. doi: 10.1039/c1cp21790a. [DOI] [PubMed] [Google Scholar]
- Zhao ZJ, Husainy S, Smith GD. Kinetics Studies of the Gas-Phase Reactions of NO3 Radicals with Series of 1-Alkenes, Dienes, Cycloalkenes, Alkenols, and Alkenals. J Phys Chem A. 2011b;115:12161–12172. doi: 10.1021/jp206899w. [DOI] [PubMed] [Google Scholar]
- Zheng J, Zhang R, Fortner EC, Volkamer RM, Molina L, Aiken AC, Jimenez JL, Gaeggeler K, Dommen J, Dusanter S, Stevens PS, Tie X. Measurements of HNO3 and N2O5 using ion drift-chemical ionization mass spectrometry during the MILAGRO/MCMA-2006 campaign. Atmos Chem Phys. 2008;8:6823–6838. doi: 10.5194/acp-8-6823-2008. [DOI] [Google Scholar]
- Zheng Y, Unger N, Hodzic A, Emmons L, Knote C, Tilmes S, Lamarque JF, Yu P. Limited effect of anthropogenic nitrogen oxides on secondary organic aerosol formation. Atmos Chem Phys. 2015;15:13487–13506. doi: 10.5194/acp-15-13487-2015. [DOI] [Google Scholar]
- Zhou S, Wenger JC. Kinetics and products of the gas-phase reactions of acenaphthene with hydroxyl radicals, nitrate radicals and ozone. Atmos Environ. 2013;72:97–104. doi: 10.1016/j.atmosenv.2013.02.044. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.