

Abstract
The mammalian brain is a very complex organ containing an estimated 200 billion cells in humans. Therefore, studying human brain development has become very challenging given all the data that are available from different approaches, notably genetic studies.
Recent pluripotent stem cell methods have given rise to the possibility of modeling neurodevelopmental diseases associated with genetic defects. Fibroblasts from patients have been reprogrammed into pluripotent stem cells to derive appropriate neuronal lineages. They specifically include different subtypes of cortical neurons that are at the core of human-specific cognitive abilities. The use of neurons derived from induced pluripotent stem cells (iPSC) has led to deciphering convergent and pleiotropic neuronal synaptic phenotypes found in neurodevelopmental disorders such as autism spectrum disorders (ASD) and their associated syndromes. In addition to these initial studies, remarkable progress has been made in the field of stem cells, with the major objective of reproducing the in vivo maturation steps of human neurons. Recently, several studies have demonstrated the ability of human progenitors to respond to guidance cues and signals in vivo that can direct neurons to their appropriate sites of differentiation where they become fully mature neurons.
We provide a brief overview on research using human iPSC in ASD and associated syndromes and on the current understanding of new theories using the re-implantation of neural precursors in mouse brain.
Keywords: Human induced pluripotent stem cells, Autism, Developmental disorders, Transplantation, Brain circuits
Background
The use of induced pluripotent stem cells (iPSC) has provided new opportunities for analyzing brain development and the consequences of its dysfunctions in neurodevelopmental disorders. The iPSC approach has been particularly useful for neurodevelopmental diseases for which major genes are considered responsible. One important aspect resides in the fact that the reprogrammed iPSC studied so far carry single genetic deficits which were initially identified in the genomes of patients. This is the case for autism spectrum disorders (ASD) and their related disorders, which include Rett syndrome, Timothy syndrome, fragile X syndrome, and Phelan-McDermid syndrome (PMS). Causative genes are mostly related to synaptic functions. Among others, traditional approaches include large-scale genomic data analysis and engineered animal models [1–3]. The discovery of human iPSC has enabled the analysis of neuronal phenotypes after the derivation of patients’ somatic cells into neurons. One limitation of such an in vitro approach is the inability to grow the cells for periods long enough to reproduce the postnatal development and maturation of human iPSC-derived neurons. Alternative techniques are based on the use of neural progenitors that are patterned after their re-implantation in mouse brain to undergo differentiation via specific neural pathways in vivo.
In vitro use of human iPSC-derived neurons in neurodevelopmental disorders: the case of syndromic and non-syndromic forms of ASD
A common understanding of the pathogenesis of neurodevelopmental disorders, including ASD, has still not been achieved. This remains an important issue, as it is a prerequisite for the development of pharmacological drugs for the treatment of the core symptoms of such disorders. Reprogramming of iPSC from patients into neuronal cell types was first used to further elucidate the phenotypes related to pathologies such as Alzheimer’s disease [4], Parkinson’s disease [5], epilepsy [6], and schizophrenia [7, 8]. Peripheral neurodegenerative disorders such as amyotrophic lateral sclerosis [9] have also been investigated. For all these pathological conditions, human iPSC have been reprogrammed into selective neuronal cell types by considering the neuronal phenotypes that are damaged as predicted by available animal models and clinical investigations in patients. The iPSC model has enabled further insights into the cellular and molecular mechanisms that are affected during brain development. This model was first developed in the case of monogenic diseases such as Rett syndrome, fragile X syndrome, Timothy syndrome, PMS, and various forms of schizophrenia that display symptoms in common with ASD. The common features between such monogenic diseases and ASD include cognitive dysfunctions with mental retardation and dysmorphia. Other features comprise epilepsy (atypical forms of Rett syndrome), cardiac dysfunction (Timothy syndrome), motor coordination, and sensorial hypersensitivity (fragile X syndrome). Studies using iPSC technology have been focused both on neural progenitors and on mature neurons derived mostly from the fibroblasts (or less often from blood) of patients. The identification of phenotypes in human iPSC-derived neurons was first studied in four syndromic forms of ASD with well-known identified causal genes.
Rett syndrome is a severe X-linked neurodevelopmental disorder. The main form of this disorder is characterized by defects in the MECP2 gene coding for the transcriptional regulator methyl CpG binding protein 2, which is expressed in a wide variety of tissues, including the brain. The most extensive work using the iPSC model has been devoted to Rett syndrome, with the phenotypic characterization of these cell lines obtained from patients’ fibroblasts. Decreased cell soma size and neuritogenesis [10], reduced expression of the adhesion molecule L1 [10], and synaptic alterations [11, 12] have been observed by using this model, supporting the expected defects in neuronal connectivity.
Timothy syndrome is a very rare autosomal dominant disorder which results from mutations in the CACNA1C gene coding for the alpha-1 subunit of the L-type voltage-gated calcium channel Cav1.2. To date, only a few studies have analyzed the neuronal phenotypes of human iPSC-derived neurons, which have shown specific alterations in calcium signaling [13] and dendritic plasticity [14]. One of the initial studies reported a reduced number of cells expressing the DNA-binding protein SATB2 [15]. Satb2 regulates the fate of upper and deeper layers of cortical neurons as shown in mouse brain models [16]. It is therefore reasonable to suggest the existence of deleterious effects on neural development which are due to altered calcium signaling in parallel with the absence of SATB2 protein, and more specifically on cortico-cortical connections. Nevertheless, it should be noted that in vitro developmental neuronal patterns differ from in vivo ones, especially when the deleterious mutations target pleiotropic genes, such as CACNA1C, that coordinate organ functions in a non-independent manner.
PMS is a neurodevelopmental disorder strongly associated with ASD that is caused by a deletion of the SHANK3 gene at the 22q13 locus, identified as a 22q13 deletion syndrome. Shcheglovitov and colleagues [17] used iPSC-derived cortical neurons from two patients and observed significant deficits in the excitatory transmission of reprogrammed neurons which were rescued by IGF1 exposure.
Fragile X syndrome is considered as one of the most common cause of syndromic ASD. This syndrome results from an expansion of a CGG repeat within the fragile X mental retardation 1 (FMR1) gene on the X chromosome. This gene is required for neuronal development and a deficiency in its corresponding protein leads to altered neuronal connectivity. It has been difficult so far to find a model to study this syndrome using iPSC technology. The more recent study by Doers and colleagues [18] clearly demonstrates reduced neurite outgrowth in neurons from patients. Reduced neurite outgrowth may alter axonal growth as well as the differentiation of presynaptic and postsynaptic components, and consequently may lead to alteration of short-range and long-range neuronal connectivity.
ASD are neurodevelopmental disorders characterized by deficits in social cognition, communication, and behavior as well as moderate to severe mental retardation. ASD have a complex genetic basis, with hundreds of identified candidate genes which cannot be individually responsible for ASD clinical features and cellular phenotypes. This complexity has led to considerable effort to identify functional pathways that may reveal cellular connections between the candidate genes. Among these, the glutamatergic pathway includes genes that, once mutated, are thought to be responsible for both syndromic and non-syndromic ASD [19]. Indeed, ASD present with alterations in the brain cortex and its development and morphological organization and the brain’s short-range and long-range connectivity [20]. For example, an abnormal connectivity between the cerebellum and the entire cerebral cortex has recently been shown using fMRI brain imaging [21]. The development and maintenance of neuronal networks depend on differentiation of neurons and axonal outgrowth as well as dendrite branching. We have shown that these processes are controlled differently by cell-adhesion molecules such as contactin 4 (CNTN4), contactin 5 (CNTN5), and contactin 6 (CNTN6) proteins [22]. Among the genes which code for CNTN4–6, we have demonstrated that CNTN6 is a susceptibility gene for ASD [23]. Alterations in the formation of neural networks that are controlled by CNTN6 may underlie the cognitive, sensory, and motor deficits that we observe in autistic patients carrying CNTN6 coding variants [22].
Modeling shankopathies using human iPSC-derived neurons
Among the mutated genes in ASD, the SHANK genes offer one of the best possibilities to explore the core symptoms of these disorders by analyzing the cellular defects that are associated with mutations in them. The major SHANK family genes involved in ASD include SHANK1, SHANK2, and SHANK3. SHANK genes encode scaffolding proteins present at the postsynaptic density of excitatory synapses. The first report on specific SHANK3 mutations was provided by our laboratory [24]. Further investigations have clearly demonstrated the role of all SHANK genes in ASD [1]. The involvement of SHANK3 as a causal gene in ASD was observed in 0.7% of patients in large cohorts, with different types of mutations including microdeletions, point mutations, and stop mutations [1]. SHANK3 is also associated with behavioral phenotypes of the 22q13 deletion syndrome [25, 26]. Human iPSC have been generated from patients with heterozygous deletions of chromosome 22q13.3. Derived neurons displayed reduced SHANK3 expression and major defects in their excitatory but not inhibitory synaptic transmission [17]. These findings strongly suggest that a disruption of the excitatory/inhibitory balance occurs in the brain of patients with PMS. Kathuria and colleagues [27] differentiated iPSC from two patients with ASD carrying microdeletions of SHANK3 into either cortical or olfactory placodal neurons. These authors showed that placodal neurons had a reduced number of synapses compared with control neurons. The young postmitotic neurons also had reduced cell bodies with higher neuronal arborization. These two developmental phenotypes were specific to placodal neurons and were not observed in iPSC-derived cortical neurons. The morphogenetic deficits were rescued by genome editing techniques [27]. The iPSC model has also been used in patients with ASD presenting de novo point truncating mutations in the SHANK3 gene; under the experimental conditions, pyramidal excitatory neurons accounted for more than 80% of cortical neurons [28]. In a subsequent study and under similar experimental conditions, these authors were able to evaluate and reverse the neuronal dysfunctions in two individuals with de novo point mutations in the SHANK3 gene [29], namely decreased neurite length and branching and spontaneous calcium oscillations. In addition, the authors found that lithium as well as valproic acid and fluoxetine increased the SHANK3 mRNA and protein levels in a concentration-dependent manner [29]. SHANK3 is expressed at neuronal excitatory synapses and forms protein complexes [30] which may contribute to ASD phenotypes when they are broken up. SHANK3 interactomes are expressed at single-spine level [31], which also receive neuronal excitation inputs. For the analysis of spine densities, most studies using animal models have been performed in two dimensions, which may not totally reflect the asymmetric morphology of diverse spine categories. Regarding ASD and human iPSC models, none of the published data have so far described a quantitative analysis of spinogenesis in patients with SHANK3 mutations. Using the same protocol and human iPSC lines described by Darville and colleagues [29], we established a method which allows the quantification of spine morphology in three dimensions [32]. The shape of dendritic spines and volume vary according to the stage of their maturation. Mature spines are usually characterized by a larger head and a thin neck, whereas immature spines are thinner with a poorly defined head with small postsynaptic densities. We found the latter category to be predominant in human iPSC-derived pyramidal neurons from individuals without ASD-related disorders (unpublished observations). We are undertaking an extensive analysis of spinogenesis in a subset of patients carrying different de novo SHANK3 point mutations to evaluate the inter-individual variability. This aspect is still lacking so far, since most studies have been conducted using cells reprogrammed from a few patients only.
Main pitfalls using human iPSC cells in vitro
iPSC culture systems can offer an almost unlimited source of neurons for fundamental research on the first stages of neural development and for pharmacological screening. However, this model presents some limitations. Indeed, reprogramming of somatic cells through the expression of the four Yamanaka transcription factors, OCT4, KLF4, SOX2, and cMYC, has been shown to be asynchronous and have low efficiency. The rate of cell reprogramming also depends on donor cell types and culture conditions [33]. Different models have been proposed to analyze the reprogramming processes and the roles of transcription factors and epigenetic regulators [34]. To circumvent these problems, various methods have been developed in order to study reprogramming dynamics under more unified frameworks [35]. Alternative reprogramming protocols have also been proposed that are based on the use of synthetic capped mRNAs containing modified nucleobases (mod-mRNA) [36]. However, these methods do not seem to be efficient enough for the accurate reprogramming of human primary fibroblasts. A new, optimized method [37] which combines mod-mRNA with reprogramming factors together with improved cell culture conditions is encouraging and seems to provide an alternative approach for reprogramming of human fibroblasts in the case of ASD and related syndromes. New protocols have also been developed for improving the differentiation and maturation of iPSC-derived neurons [38].
When analyzing data from human iPSC-derived neurons in vitro, focusing on two-dimensional cell cultures often derived from one single cell type at a time may lead to an underestimation of cellular defects. For example, recent findings using the human iPSC model clearly indicate that astrocytes play important role in synaptogenesis and neuronal morphology [39]. The low efficiency of cell reprogramming observed so far has rendered the simultaneous derivation of distinct isogenic cell types from the same human iPSC much more difficult. The co-culture of several isogenic cell types, including distinct neuronal and microglial cells, would represent a significant improvement for studying ASD and its related disorders.
Finally, in vitro systems do not allow the reproduction of global cellular homeostasis and cell orientation and projections within the distinct cortical layers. It is also not clear to what extent the immature neurons that are produced in vitro recapitulate the diverse steps of neurogenesis. Interestingly, Imaizumi and colleagues [40] developed specific culture systems to control the identity of derived neuronal cells along the anteroposterior and dorsoventral axes. New protocols including three-dimensional culture systems [41] and brain organoids [42, 43] have been developed for iPSC models. Brain organoids consist of cellular aggregates derived from human embryonic stem cells (ESC) and iPSC. They may represent new in vitro systems with an oriented cell organization. Depending on the cell line and the number of passages, however, the brain organoids can be variable. Consequently, the development of brain organoids remains a challenging process due to the complexity of neuronal phenotypes and circuitry. So far, no method can provide a full reproduction of the phenotypic brain environment in vivo and the exact characteristics of developmental disorders due to the absence of a wide variety of conditions, including vascularization, nutrients, and specific developmental cues and signals. Human brain organoids are reviewed elsewhere in more detail [44, 45].
Reconstruction of brain circuitry using neural transplants generated from iPSC
As discussed above, the main features of brain cortical development cannot be reproduced using in vitro models. Neurodevelopmental disorders are currently associated with cognitive dysfunctions, with the neocortex underlying high cognitive functions in humans. For this reason, cortical neuronal subtypes such as pyramidal glutamatergic cells have been predominantly used in vitro [15, 17, 28, 29, 46].
For neurodevelopmental disorders, including ASD, defects in neuronal connectivity have been associated with increased local and reduced long-range connectivity as discussed above. One interesting aspect is the fact that an early neurodevelopmental dysfunction in single subcortical regions may modify the cerebral networks underlying early sensory-motor impairments and social deficits, including those observed in ASD [47]. The reconstruction of brain circuitry can be partially achieved by transplantation of human neurons generated from iPSC into mouse brain [48–52]. Since the early studies, significant progress has been made. Espuny-Camacho and colleagues [48] showed that ESC and iPSC can recapitulate corticogenesis and lead to sequential generation of functional pyramidal cells when grafted into mouse brain in vivo. In addition, these authors have demonstrated that, with regard to differentiation and connectivity, transplanted cells extend their ramifications over several months and constitute functional synapses with the host neuronal circuits [48]. Transplantation of neural precursors in mouse brain can be performed without a preliminary period of in vitro culture to allow cells to fully differentiate. Under such conditions, a post-characterization of cell phenotypes is necessary to clearly identify neurons from other iPSC-derived cells, such as oligodendrocytes and astrocytes. One key aspect resides in the fact that cortical pyramidal neurons derived from mouse or human iPSC follow species-specific maturation processes after their transplantation into mouse brain. Michelson and colleagues [49] also demonstrated that maturation of human ESC/iPSC takes 9 months post-transplantation and maintains the chronology of developmental steps for a given species. Using a similar approach, these authors derived neurons from mouse ESC in vitro which could be identified as those from visual cortex. The resulting neurons were then transplanted successfully into lesioned adult mouse visual cortex [49]. A possible rescue of the damaged pathways, including long-range and reciprocal axonal projections with appropriate synapses, was also observed [49]. Moreover, electrophysiological recordings were used to show that grafted neurons were responsive to visual stimuli [49]. Such an approach has not been tested with human iPSC. Nagashima and colleagues [50] developed a method consisting of an in utero transplantation system of pluripotent ESC based on a mild dissociation of adherens junctions in neuroepithelial tissue. Transplanted cells migrated from the subventricular zone to the cortical plate and, after only several days, presented the morphology of immature pyramidal cells [50]. To our knowledge, this method has not yet been used for the iPSC model. In their recent study, Falkner and colleagues [51] used chronic in vivo two-photon imaging to study the integration of mouse transplanted neurons into existing circuits of the mouse visual cortex [51]. After 2–3 months, the transplanted neurons were fully integrated, with functional properties indistinguishable from those of the pre-existing neuronal networks [51]. Functional imaging of grafted neurons into mouse brain represents an advantage compared to anatomical methods. This approach has not yet been used in the case of human neurons derived from patients with neurodevelopmental disorders. Figure 1 illustrates disease modeling using both in vitro and in vivo human iPSC models, independently or in a complementary manner. Figure 1 also represents the possible introduction of a genetic mutation by genome editing techniques such as CRISPR/Cas9 [53, 54] and/or the reversion of cellular phenotypic alterations found in monogenic disorders. The recent work by Wuttke and colleagues [52] is promising. Using an optogenetics-based electrophysiology approach, these authors demonstrated that developmentally “primed” cortical neurons can maintain their precise pattern of differentiation and regional connectivity after transplantation, with the development of appropriate long-distance projections and synapses. Reconstruction of the neonatal circuitry may be possible by the micro-transplantation of primed cortical neurons. Finally, methods for volume imaging of optically transparent brain tissues should offer new possibilities for the analysis of brain circuits.
Fig. 1.
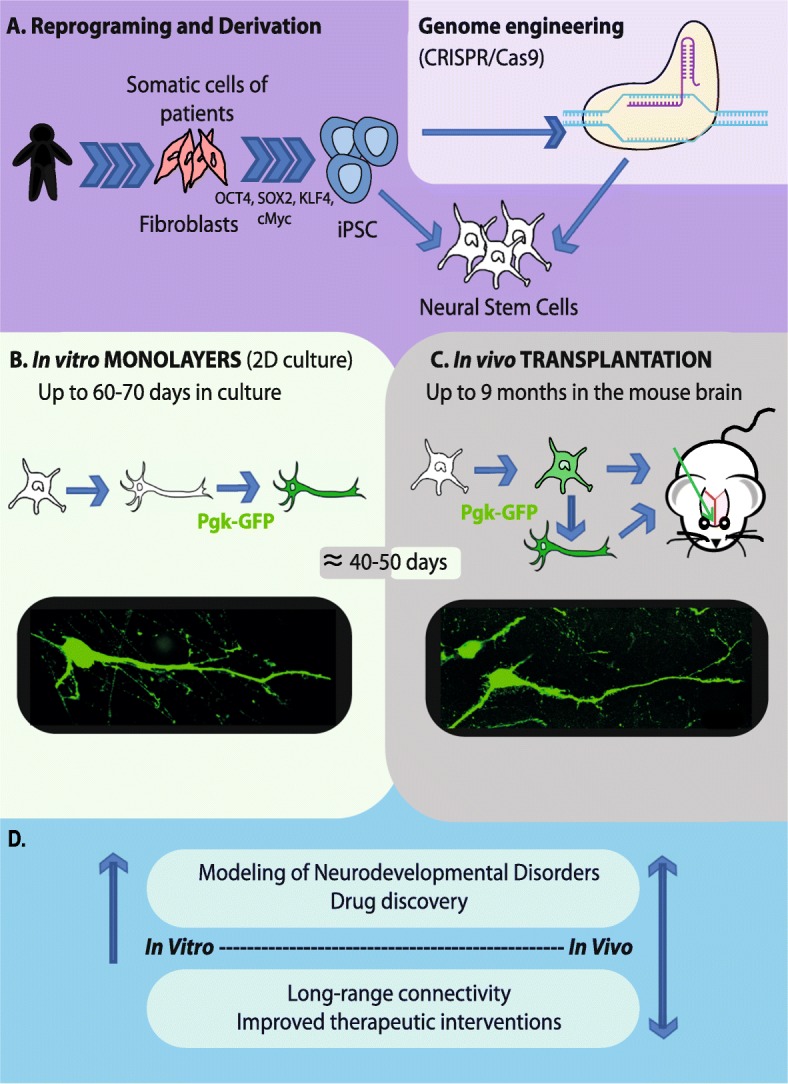
Main experimental designs for human iPSC models of monogenic neurodevelopmental disorders. a Patient’s specific iPSC are derived from fibroblasts using the four Yamanaka’s factors. Genome engineering using the CRISPR/Cas9 method allows the reversion of phenotypic defects by re-introducing the wild-type allele into the genome of iPSC lines. The CRISPR/Cas9 method also allows introduction of the mutation under study directly into the genome of control iPSC lines in order to compare visually similar phenotypes to those seen in the iPSC from patients. Both non-edited and isogenic iPSC are differentiated into the affected neuronal subtypes, mostly pyramidal cortical neurons in the case of cognitive disorders. b Viable neurons can be maintained in culture up to 70 days after the differentiation of neural stem cells (NSC). The transduction of neuronal cells with a green fluorescent protein (GFP)-lentivirus allows their visualization and phenotypic characterization using fluorescence microscopy. A GFP-labeled pyramidal neuron 40-45 days after the differentiation of NSC. c Neuronal precursors or neurons fully differentiated in vitro are transplanted into the brain of mouse neonates. The visualization of fluorescent neurons is done using fluorescent microscopy on brain slices. A transplanted GFP-labeled pyramidal neuron is illustrated at 40–50 days post-injection (picture from our experiments). Mice are maintained up to 9 months of age after grafting. d Comparative information and main therapeutics perspectives provided by the use of iPSC-derived neurons in vitro vs in vivo
The recent work by Mansour and colleagues is quite [54] promising. These authors have developed a method which consists of grafting human brain organoids into the adult mouse brain. They observed vascularized and functional intra-graft neuronal networks as well as graft-to-host synaptic connectivity. Together with ongoing technological improvements [45], grafting human brain organoids into mouse brain should represent an accurate alternative method to model a wide range of neurodevelopmental disorders, including ASD.
Conclusions
The effective integration of transplanted cells that mature into neuronal subtypes together with appropriate long-range connectivity within critical regions such as brain cortex should allow the functional reconstruction of cortical circuitry over time. In addition, advances in genome editing technologies allow the genetic manipulation of iPSC in a site-specific manner. Combined with a circuit level analysis, such approaches should provide new plausible models to study human neurodevelopmental diseases and additional opportunities for future drug development.
Acknowledgements
Data in Fig. 1 used samples from the NINDS Human Genetics Resource Center DNA and Cell Line Repository; NINDS repository sample numbers corresponding to the samples used are GM01869 and GM04603.
Funding
The French Ministry of Education provided the funding for AV’s PhDs.
Abbreviations
- ASD
Autism spectrum disorders
- CNTN
Contactin
- ESC
Embryonic stem cell
- GFP
Green fluorescent protein
- iPSC
Induced pluripotent stem cell
- NSC
Neural stem cell
- PMS
Phelan-McDermid syndrome
Authors’ contributions
AV and ICT were responsible for conceptualization of the review article. AV designed the figure. AV and ICT wrote the manuscript and ICT finalized the manuscript. Both authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
The graphic figure in this article is original and is subject to the copyright policy of the journal.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Aline Vitrac, Email: aline.vitrac@pasteur.fr.
Isabelle Cloëz-Tayarani, Phone: 33 (0)1 45 68 88 04, Email: isabelle.cloez-tayarani@pasteur.fr.
References
- 1.Leblond CS, Nava C, Polge A, Gauthier J, Huguet G, Lumbroso S, et al. Meta-analysis of SHANK mutations in autism Spectrum disorders: a gradient of severity in cognitive impairments. PLoS Genet. 2014;10:e1004580. doi: 10.1371/journal.pgen.1004580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huguet G, Benabou M, Bourgeron T. The genetics of autism spectrum disorders. In: Sassone-Corsi P, Christen Y, editors. A time for metabolism and hormones. 2016. pp. 101–129. [PubMed] [Google Scholar]
- 3.Ferhat AT, Halbedl S, Schmeisser MJ, Kas MJ, Bourgeron T, Ey E. Behavioural phenotypes and neural circuit dysfunctions in mouse models of autism Spectrum disorder. Adv Anat Embryol Cell Biol. 2017;224:85–101. doi: 10.1007/978-3-319-52498-6_5. [DOI] [PubMed] [Google Scholar]
- 4.Lee HK, Velazquez Sanchez C, Chen M, Morin PJ, Wells JM, Hanlon EB, et al. Three dimensional human neuro-spheroid model of Alzheimer’s disease based on differentiated induced pluripotent stem cells. PLoS One. 2016;11:e0163072. doi: 10.1371/journal.pone.0163072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fernandez-Santiago R, Carballo-Carbajal I, Castellano G, Torrent R, Richaud Y, Sanchez-Danes A, et al. Aberrant epigenome in iPSC-derived dopaminergic neurons from Parkinson’s disease patients. EMBO Mol Med. 2015;7:1529–1546. doi: 10.15252/emmm.201505439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parent JM, Anderson SA. Reprogramming patient-derived cells to study the epilepsies. Nat Neurosci. 2015;18(3):360–66. 10.1038/nn.3944. [DOI] [PMC free article] [PubMed]
- 7.Wen Z, Nguyen HN, Guo Z, Lalli MA, Wang X, Su Y, et al. Synaptic dysregulation in a human iPS cell model of mental disorders. Nature. 2015;515:414–418. doi: 10.1038/nature13716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wright R, Réthelyi JM, Gage FH. Enhancing induced pluripotent stem cell models of schizophrenia. JAMA Psychiatry. 2014;71:334–335. doi: 10.1001/jamapsychiatry.2013.4239. [DOI] [PubMed] [Google Scholar]
- 9.Jaiswal MK. Therapeutic opportunities and challenges of induced pluripotent stem cells-derived motor neurons for treatment of amyotrophic lateral sclerosis and motor neuron disease. Neural Regeneration Research. 2017;12:723-736. 10.4103/1673-5374.206635. [DOI] [PMC free article] [PubMed]
- 10.Yoo M, Carromeu C, Kwon O, Muotri A, Schachner M. Biochemical and biophysical research communications the L1 adhesion molecule normalizes neuritogenesis in Rett syndrome-derived neural precursor cells. Biochem Biophys Res Commun. 2017;494:504–510. doi: 10.1016/j.bbrc.2017.10.073. [DOI] [PubMed] [Google Scholar]
- 11.Marchetto MCN, Carromeu C, Acab A, Yu D, Yeo G, Mu Y, et al. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell. 2010;143:527–539. doi: 10.1016/j.cell.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patriarchi T, Amabile S, Frullanti E, Landucci E, Lo Rizzo C, Ariani F, et al. Imbalance of excitatory/inhibitory synaptic protein expression in iPSC-derived neurons from FOXG1 patients and in foxg1 mice. Eur J Hum Genet. 2016;24:871–880. doi: 10.1038/ejhg.2015.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tian Y, Voineagu I, Paşca SP, Won H, Chandran V, Horvath S, et al. Alteration in basal and depolarization induced transcriptional network in iPSC derived neurons from Timothy syndrome. Genome Med. 2014;6:1–16. doi: 10.1186/s13073-014-0075-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krey JF, Paşca SP, Shcheglovitov A, Yazawa M, Schwemberger R, Rasmusson R, et al. Timothy syndrome is associated with activity-dependent dendritic retraction in rodent and human neurons. Nat Neurosci. 2013;16:201–209. doi: 10.1038/nn.3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paşca SP, Portmann T, Voineagu I, Yazawa M, Shcheglovitov A, Paşca AM, et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat Med. 2011;17:1657–1662. doi: 10.1038/nm.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leone DP, Heavner WE, Ferenczi EA, Dobreva G, Huguenard JR, Grosschedl R, et al. Satb2 regulates the differentiation of both callosal and subcerebral projection neurons in the developing cerebral cortex. Cereb Cortex. 2015;25:3406–3419. doi: 10.1093/cercor/bhu156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shcheglovitov A, Shcheglovitova O, Yazawa M, Portmann T, Shu R, Sebastiano V, et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature. 2013;503:267–271. doi: 10.1038/nature12618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doers ME, Musser MT, Nichol R, Berndt ER, Baker M, Gomez TM, et al. iPSC-derived forebrain neurons from FXS individuals show defects in initial neurite outgrowth. Stem Cells Dev. 2014;23:1777–1787. doi: 10.1089/scd.2014.0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rojas DC. The role of glutamate and its receptors in autism and the use of glutamate receptor antagonists in treatment. J Neural Transm. 2014;121:891–905. doi: 10.1007/s00702-014-1216-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khan S, Gramfort A, Shetty NR, Kitzbichler MG, Ganesan S, Moran JM, et al. Local and long-range functional connectivity is reduced in concert in autism spectrum disorders. Proc Natl Acad Sci U S A. 2013;110:3107–3112. doi: 10.1073/pnas.1214533110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khan AJ, Nair A, Keown CL, Datko MC, Lincoln AJ, Müller RA. Cerebro-cerebellar resting-state functional connectivity in children and adolescents with autism spectrum disorder. Biol Psychiatry. 2015;78:625–634. doi: 10.1016/j.biopsych.2015.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mercati O, Danckaert A, Andre-Leroux G, Bellinzoni M, Gouder L, Watanabe K, et al. Contactin 4, −5 and −6 differentially regulate neuritogenesis while they display identical PTPRG binding sites. Biol Open. 2013;2:324–334. doi: 10.1242/bio.20133343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mercati O, Huguet G, Danckaert A, André-Leroux G, Maruani A, Bellinzoni M, et al. CNTN6 mutations are risk factors for abnormal auditory sensory perception in autism spectrum disorders. Mol Psychiatry. 2016;22:625–633. doi: 10.1038/mp.2016.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bourgeron T. Current knowledge on the genetics of autism and propositions for future research. C R Biol. 2016;339:300–307. doi: 10.1016/j.crvi.2016.05.004. [DOI] [PubMed] [Google Scholar]
- 25.Harony-Nicolas H, De Rubeis S, Buxbaum JD. Phelan McDermid syndrome: from genetic discoveries to animal models and treatments. J Child Neurol. 2015;30:1861–1870. doi: 10.1177/0883073815600872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tabet AC, Rolland T, Ducloy M, Lévy J, Buratti J, Mathieu A, et al. A framework to identify contributing genes in patients with Phelan-McDermid syndrome. NPJ Genomic Med. 2017;2:32. doi: 10.1038/s41525-017-0035-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kathuria A, Nowosiad P, Jagasia R, Aigner S, Taylor RD, Andreae LC, et al. Stem cell-derived neurons from autistic individuals with SHANK3 mutation show morphogenetic abnormalities during early development. Mol Psychiatry. 2017;23:735–746. doi: 10.1038/mp.2017.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boissart C, Poulet A, Georges P, Darville H, Julita E, Delorme R, et al. Differentiation from human pluripotent stem cells of cortical neurons of the superficial layers amenable to psychiatric disease modeling and high-throughput drug screening. Transl Psychiatry. 2013;3:e294. doi: 10.1038/tp.2013.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Darville H, Poulet A, Rodet-Amsellem F, Chatrousse L, Pernelle J, Boissart C, et al. Human pluripotent stem cell-derived cortical neurons for high throughput medication screening in autism: a proof of concept study in SHANK3 Haploinsufficiency syndrome. EBioMedicine. 2016;9:293–305. doi: 10.1016/j.ebiom.2016.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee Y, Kang H, Lee B, Zhang Y, Kim Y, Kim S, et al. Integrative analysis of brain region-specific Shank3 Interactomes for understanding the heterogeneity of neuronal pathophysiology related to SHANK3 mutations. Front Mol Neurosci. 2017;10:1–13. doi: 10.3389/fnmol.2017.00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sarowar T, Grabrucker AM. Actin-dependent alterations of dendritic spine morphology in Shankopathies. Neural Plast. 2016;2016:8051861. doi: 10.1155/2016/8051861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gouder L, Tinevez J-Y, Goubran-Botros H, Benchoua A, Bourgeron T, Cloëz-Tayarani I. Three-dimensional quantification of dendritic spines from pyramidal neurons derived from human induced pluripotent stem cells. J Vis Exp. 2015;(104):1–8. 10.3791/53197. https://www.jove.com/video/53197/three-dimensional-quantification-dendritic-spines-from-pyramidal. [DOI] [PMC free article] [PubMed]
- 33.Nefzger CM, Rossello FJ, Chen J, Liu X, Knaupp AS, Firas J, et al. Cell type of origin dictates the route to pluripotency. Cell Rep. 2017;21:2649–2660. doi: 10.1016/j.celrep.2017.11.029. [DOI] [PubMed] [Google Scholar]
- 34.Theunissen TW, Jaenisch R. Molecular control of induced pluripotency. Cell Stem Cell. 2014;14:720–734. doi: 10.1016/j.stem.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu LL, Brumbaugh J, Bar-Nur O, Smith Z, Stadtfeld M, Meissner A, et al. Probabilistic modeling of reprogramming to induced pluripotent stem cells. Cell Rep. 2016;25:3395–3406. doi: 10.1016/j.celrep.2016.11.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Warren L, Manos PD, Ahfeldt T, Loh Y, Li H, Daley Q, et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells using synthetic modified mRNA. Cell Stem Cell. 2010;7:618–630. doi: 10.1016/j.stem.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kogut I, McCarthy SM, Pavlova M, Astling DP, Chen X, Jakimenko A, et al. High-efficiency RNA-based reprogramming of human primary fibroblasts. Nat Commun. 2018;9:745. doi: 10.1038/s41467-018-03190-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gunhanlar N, Shpak G, van der Kroeg M, Gouty-Colomer LA, Munshi ST, Lendemeijer B, et al. A simplified protocol for differentiation of electrophysiologically mature neuronal networks from human induced pluripotent stem cells. Mol Psychiatry. 2018;23:1336–1344. doi: 10.1038/mp.2017.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Russo FB, Freitas BC, Pignatari GC, Fernandes IR, Sebat J, Muotri AR, et al. Modeling the interplay between neurons and astrocytes in autism using human induced pluripotent stem cells. Biol Psychiatry. 2017;83:569–578. doi: 10.1016/j.biopsych.2017.09.021. [DOI] [PubMed] [Google Scholar]
- 40.Imaizumi K, Sone T, Ibata K, Fujimori K, Yuzaki M, Akamatsu W, et al. Controlling the regional identity of hPSC-derived neurons to uncover neuronal subtype specificity of neurological disease phenotypes. Stem Cell Rep. 2015;5:1010–1022. doi: 10.1016/j.stemcr.2015.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park J, Wetzel I, Marriott I, Dréau D, D’Avanzo C, Kim DY, et al. A 3D human triculture system modeling neurodegeneration and neuroinflammation in Alzheimer’s disease. Nat Neurosci. 2018;21:941–951. doi: 10.1038/s41593-018-0175-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paşca AM, Sloan SA, Clarke LE, Tian Y, Makinson CD, Huber N, et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat Methods. 2015;12:671–678. doi: 10.1038/nmeth.3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Birey F, Andersen J, Makinson CD, Islam S, Wei W, Huber N, et al. Assembly of functional forebrain spheroids from human pluripotent cells. Nature. 2017;545:54–59. doi: 10.1038/nature22330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heide M, Huttner WB, Mora-Bermúdez F. Brain organoids as models to study human neocortex development and evolution. Curr Opin Cell Biol. 2018;55:8–16. doi: 10.1016/j.ceb.2018.06.006. [DOI] [PubMed] [Google Scholar]
- 45.Wang H. Modeling neurological diseases with human brain organoids. Front Synaptic Neurosci. 2018;10:1–14. doi: 10.3389/fnsyn.2018.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chailangkarn T, Muotri AR. Modeling Williams syndrome with induced pluripotent stem cells. Neurogenesis. 2017;4:e1283187. doi: 10.1080/23262133.2017.1283187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Piven J, Elison JT, Zylka MJ. Toward a conceptual framework for early brain and behavior development in autism. Mol Psychiatry. 2017;22:1385–1394. doi: 10.1038/mp.2017.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Espuny-Camacho I, Michelsen K a, Gall D, Linaro D, Hasche A, Bonnefont J, et al. Pyramidal neurons derived from human pluripotent stem cells integrate efficiently into mouse brain circuits. In Vivo Neuron. 2013;77:440–456. doi: 10.1016/j.neuron.2012.12.011. [DOI] [PubMed] [Google Scholar]
- 49.Michelsen KA, Acosta-Verdugo S, Benoit-Marand M, Espuny-Camacho I, Gaspard N, Saha B, et al. Area-specific reestablishment of damaged circuits in the adult cerebral cortex by cortical neurons derived from mouse embryonic stem cells. Neuron. 2015;85:982–997. doi: 10.1016/j.neuron.2015.02.001. [DOI] [PubMed] [Google Scholar]
- 50.Nagashima F, Suzuki IK, Shitamukai A, Sakaguchi H, Iwashita M, Kobayashi T, et al. Novel and robust transplantation reveals the acquisition of polarized processes by cortical cells derived from mouse and human pluripotent stem cells. Stem Cells Dev. 2014;23:2129–2142. doi: 10.1089/scd.2013.0251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Falkner S, Grade S, Dimou L, Conzelmann KK, Bonhoeffer T, Götz M, et al. Transplanted embryonic neurons integrate into adult neocortical circuits. Nature. 2016;539:248–253. doi: 10.1038/nature20113. [DOI] [PubMed] [Google Scholar]
- 52.Wuttke TV, Markopoulos F, Padmanabhan H, Wheeler AP. Developmentally primed cortical neurons maintain fidelity of differentiation and establish appropriate functional connectivity after transplantation. Nat Neurosci. 2018;21:517–529. doi: 10.1038/s41593-018-0098-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Park CY, Halevy T, Lee DR, Sung JJ, Lee JS, Yanuka O, et al. Reversion of FMR1 methylation and silencing by editing the triplet repeats in fragile X iPSC-derived neurons. Cell Rep. 2015;13:234–241. doi: 10.1016/j.celrep.2015.08.084. [DOI] [PubMed] [Google Scholar]
- 54.Mansour AA, Goncalves JT, Bloyd CW, Li H, Fernandes S, Quang D, Johnston S, Parylak SL, Jin X, Gage FH. An in vivo model of functional and vascularized human brain organoids. Nature Biotechnology, 2018;36:432–441. [DOI] [PMC free article] [PubMed]