

Abstract
BACKGROUND
Variation in response to the most commonly used class of asthma controller medication, inhaled corticosteroids (ICS), presents a serious challenge in asthma management, particularly for steroid-resistant patients who display little or no response to treatment.
OBJECTIVE
We applied a systems-biology approach to primary clinical and genomic data to identify and validate genes that modulate steroid response in children with asthma.
METHODS
We selected 104 ICS-treated asthmatic non-Hispanic white children and determined a Steroid Responsiveness Endophenotype (SRE) using observations of six clinical measures over four years. We modeled each subject’s cellular steroid response using data from a previously published study of immortalized lymphoblastoid cell lines (LCL) under dexamethasone (DEX) and sham treatment. We integrated SRE with LCL DEX response and genotypes to build a genome-scale network using the Reverse Engineering, Forward Simulation (REFS) modeling framework, identifying seven genes modulating SRE.
RESULTS
Three of these genes were functionally validated using a stable NFκB Luc reporter in A549 Human lung epithelial cells, IL1β cytokine stimulation, and dexamethasone treatment. Using siRNA transfection, knockdown of Family With Sequence Similarity 129 Member A (FAM129A) produced a reduction in steroid treatment response (p<0.001).
CONCLUSION
With this systems-based approach, we have shown that FAM129A is associated with variation in clinical asthma steroid responsiveness and that FAM129A modulates steroid responsiveness in lung epithelial cells.
Keywords: Inhaled Corticosteroids, Steroid Response Endophenotype, Genomics, FAM129A, REFS, Systems Biology
Graphical abstract
Introduction
Asthma affects approximately 23 million individuals in the United States and approximately 300 million individuals worldwide.(1) Inhaled corticosteroids (ICS) are the most commonly prescribed medications to control asthma. However, not all individuals with asthma respond to these treatments; some are well-controlled on ICS, while others require additional or alternative medications.(2, 3) While asthma is a heterogeneous disease, even among a relatively similar cohort of children with mild-to-moderate asthma, there are noticeable differences in the efficacy of ICS at maintaining asthma control.(2) Several genetic markers have been found to be associated with the efficacy of ICS therapy, such as the glucocorticoid induced transcript 1 (GLCCI1)(4) and the corticotrophin releasing hormone receptor 1 (CRHR1).(2, 5) Although these variants have fairly large effect sizes, in the case of GLCCI1, GWAS indicated up to 6% of variation in response to ICS treatment,(4) but attempts at clinical validation have produced mixed results.(6–9) These findings suggests that additional genes may be important for determining ICS response in asthma.(10)
While studies reviewed in a recently published systematic review of over 33 publications of ICS responsiveness have used particular asthma-related outcomes as proxies for response to ICS,(11) we previously demonstrated that a combination of multiple clinical outcomes measures steroid responsiveness with higher accuracy, higher stability across populations, and higher robustness to missing data.(12) We refer to this herein as the Steroid Responsiveness Endophenotype (SRE), and it is a combination of six clinical factors: baseline lung function (forced expiratory volume in 1 second, FEV1); bronchodilator response (change in FEV1 after administration of albuterol); frequency of asthma-related emergency department and hospital visits; frequency of supplemental courses of oral corticosteroids required to maintain asthma control; airway hyperreactivity measured by a methacholine challenge test; and daily symptom diary cards. Integrating these factors provides a more complete picture of the response to ICS treatment, which may manifest differently in distinct individuals with asthma, and has been shown to measure steroid responsiveness more accurately than any of these clinical factors alone.(12)
Genomic systems biology approaches – defined as those that take a holistic approach to disease understanding informed by multiple disciplines, genomics, and bioinformatic data sources(13, 14) – have previously been effective at identifying biological processes contributing to the etiology of complex genetic diseases.(15, 16) Here, we take a systems approach to address the genomics of ICS response in individuals with asthma, building on our previous Bayesian-network based methodology, Reverse Engineering and Forward Simulation (REFS).(17) This method uses patient genomic and outcome data to identify genes driving the associations of interest, enriching for likely causal importance of such associations. REFS uses Markov Chain Monte Carlo sampling to construct a collection of Bayesian networks. This collection of networks is termed an ensemble, and by querying the ensemble as a whole, questions can be answered such as: how do SNPs and genes interact, and which genes have the greatest effect on patient outcomes?
We hypothesized that it should be possible to integrate the ICS responsiveness signal measured in clinical trial subjects with the dexamethasone response signal measured in immortalized lymphoblastoid cell lines (LCL DEX response) from those subjects to create an informative steroid response network. Thus we expected to determine network edges that are indicative of interactions in steroid biology. We further hypothesized that this integrative network will not just represent steroid biology, but that secondary sources of variation in LCL DEX response are due to the variation in the donor’s genotypes and steroid responsiveness endotype. Thus we expected, if relevant signals were detected with sufficient power, to determine network edges indicative of variations in genotypes and SRE, and for the first neighbors of SRE to be genes or SNPs that modulate ICS response.
Methods
Our study design is shown in Figure 1. We selected a cohort from mild-to-moderate childhood asthmatics who were on ICS therapy in the Childhood Asthma Management Program (CAMP)(18, 19) trial and for which LCL DEX response and genotypes were available. These data were filtered and combined in the REFS framework to identify genes associated with SRE. To guard against false positives, we carried genes forward for functional validation in human airway epithelial cells where we measured the effect of gene knockdown on NFκB response to treatment with dexamethasone (DEX) and interleukin-1 beta (IL1β).
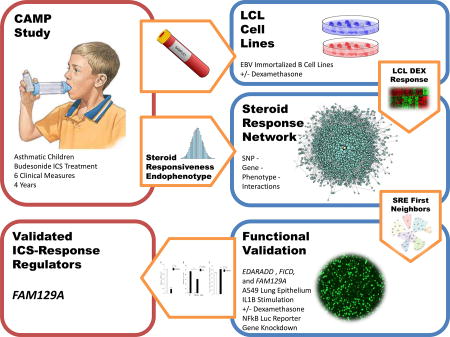
Figure 1.

Study Design Diagram. We used 104 participants of the CAMP clinical trial all randomized to the budesonide ICS treatment arm. Each was assessed along 6 longitudinal measures of steroid treatment for the duration of the original CAMP trial (4 years) to obtain their Steroid Responsiveness Endophenotype (SRE) value, measuring the effectiveness of ICS therapy at managing asthma. These subjects further provided blood samples which were immortalized and treated with both a steroid, Dexamethasone, and a sham treatment. LCL DEX response and genome-wide SNPs from these participants were used in the REFS network modeling framework to identify a number of genes that interacted with SRE. Three of these seven genes were validated in lung epithelial cell lines, and FAM129A was shown to be significantly associated with IL1B under steroid treatment.
CAMP
The Childhood Asthma Management Program was a randomized clinical trial of the efficacy of long-term daily ICS treatments at managing asthma symptoms and improving lung function.(19) CAMP enrolled 1,041 children ages 5 to 12 years with mild to moderate persistent asthma over a 23-month period, and followed them for an additional 4 years. Recruitment criteria and study characteristics have been described previously.(18, 19) During the original trial, CAMP participants were randomized into three treatment arms: budesonide (ICS), nedocromil, or placebo. We utilized three major types of data from selected CAMP participants: clinical data (from which we calculated SRE); genotype SNP data; and transcriptomic profiling from LCLs. CAMP participants were selected for the present study cohort as described below.
Expression Data, Preprocessing, and QC
After the main CAMP trial concluded, subjects were followed for at least yearly observation. CAMP participants were recontacted for additional blood draw roughly 6 years after study conclusion. Of participants randomized to ICS treatment, 161 non-Hispanic white subjects provided blood samples from which CD4+ lymphocytes were isolated. These samples were immortalized and transcriptomically profiled under two treatment conditions: dexamethasone treatment (10−6 M) and a sham (ethanol) control. After six hours, expression levels were measured using the Illumina HumanRef8 v2 BeadChip (Illumina, San Diego, CA). Bead-level data was stored using BeadStudio (Illumina) and arrays which had low detection thresholds or low dynamic range were marked as failed. Differential and eQTL analysis of this expression set has been previously described,(20) although we used a different QC and filtering process for the present study, as described below.
We used the beadarray package(21) for the R statistical language to read the previously saved bead-level data of passing arrays and to produce QC plots, Log-2 transformed bead-level data, bead-type statistics, bead summary data, and to perform differential expression analysis. We assembled an expression set of DEX and sham arrays, filtered out low quality probes based on the R illuminaHumanv2.db annotation package, and quantile normalized the data. We used the Bioconductor limma package(22) to examine the multidimensional scaling (MDS) clustering of all arrays including replicates. We identified and discarded defective arrays and sample mix-ups. We identified arrays with treatment condition mislabeling and either discarded the array or corrected the label. We created a paired set of arrays by discarding subjects with only one treatment condition and remaining replicates in such a way as to favor array pairs on the same chip or processed on the same day. We batch-corrected this set using the sva package ComBat function(23) and examined the MDS plot of DEX-Sham differential expression. We identified and removed outliers to produce the final set of paired DEX and Sham arrays.
We selected probes for the training set using the intra-class correlation (ICC) method provided by the R beadarrayFilter package.(24) We used a cutoff of 0.5 to select probes for which the between array variability exceeded the within array variability. This package and cutoff value have been demonstrated to produce more powerful analysis of differentially expressed genes on a publicly available Illumina Spike-in expression set.(25) We selected probes separately for the DEX and Sham treatment conditions and, using probes that were informative in both conditions, took the (Log2(DEX) − Log2(sham)) difference to produce the LCL DEX response data for the training set.
SRE phenotype
In this study we used the Corticosteroid Responsiveness Endophenotype Model previously described.(12) Briefly, it allows the combination of multiple observations of six clinical measures to assess a subject’s responsiveness to ICS. This results in one continuous variable that measures the effectiveness of steroid therapy for an individual. A more positive value indicates a stronger response to ICS. SRE has been shown to produce more accurate measurements of ICS response than individual phenotypes in white replication populations of children and adults with mild to moderately severe asthma and, for children, of multiple races.(12) We applied it throughout the four years of the clinical trial portion of CAMP to produce SRE and its 6 components for each subject.
Genotypes
Genome-wide genotyping was previously performed at the Channing Division of Network Medicine (CDNM), using Illumina HumanHap 610 bead chips (Illumina, Inc., San Diego, CA).(26) After pruning for minor allele frequency (less than 5%) and probability of Hardy-Weinberg equilibrium (less than 0.1%), and removing discordant probes identified by Illumina, 471,851 SNPs remained genotyped among 581 non-Hispanic white CAMP participants in 551 families with a genotyping rate of .998. We imputed missing genotypes using MaCH.(27)
SNP Selection
To identify potentially informative SNPs for REFS modeling, we performed an SRE genome-wide association study (GWAS) within the CAMP Budesonide ICS treatment arm (N=172) using the R GenABLE package.(28) We selected one sibling from each family for inclusion based on which sibling had LCL DEX response data, or randomly if no sibling had response data, excluded subjects with too high autosomal heterozygosity, and excluded markers with <1.5% minor allele frequency. We performed statistical tests for each SNP’s association with SRE based on an additive SNP-model using the GenABLE qtscore function.(28) We used the R fdrtool package(29) to filter GWAS results using the Higher Criticism Statistic(30) (function hc.thresh) and a p-value threshold of p < 0.001 to select SNPS for the training set. This was expected to produce a tractable number of SNPs containing mostly GWAS false positives from which the REFS modeling system could select significant predictors.
Integrative Training Set
We selected the cohort of non-Hispanic white CAMP participants from the ICS treatment arm who had 1) computable SRE measures, 2) genotype data, and 3) LCL DEX response.
REFS
We modeled the training set using GNS Healthcare’s REFS (GNS Healthcare, Cambridge, MA) modeling pipeline. This method has been previously described.(17) Briefly, REFS learns directly from data without need for a pre-specified hypothesis, producing an “ensemble” or set of Bayesian networks (BNs). Because REFS uses the stochastic Markov chain Monte Carlo (MCMC)(31) and simulated annealing methods, each network in the ensemble is different. The number of networks to be generated is specified as an input parameter. These networks are generated and converged in parallel to produce a representative sample of all possible Bayesian networks that could have generated the observed data.
We configured REFS to model our training set variables as continuous values and to allow each variable to act as an input to or output from any other variable, with the exception of SNP genotypes which were constrained to act as inputs only. After generation, the ensemble was checked to confirm that the process had converged (Supplemental Figures 7 & 8) and predicted the training data reasonably well.
The REFS ensemble consists of possible network topologies that might have generated the observed data. In order to integrate the predictions of the ensemble and ascertain statistical relevance of individual relationships, as well as to identify functional indirect relationships, it is necessary to perform in silico simulations. Using such simulations, we perturbed all possible input variables and measured the effect on all possible downstream targets.
The stochastic nature of the REFS modeling approach causes variability in results. We performed the REFS modeling step twice, each with an ensemble size of 256 networks, using the same input parameters but different random seeds, to limit dependence of our results on a single stochastic simulation.
Interaction Network
We created a secondary “interaction network” to summarize each REFS ensemble. In this network we included a vertex for every training set variable and a directed edge for every downstream target for which REFS in silico simulation produced a P-value of less than 0.05. This is an empirical p-value computed using a permutation-based procedure.(17) This produced networks potentially containing multiple isolated subgraphs. We stored the REFS statistics characterizing the ensemble and simulation results as edge and vertex attributes. We assembled, manipulated, and plotted the interaction network using the R igraph package. We queried the interaction network to identify SNPs and genes that interact directly or indirectly with SRE. We focused our analysis on the subgraph containing SRE and characterized the size of each interaction network by the size of this SRE subgraph.. We examined the immediate vicinity of SRE by selecting vertices within 3 hops of SRE (using R igraph package ego function) and identifying neighborhoods based on community structure using the igraph cluster_walktrap function to find densely connected subgraphs via random walks. We simplified plots of these neighborhoods by removing neighborhoods not containing a first neighbor gene.
Cell culture, siRNA Transfection and Chemical Treatment
A549/NFκB-Luc human lung epithelial cells were maintained in high-glucose Dulbecco's Modified Eagle Medium (DMEM) containing 10% Fetal Bovine Serum (FBS). Transfection of target gene small interfering RNA (siRNA) and non-targeting siRNA (ON-TARGETplus pools; GE Dharmacon, Lafayette, CO) was performed using DharmaFECT 1 reagent according to the recommended protocol from the manufacturer (GE Dharmacon). The final concentration of siRNA was 25 nM. 72 hours after transfection, cells were washed and switched to F-12 medium supplemented with 5% dialyzed FBS. Cells were treated with 5 nM DEX (Sigma-Aldrich, Natick, MA) or 5 ng/mL IL1β (ThermoFisher Scientific, Waltham, MA) in the medium for 16 hours.
Luciferase Assay
Cells were washed with phosphate buffered saline (PBS) and lysed with Glo Lysis Buffer (Promega, Fitchburg, WI). Protein concentration was measured using bicinchoninic acid (BCA) agent (Promega). Luciferase assays were performed using Steady-Glo luciferase agent (Promega). Luminescence was measured by using Synergy 2 plate reader (BioTek Instruments, Winooski, VT). Each sample was measured in triplicates. Statistical significance was assessed using a double-sided t-test.
Quantitative Real-Time PCR (qRT-PCR)
Total RNA was isolated from cells by using QIAshredder and RNeasy kits (Qiagen, Germantown, MD). Oligo(dT)-primed cDNA was prepared from 50 ng of total RNA by using SuperScript III First-strand Synthesis System (ThermoFisher). qRT-PCR was set up in the presence of 0.5 µM primers by using QuantiTect SYBR Green PCR kit (Qiagen). qRT-PCR was performed on an StepOne Plus real time PCR machine (Applied Biosystems, Foster City, CA) and analyzed using Ct method. β-actin was used as an internal control for data normalization. Each sample was measured in triplicates.
Study Approval
Written informed consent for CAMP participants was obtained at CAMP enrollment by the child’s parent or guardian, and again at blood draw for the collection of biological materials for the purpose of genetic and genomic investigation. These studies were approved by the Partners Human Research Committee, the institutional review board for Brigham and Women’s Hospital.
Results
LCL DEX response
We used bead-level data from 362 previously passed arrays for 151 CAMP non-Hispanic white subjects. MDS clustering (Supplemental Figure 1) indicated that additional QC was required. We discarded 5 defective arrays, discarded nine sample mixups, discarded three mislabeled arrays, relabeled the treatment condition for seven arrays, and discarded unpairable arrays and unneeded replicates to produce a set of paired Dex-Sham arrays for 148 subjects (Supplemental Figure 2). We discarded five subjects for which batch corrected differential expression exhibited outlier behavior. We identified 3732 probes that were informative in both DEX and Sham conditions for which we took the (Log2(DEX) − Log2(sham)) difference to determine LCL DEX response in 3732 genes for 143 subjects.
SNP selection
After selection of subjects and exclusion of markers with low MAF, our SRE GWAS panel contained 165 subjects (74 female, 91 male) with 471,322 markers. After SRE GWAS and selection using higher criticism we retained 360 SNPs (with p < 0.001) to carry forward to the training set.
Cohort characteristics
Following quality control (see Methods), we identified a cohort of 104 participants with complete genotypes, LCL DEX response, and SRE (Table 1). In the cohort, SRE exhibited a reasonably normal distribution (Supplemental Figure 3), and, with Area Under the receiver-operator characteristic Curve (AUC) of 0.75, measured ICS responsiveness significantly more accurately than any of the individual clinical measures (AUCs of 0.58 to 0.68, p-values for difference with SRE of <0.001 to 0.018. Supplemental Table 2). After removing 5 SNPS for which MAF fell below 0.05 in the cohort population, we finalized the REFS training set with 1 phenotype (SRE), 255 SNPS, and 3732 LCL DEX response genes for 104 subjects. We performed the REFS modeling step twice and produced two REFS ensembles of 256 networks each.
Table 1. Population characteristics.
Characteristics of the study cohort. Subjects were children with asthma selected from the CAMP trial ICS treatment group.
| Study | CAMP ICS Treatment Arm | P Value | |
|---|---|---|---|
| Sample Size | 104 | 311 | |
| Race (%) | White(100%) | White(65%), Black(14%), Hispanic(10%), Other(11%) | |
| Age (years, SD) | 8.6 (2.1) | 9 (2.1) | 0.331 |
| Sex, male (n, %) | 60 (58%) | 181 (58%) | 1 |
| FEVPPB (SD) | 95% (15%) | 94% (14%) | 0.478 |
| LNPC20B (SD) | 0.066 (1.2) | 0.085 (1.2) | 0.967 |
| BDRB (SD) | 12% (0.11%) | 11% (0.11%) | 0.986 |
| SRE | 0.79 (1.3) | 0.59 (1.4) | 0.207 |
(SD) = standard deviation; FEVPPB = FEV1 percent predicted at baseline; LNPC20B = natural log provocative concentration of methacholine required to effect a 20% reduction in FEV1 (PC20) at baseline; BDRB = bronchodilator percent change at baseline. For comparison, the full CAMP budesonide treatment group and p-values for the null hypothesis that both populations were drawn from the same distribution are presented. This comparison shows that our study cohort reasonably represented the full CAMP budesonide treatment group.
Interaction Network
Following our first hypothesis, we created an interaction network for each of the two REFS ensembles. These contained SRE subgraphs with 3319 and 3313 vertices connected to SRE either directly or indirectly using 7958 and 8028 edges, respectively. The second REFS ensemble SRE subgraph contained 3302 (88%) of training set genes and 10 (2.8%) of training set SNPs as shown in Figure 2. We inspected the networks to identify candidate steroid response controller genes, in the present analysis choosing genes that were first-degree neighbors of SRE in either network. Six genes appeared in both interaction networks (CEP152, EDARADD, FAM129A, FAM174B, FICD, YY1) and FAF1 appeared in only one (Table 2); that FAF1 appears in some but not all networks is not surprising given the stochastic modeling process and the potentially nascent signals associated with SRE. Gene neighborhoods from the second REFS ensemble are shown in Figure 3. Detailed views of the FAM129A and other gene neighborhoods are shown, respectively, in Figure 4 and Supplemental Figure 9. REFS simulation statistics for the firstdegree SRE neighbors in the second REFS network are shown in Supplemental Table 1. As predicted in our second hypothesis, all seven first-neighbor genes showed significant changes with SRE (p < 0.05) and high correlation between the predicted expression levels and the LCL DEX expression training values (Spearman correlations 0.657 to 0.894). We examined the association of LCL DEX response with SRE for these genes and found uncorrected p-values of .0004 to .02 (Supplemental Figure 5).
Figure 2.

Interaction network of Gene-SNP-phenotype associations for SRE. A plot of the interaction network SRE subgraph taken from the second ensemble of 256 REFs networks. Vertex type is indicated by: red star = SRE,. blue circle = gene, and green square = SNP. In the network, genes are seen to play a major role, and SNPs a minor role. The size of symbols is proportional to the number of edges incident on the vertex. The sizes of SRE and SNPs are exaggerated by factor of 3 and 2 respectively for readability. Vertices are located using multidimensional scaling of the network shortest path matrix using the R igraph layout_with_multidimentional function.
Table 2.
Direct neighbor genes of Steroid Response found in the REFS ensembles.
| SRE 1st Neighbor Gene |
# of Ensembles |
PubMed Gene Hits |
PubMed Gene + asthma Hits |
PubMed Gene + NFκB Hits |
Selected for Validation |
Genomic Location | Illumina Probe ID |
|---|---|---|---|---|---|---|---|
|
| |||||||
| CEP152 | 2 | 42 | 0 | 0 | chr15:49033573:49033622:− | ILMN_1732311 | |
|
| |||||||
| EDARADD | 2 | 63 | 0 | 16 | X | chr1:236647855:236647904:+ | ILMN_1761820 |
|
| |||||||
| FAF1 | 1 | 88 | 0 | 6 | chr1:51061793:51061831:− | ILMN_1666634 | |
| chr1:51050473:51050483:− | |||||||
|
| |||||||
| FAM129A | 2 | 16 | 2 | 0 | X | chr1:184763475:184763524:− | ILMN_1810725 |
|
| |||||||
| FAM174B | 2 | 2 | 0 | 0 | chr15:93160740:93160789:− | ILMN_1652797 | |
|
| |||||||
| FICD | 2 | 10 | 0 | 0 | X | chr12:108913110:108913159:+ | ILMN_1778064 |
|
| |||||||
| YY1 | 2 | 1148 | 11 | 44 | X | chr14:100743950:100743999:+ | ILMN_1770892 |
Figure 3.

Gene neighborhoods from the second REFS Interaction Network. Vertex type is indicated by: red star = SRE, circle = gene. Neighborhoods, indicated by colored outlines, were determined as described in Methods. Symbol size is proportional to the number of all interaction network edges incident on the vertex, some of which may not be shown. The figure was plotted using the Kamada-Kawai layout algorithm. SRE appears in a central location as a result of the neighborhood and layout selection.
Figure 4.

Neighborhood of the FAM129A gene, a direct neighbor of SRE. This shows that action of FAM129A on SRE is likely mediated by a number of genes including AGO2 and RAD21. Edge width is proportional to, and edges are labeled with, the proportion of the 256 ensemble networks that contain the edge. Vertex type is indicated by: red star = SRE, circle = gene. The FAM129A neighborhood, indicated by the colored outline, was determined as described in Methods. Symbol size is proportional to the number of all interaction network edges incident on the vertex, some of which may not be shown. The figure was plotted using the same layout as Figure 2.
We performed PubMed literature searches to determine if these genes had previously been associated with asthma or nuclear factor kappa-light-chain-enhancer of activated B-cells (NFκB). We found that CRP152, EDARADD, FAF1 and YY1 had a higher number of publications, FAM129A, FAM174B, and FICD had fewer publications; FAM129A and YY1 were reported to be associated with asthma; and EDARADD and YY1 were reported to be associated with NFκB.
Resource limits constrained us to limit our cellular validation experiments to four genes. We chose EDARADD, FAM129A, FICD, and YY1, in order to include genes that were better represented in the literature (YY1, EDARADD), less well represented (FAM129A, FICD), associated with asthma (FAM129A, YY1) and associated with NFκB (YY1, EDARADD).
Differential Expression Analysis
We analyzed differential expression in the training set genes (Supplemental Figure 4). We found that, had we used a typical differential expression cutoff such as fold change > 2 to select genes for the training set, 6 of the 7 SRE 1st neighbor genes would not have been included in the training set and thus would not have been found. While only a few genes were connected directly to SRE, gene-gene interactions were reported for 96% of fold change > 2 genes and 91% of fold change < 2 genes with p-value for differential expression <= 0.05. Interactions were also reported for 87% of the 2682 genes with p > 0.05 that would be consider not significant in differential expression analysis (Supplemental Table 3).
NFκB activation
Our functional validation strategy is based on NFκB, a master transcription factor that controls the expression of a large number of inflammation factors, including cytokines and chemokines known to be mediators of asthma. Crucially, NFκB is a transcription factor central to the modulation of glucocorticoid induced transrepression, a mechanism underlying the anti-inflammatory effects of corticosteroids.(32, 33) Accordingly, we used NFκB as a functional read-out of inflammation response and anti-inflammation activity. We used A549 human lung epithelial NFκB-Luc reporter cells for functional validation. We created an inflammatory-response with cytokine IL1β treatment and compared the response to IL1β + dexamethasone treatment to determine the anti-inflammation treatment effect of glucocorticoids.
We used targeted small interfering RNA (siRNA) transfection to knockdown the expression of candidate genes. We achieved efficient knockdown of mRNA for FAM129A (Fig. 5A), EDARADD (Supplemental Figure 6A) and FICD (Supplemental Figure 6A). YY1 knockdown failed and is not reported further.
Figure 5.

Effects of FAM129A on NFκB activation and anti-inflammation effect of glucocorticoid. (A) A549/NFκB-Luc cells were transfected with non-targeting (NT) siRNA or siRNA targeting FAM129A, and 88 h later mRNA levels of FAM129A gene were measured using qPCR and normalized to the level in control siRNA transfected cells. (B) A549/NFκB-Luc cells were transfected with siRNA as in (A). 72 h after transfection, cells were treated with 5 ng/mL IL1β for 16h. The luciferase (Luc) activity was measured and normalized with the amount of total protein. The Luc level was shown relative to that of untreated NT siRNA transfected cells. (C) In the same experiment as shown in (B), additional groups of cells were treated with 5 ng/mL IL1β+100 nM DEX and the Luc level normalized with that upon 5 ng/mL IL1β treatment to determine the percentage values shown. **, P<0.01; ***, P<0.005.
Knockdown of the FAM129A gene reduced IL1β-induced NFκB activity and (Figure 5B), more importantly, produced a moderate but statistically significant (P<0.001) reduction in steroid treatment response (Figure 5C). EDARADD and FICD knockdown slightly affected IL1β-induced NFκB activity (Supplemental Figure 6B) but did not alter the effect of DEX. (Supplemental Figure 6C).
Post-hoc investigation of the 355 SNPs included in REFS and FAM129A revealed no SNPs that were identified as expression quantitative trait loci (eQTLs) for FAM129A in GTEx data. FAM129A was differentially expressed in asthmatics vs. healthy controls in the Unbiased Biomarkers in Prediction of Respiratory Disease Outcomes (U-BIOPRED) cohort Gene Expression Omnibus (GEO) data (p = 5.9*10−6).(34)
Discussion
We hypothesized that we could identify a network representative of steroid biology using LCL DEX response. Using systems biology, detailed clinical observations, and blood drawn from children with asthma, we identified seven genes associated with steroid response. A key finding of our study is that FAM129A modulates the in vitro steroid anti-inflammation response in A549 human lung epithelial cells from the three genes successfully tested in knockout experiments. Thus both hypotheses, that the network would represent steroid biology and individual variations in SRE, were confirmed. The direction of effect of FAM129A in our airway epithelial cell model supports FAM129A as augmenting the anti-inflammatory effects of ICS in asthma. In addition, we found that the systems biology model helps to ensure the biologic validity of these genes through the multiple REFS iterations resulting in carefully annotated interaction networks.
Our finding that FAM129A modulates steroid response is supported by a study by King et al. on glucocorticoid repression of inflammatory genes, also in A549 cells. They found 34 genes, including FAM129A that, at 6 hours, exhibited more than 10-fold induction by IL1β and significant repression by the addition of dexamethasone.(35) FAM129A mRNA was measured at 1, 2, 6, and 18 hours for cells treated with IL1β, IL1β + DEX, DEX alone, or no treatment. FAM129A mRNA was observed to remain at basal levels at 1 and 2 hours; be repressed to 47% (P<0.001) of the IL1β induction level by the addition of DEX at 6 hours, and returned to basal levels at 18 hours independent of IL1β and/or DEX treatment. Thus, IL1β induced FAM129A expression, and the induction was repressed with DEX. Collectively, our and King et al.'s results point to FAM129A's active role in airway epithelial cell inflammatory response and steroid treatment effect, both as control variable and controlled variable. This suggests the existence of a FAM129A regulatory circuit playing a central role in airway epithelial cell inflammatory response, where FAM129A seems to promote inflammation but enhance the anti-inflammation effect of DEX. Additionally, FAM129A has been reported as a marker(36) and modulator of thyroid tumors.(37) It is possible that dysregulation of FAM129A leads to cell cycle abnormalities and aberrant cell growth, which could cause both cancers and airway remodeling.
Our network indicates an interaction between FAM129A product and steroid responsiveness in children with asthma. Other work shows that the FAM129A regulatory circuit observed in airway epithelial cells is active in asthma inflammation and steroid treatment response: in a study of 12 asthmatics, Yick et al. found that prednisolone upregulated 85 genes, one of which was FAM129A in laser microdissected airway smooth muscle.(38) In a subsequent study, Yick et al. found FAM129A to be one of eight genes exhibiting differential expression in asthma.(39)
Our study demonstrates how a systems biology model can help identify biologically relevant genes. Such methods have been successfully applied in other studies of lung disease, identifying gene groups associated with asthma control,(40) gene groups associated with severity of chronic obstructive pulmonary disease,(41) and identifying GAB1 as a novel regulator of inflammatory response in asthma.(42) Furthermore, the REFS modeling technique has been effective in a variety of other applications, including novel gene identification for rheumatoid arthritis,(17) type 2 diabetes,(43) and breast cancer.(44) Interestingly, GLCCI1(4), a GWAS hit found in CAMP, was not a SRE 1st neighbor. This may be related to different phenotypes used: the GWAS phenotype which identified GLCCI1 was change in FEV1 after 4–8 weeks of ICS treatment; whereas the phenotype used in the current work, SRE, is a composite of 6 phenotypes and is not strongly correlated with FEV1. These studies are thus likely to select different genomic features. CRHR1(2, 5) may not have been identified in the current study for similar reasons.
We expected the REFS modeling system to connect genes and SNPs with SRE that would not otherwise be identified by traditional differential expression analysis. This is evidenced by the divergence between genes that would be judged important in a differential expression volcano plot (Supplemental Figure 4) and the SRE 1st neighbor genes we identified. Most genes included in the REFS modeling step were connected or indirectly connected to SRE in the consensus network ensemble (88%, see Supplemental Table 3), most of which would typically be ignored in differential expression analysis. This suggests the potential of the study methodology to extract additional information from existing expression sets.
Our study has several strengths. We used a composite measure of steroid-responsiveness, the SRE, which allowed us to interrogate a general, asthma-related steroid response phenotype. We used a powerful machine-learning and network-based methodology to identify interactions of several genes with SRE in children with mild-to-moderate asthma. We also provided strong functional evidence that FAM129A is an in vitro modulator of steroid efficacy in subsiding inflammation. However, our study also has several limitations. We had a modest sample size of LCLs. Although we did identify some steroid-response genes, we may have been able to identify more if the LCLs were stimulated with an inflammatory cytokine in addition to dexamethasone. This may provide a more direct read out of the cellular response to steroid remediation of inflammation. Alternatively, other steroids besides dexamethasone could be investigated; although our previous work has shown that clinical response to various ICS’s is distributionally similar,(5, 12) there may be differences in cellular response by the type of ICS used. Additionally, our study may have been weakened because LCLs were derived from blood drawn six years past the conclusion of the CAMP study trial rather than during the trial. There are also parameters of the REFS methodology that could be altered, with the possibility of identifying additional steroid-response modulating genes, by, for example, altering the p-value threshold (p<0.05) for edge inclusion in REFS networks. Although our previous work on the SRE showed no significant age effect within the CAMP population and validated its accuracy in heterogeneous study populations with mean ages from 9 to 34 years,(12) it is possible that steroid responsiveness changed somewhat from CAMP enrollment to blood draw. Additionally, testing of in vitro steroid response in primary epithelial cells instead of the A549 Human epithelial cell line could improve the relevance of validation results. The observed change in steroid response with the FAM129A knockdown was small, and a change to primary cells may result in an increased observed effect size, since A549 cells have intact NFκB activation and glucocorticoid receptor responsive mechanisms, and thus may underestimate the inflammatory response in asthma patients. Figure 5C may therefore underestimate the effect of FAM129A. Further functional validation would also be important, including testing of FAM129A in an independent cohort of children with asthma.
In conclusion, previous findings by others have shown that FAM129A responds to steroid treatment in asthma and in lung epithelial cells and that FAM129A is differentially expressed in asthma. We have shown that FAM129A is associated with variation in clinical asthma steroid responsiveness and that FAM129A modulates steroid responsiveness in lung epithelial cells. Further work is needed to validate the other SRE neighbor genes identified by the network (CEP152, FAF1, FAM174B & YY1). At the cellular level, additional investigations are needed to characterize the FAM129A circuit. At the clinical level, further work is needed to investigate the potential for translation of FAM129A to the management of steroid resistant asthmatics. Finally, the relevance of the FAM129A circuit to other inflammatory diseases needs to be investigated.
Supplementary Material
Key Message.
FAM129A is a modulator of inhaled corticosteroid response in asthmatic patients.
Steroid response is usefully measured as a combination of asthma control outcomes.
Systems biology methodologies can identify biologically relevant genes that would go undetected by traditional analysis.
Capsule Summary.
A systems biology investigation of gene expression data from a cohort of mild-to-moderate persistent asthmatic children identified FAM129A as a regulator of inhaled corticosteroid efficacy. Differences in steroid response are at least partially due to genomic factors and future dosing and treatment recommendations will consider these.
Acknowledgments
We thank Paul McDonagh for his help with an earlier version of this analysis. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the eQTL analysis described in this manuscript were obtained from the GTEx Portal on 09/25/2017.
Funding: This project was supported by U01 HL65899 and R01 HL092197. MJM is supported by a grant from the Parker B Francis Foundation. KGT is supported by R01 HL127332 and R01 HL129935. ACW is supported by R01 HD085993. QL is supported by R01 HL114769.
Abbreviations Used
- AUC
Area Under receiver operator characteristic Curve
- CAMP
Childhood Asthma Management Program
- CD4+
Cluster of Differentiation 4
- CEP152
Centrosomal Protein 152
- CRHR1
Corticotropin Releasing Hormone Receptor 1
- DEX
Dexamethasone
- EDARADD
Ectodysplasia A Receptor Associated Death Domain
- eQTL
expression Quantitative Trait Loci
- FAF1
FAS-Associated Factor 1
- FAM129A
Family With Sequence Similarity 129 Member A
- FAM174B
Family With Sequence Similarity 174 Member B
- FEV1
Forced Expiratory Volume in 1 second
- FICD
FIC Domain Containing
- GEO
Gene Expression Omnibus
- GLCCI1
Glucocorticoid Induced 1
- GWAS
Genome Wide Association Study
- ICS
Inhaled Corticosteroid
- IL1B
Interleukin 1 Beta
- LCL
immortalized Lymphoblastoid Cell Lines
- MAF
Minor Allele Frequency
- MCMC
Markov Chain Monte Carlo
- MDS
Multidimensional Scaling
- NFκB
Nuclear Factor kappa-light-chain-enhancer of activated B-cells
- QC
Quality Control
- qRT-PCR
quantitative Reverse Transcription Polymerase Chain Reaction
- REFS
Reverse Engineering, Forward Simulation methodology
- RNA
Ribonucleic Acid
- SNP
Single Nucleotide Polymorphism
- SRE
Steroid Responsiveness Endophenotype
- YY1
Yin Yang 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Statement of Competing Interests: BH, KR, BC, and IK are employed by GNS Healthcare which developed the REFS methodology.
References
- 1.Masoli M, Fabian D, Holt S, Beasley R. The global burden of asthma: executive summary of the GINA Dissemination Committee report. Allergy. 2004;59(5):469–78. doi: 10.1111/j.1398-9995.2004.00526.x. [DOI] [PubMed] [Google Scholar]
- 2.Tantisira KG, Lake S, Silverman ES, Palmer LJ, Lazarus R, Silverman EK, Liggett SB, Gelfand EW, Richter B, Israel E, et al. Corticosteroid Pharmacogenetics: Association of sequence variants in CRHR1 with improved lung function in asthmatics treated with inhaled corticosteroids. Hum Mol Genet. 2004 doi: 10.1093/hmg/ddh149. [DOI] [PubMed] [Google Scholar]
- 3.Drazen JM, Silverman EK, Lee TH. Heterogeneity of therapeutic responses in asthma. Br Med Bull. 2000;56(4):1054–70. doi: 10.1258/0007142001903535. [DOI] [PubMed] [Google Scholar]
- 4.Tantisira KG, Lasky-Su J, Harada M, Murphy A, Litonjua AA, Himes BE, Lange C, Lazarus R, Sylvia J, Klanderman B, et al. Genomewide association between GLCCI1 and response to glucocorticoid therapy in asthma. N Engl J Med. 2011;365(13):1173–83. doi: 10.1056/NEJMoa0911353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tantisira KG, Lazarus R, Litonjua AA, Klanderman B, Weiss ST. Chromosome 17: association of a large inversion polymorphism with corticosteroid response in asthma. Pharmacogenet Genomics. 2008;18(8):733–7. doi: 10.1097/FPC.0b013e3282fe6ebf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chiba S, Nakamura Y, Mizuno T, Abe K, Horii Y, Nagashima H, Sasaki N, Kanno H, Tanita T, Yamauchi K. Impact of the genetic variants of GLCCI1 on clinical features of asthmatic patients. Clin Respir J. 2017 doi: 10.1111/crj.12647. [DOI] [PubMed] [Google Scholar]
- 7.Hu C, Xun Q, Li X, He R, Lu R, Zhang S, Hu X, Feng J. GLCCI1 Variation Is Associated with Asthma Susceptibility and Inhaled Corticosteroid Response in a Chinese Han Population. Arch Med Res. 2016;47(2):118–25. doi: 10.1016/j.arcmed.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 8.Vijverberg SJ, Tavendale R, Leusink M, Koenderman L, Raaijmakers JA, Postma DS, Koppelman GH, Turner SW, Mukhopadhyay S, Palmer CN, et al. Pharmacogenetic analysis of GLCCI1 in three north European pediatric asthma populations with a reported use of inhaled corticosteroids. Pharmacogenomics. 2014;15(6):799–806. doi: 10.2217/pgs.14.37. [DOI] [PubMed] [Google Scholar]
- 9.Hosking L, Bleecker E, Ghosh S, Yeo A, Jacques L, Mosteller M, Meyers D. GLCCI1 rs37973 does not influence treatment response to inhaled corticosteroids in white subjects with asthma. J Allergy Clin Immunol. 2014;133(2):587–9. doi: 10.1016/j.jaci.2013.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kersten ET, Koppelman GH. Pharmacogenetics of asthma: toward precision medicine. Curr Opin Pulm Med. 2017;23(1):12–20. doi: 10.1097/MCP.0000000000000335. [DOI] [PubMed] [Google Scholar]
- 11.Farzan N, Vijverberg SJ, Arets HG, Raaijmakers JA, Maitland-van der Zee AH. Pharmacogenomics of inhaled corticosteroids and leukotriene modifiers: a systematic review. Clin Exp Allergy. 2017;47(2):271–93. doi: 10.1111/cea.12844. [DOI] [PubMed] [Google Scholar]
- 12.Clemmer GL, Wu AC, Rosner B, McGeachie MJ, Litonjua AA, Tantisira KG, Weiss ST. Measuring the corticosteroid responsiveness endophenotype in asthmatic patients. J Allergy Clin Immunol. 2015 doi: 10.1016/j.jaci.2015.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schadt EE, Bjorkegren JL. NEW: network-enabled wisdom in biology, medicine, and health care. Sci Transl Med. 2012;4(115):115rv1. doi: 10.1126/scitranslmed.3002132. [DOI] [PubMed] [Google Scholar]
- 14.Bunyavanich S, Schadt EE. Systems biology of asthma and allergic diseases: a multiscale approach. J Allergy Clin Immunol. 2015;135(1):31–42. doi: 10.1016/j.jaci.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choi H, Song WM, Zhang B. Linking childhood allergic asthma phenotypes with endotype through integrated systems biology: current evidence and research needs. Rev Environ Health. 2017 doi: 10.1515/reveh-2016-0054. [DOI] [PubMed] [Google Scholar]
- 16.Bunyavanich S, Schadt EE, Himes BE, Lasky-Su J, Qiu W, Lazarus R, Ziniti JP, Cohain A, Linderman M, Torgerson DG, et al. Integrated genome-wide association, coexpression network, and expression single nucleotide polymorphism analysis identifies novel pathway in allergic rhinitis. BMC Med Genomics. 2014;7(48) doi: 10.1186/1755-8794-7-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xing H, McDonagh PD, Bienkowska J, Cashorali T, Runge K, Miller RE, Decaprio D, Church B, Roubenoff R, Khalil IG, et al. Causal modeling using network ensemble simulations of genetic and gene expression data predicts genes involved in rheumatoid arthritis. PLoS Comput Biol. 2011;7(3):e1001105. doi: 10.1371/journal.pcbi.1001105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Long-term effects of budesonide or nedocromil in children with asthma. The Childhood Asthma Management Program Research Group. N Engl J Med. 2000;343(15):1054–63. doi: 10.1056/NEJM200010123431501. [DOI] [PubMed] [Google Scholar]
- 19.The Childhood Asthma Management Program (CAMP): design, rationale, and methods. Childhood Asthma Management Program Research Group. Control Clin Trials. 1999;20(1):91–120. [PubMed] [Google Scholar]
- 20.Qiu W, Rogers AJ, Damask A, Raby BA, Klanderman BJ, Duan QL, Tyagi S, Niu S, Anderson C, Cahir-Mcfarland E, et al. Pharmacogenomics: novel loci identification via integrating gene differential analysis and eQTL analysis. Hum Mol Genet. 2014;23(18):5017–24. doi: 10.1093/hmg/ddu191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dunning MJ, Smith ML, Ritchie ME, Tavare S. beadarray: R classes and methods for Illumina bead-based data. Bioinformatics. 2007;23(16):2183–4. doi: 10.1093/bioinformatics/btm311. [DOI] [PubMed] [Google Scholar]
- 22.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28(6):882–3. doi: 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Forcheh AC, Verbeke G, Kasim A, Lin D, Shkedy Z, Talloen W, Göhlmann HWH, Clement L. beadarrayFilter: An R package to filter beads. The R Journal. 2013;5(1):171–80. [Google Scholar]
- 25.Forcheh AC, Verbeke G, Kasim A, Lin D, Shkedy Z, Talloen W, Gohlmann HW, Clement L. Gene filtering in the analysis of Illumina microarray experiments. Stat Appl Genet Mol Biol. 2012;11(2) doi: 10.2202/1544-6115.1710. [DOI] [PubMed] [Google Scholar]
- 26.McGeachie MJ, Wu AC, Tse SM, Clemmer GL, Sordillo J, Himes BE, Lasky-Su J, Chase RP, Martinez FD, Weeke P, et al. CTNNA3 and SEMA3D: Promising loci for asthma exacerbation identified through multiple genome-wide association studies. J Allergy Clin Immunol. 2015;136(6):1503–10. doi: 10.1016/j.jaci.2015.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34(8):816–34. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aulchenko YS, Ripke S, Isaacs A, van Duijn CM. GenABEL: an R library for genome-wide association analysis. Bioinformatics. 2007;23(10):1294–6. doi: 10.1093/bioinformatics/btm108. [DOI] [PubMed] [Google Scholar]
- 29.Strimmer K. fdrtool: a versatile R package for estimating local and tail area-based false discovery rates. Bioinformatics. 2008;24(12):1461–2. doi: 10.1093/bioinformatics/btn209. [DOI] [PubMed] [Google Scholar]
- 30.Klaus B, Strimmer K. Signal identification for rare and weak features: higher criticism or false discovery rates? Biostatistics. 2013;14(1):129–43. doi: 10.1093/biostatistics/kxs030. [DOI] [PubMed] [Google Scholar]
- 31.Friedman N, Linial M, Nachman I, Pe'er D. Using Bayesian networks to analyze expression data. J Comput Biol. 2000;7(3–4):601–20. doi: 10.1089/106652700750050961. [DOI] [PubMed] [Google Scholar]
- 32.Coutinho AE, Chapman KE. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol Cell Endocrinol. 2011;335(1):2–13. doi: 10.1016/j.mce.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Newton R. Anti-inflammatory glucocorticoids: changing concepts. Eur J Pharmacol. 2014;724:231–6. doi: 10.1016/j.ejphar.2013.05.035. [DOI] [PubMed] [Google Scholar]
- 34.Bigler J, Boedigheimer M, Schofield JPR, Skipp PJ, Corfield J, Rowe A, Sousa AR, Timour M, Twehues L, Hu X, et al. A Severe Asthma Disease Signature from Gene Expression Profiling of Peripheral Blood from U-BIOPRED Cohorts. Am J Respir Crit Care Med. 2017;195(10):1311–20. doi: 10.1164/rccm.201604-0866OC. [DOI] [PubMed] [Google Scholar]
- 35.King EM, Chivers JE, Rider CF, Minnich A, Giembycz MA, Newton R. Glucocorticoid repression of inflammatory gene expression shows differential responsiveness by transactivation- and transrepression-dependent mechanisms. PLoS One. 2013;8(1):e53936. doi: 10.1371/journal.pone.0053936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matsumoto F, Fujii H, Abe M, Kajino K, Kobayashi T, Matsumoto T, Ikeda K, Hino O. A novel tumor marker, Niban, is expressed in subsets of thyroid tumors and Hashimoto's thyroiditis. Hum Pathol. 2006;37(12):1592–600. doi: 10.1016/j.humpath.2006.06.022. [DOI] [PubMed] [Google Scholar]
- 37.Carvalheira G, Nozima BH, Cerutti JM. microRNA-106b-mediated down-regulation of C1orf24 expression induces apoptosis and suppresses invasion of thyroid cancer. Oncotarget. 2015;6(29):28357–70. doi: 10.18632/oncotarget.4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yick CY, Zwinderman AH, Kunst PW, Grunberg K, Mauad T, Fluiter K, Bel EH, Lutter R, Baas F, Sterk PJ. Glucocorticoid-induced changes in gene expression of airway smooth muscle in patients with asthma. Am J Respir Crit Care Med. 2013;187(10):1076–84. doi: 10.1164/rccm.201210-1886OC. [DOI] [PubMed] [Google Scholar]
- 39.Yick CY, Zwinderman AH, Kunst PW, Grunberg K, Mauad T, Chowdhury S, Bel EH, Baas F, Lutter R, Sterk PJ. Gene expression profiling of laser microdissected airway smooth muscle tissue in asthma and atopy. Allergy. 2014;69(9):1233–40. doi: 10.1111/all.12452. [DOI] [PubMed] [Google Scholar]
- 40.Croteau-Chonka DC, Qiu W, Martinez FD, Strunk RC, Lemanske RF, Jr, Liu AH, Gilliland FD, Millstein J, Gauderman WJ, Ober C, et al. Gene Expression Profiling in Blood Provides Reproducible Molecular Insights into Asthma Control. Am J Respir Crit Care Med. 2017;195(2):179–88. doi: 10.1164/rccm.201601-0107OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morrow JD, Qiu W, Chhabra D, Rennard SI, Belloni P, Belousov A, Pillai SG, Hersh CP. Identifying a gene expression signature of frequent COPD exacerbations in peripheral blood using network methods. BMC Med Genomics. 2015;8(1) doi: 10.1186/s12920-014-0072-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sharma A, Menche J, Huang CC, Ort T, Zhou X, Kitsak M, Sahni N, Thibault D, Voung L, Guo F, et al. A disease module in the interactome explains disease heterogeneity, drug response and captures novel pathways and genes in asthma. Hum Mol Genet. 2015;24(11):3005–20. doi: 10.1093/hmg/ddv001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anderson JP, Parikh JR, Shenfeld DK, Ivanov V, Marks C, Church BW, Laramie JM, Mardekian J, Piper BA, Willke RJ, et al. Reverse Engineering and Evaluation of Prediction Models for Progression to Type 2 Diabetes: An Application of Machine Learning Using Electronic Health Records. J Diabetes Sci Technol. 2015;10(1):6–18. doi: 10.1177/1932296815620200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gendelman R, Xing H, Mirzoeva OK, Sarde P, Curtis C, Feiler HS, McDonagh P, Gray JW, Khalil I, Korn WM. Bayesian Network Inference Modeling Identifies TRIB1 as a Novel Regulator of Cell-Cycle Progression and Survival in Cancer Cells. Cancer Res. 2017;77(7):1575–85. doi: 10.1158/0008-5472.CAN-16-0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.