

Abstract
Hepatocellular carcinoma (HCC) is one of the most prevalent malignancies worldwide. Histone-lysine N-methyltransferase SET7/9 is a protein lysine monomethylase that methylates histone H3K4 as well as various non-histone proteins. Deregulation of SET7/9 is frequently detected in human cancers. However, the role of SET7/9 in HCC development remains unclear. In the present study, upregulation of SET7/9 and E2F transcription factor 1 (E2F1) expression was detected in 68 samples of HCC tissues compared with these levels noted in the paired healthy liver samples. The expression levels of SET7/9 and E2F1 were significantly correlated with pathological stage and tumor size. Subcellular fractionation and co-immunoprecipitation analyses revealed protein-protein interaction between SET7/9 and E2F1 in the cytoplasm of HCC cells. Silencing of SET7/9, as well as treatment with 5′-deoxy-5′-methylthioadenosine (MTA), a protein methylation inhibitor, led to reduced E2F1 protein abundance in HCC cells. Using Cell Counting Kit-8 (CCK-8) assay, Transwell migration assay and wound healing assay, significantly decreased cell proliferation, migration and invasion were observed in cells exhibiting downregulation of SET7/9 and E2F1 expression, as well as in wild-type HCC cells treated with MTA. Furthermore, SET7/9 downregulation and MTA treatment resulted in reduced expression of downstream targets of E2F1, including cyclin A2, cyclin E1 and CDK2. In conclusion, the present study revealed an oncogenic function of SET7/9 in HCC and demonstrated that SET7/9 may be responsible for alterations in the proliferative ability, aggressiveness and invasive/metastatic potential of HCC cells through post-translational regulation of E2F1.
Keywords: cancer progression, E2F1, hepatocellular carcinoma, methylation, SET7/9
Introduction
Hepatocellular carcinoma (HCC) is the fourth most prevalent type of cancer and accounts for the third highest cancer-related mortality rate in China (1). One of the greatest problems preventing potential curative treatment for HCC, including ablation, transplantation and resection, is the high recurrence rate, which may reach 50% within 2 years (2). Furthermore, surgical intervention is not feasible for the majority of HCC patients with intermediate- or advanced-stage disease, while chemotherapy and radiotherapy have exhibited only limited efficacy (3). A number of different factors, including abnormal mRNA and protein expression, changes in histone modification and DNA methylation, have been implicated in the initiation and progression of HCC (4–8). However, the molecular signatures are broad and without precise overlap between studies, making them unlikely to be applied in clinical practice (2). Therefore, a better understanding of the molecular mechanisms underlying HCC development may reveal possible pathogenic and therapeutic implications and help improve the overall survival of HCC patients.
Located on human chromosome 4q28, [Su(var)3-9, Enhancer-of-zeste and Trithorax] domain-containing protein 7 (SET7/9) is a member of the protein lysine methyltransferase (PLMT) family preferentially acting on non-histone proteins (9–12). In recent years, more than 30 non-histone proteins, including tumor suppressors, membrane-associated receptors and transcription factors, have been identified as targets of SET7/9 (13–17). Changes in the expression level of SET7/9 are frequently detected in human osteosarcoma, lung, colon and cervical cancer (16–20). However, due to its promiscuous targeting of different substrates, SET7/9-mediated methylation may produce different biological outcomes under different cellular contexts, acting as a co-activator or a co-repressor (21,22). Therefore, the effects of SET7/9-mediated methylation on carcinogenesis and the related molecular mechanisms require further investigation.
Thus far, the precise role of SET7/9 in HCC development has not been well characterized. Using gene expression profiling, a previous study identified two co-factors of SET7/9 involved in HCC progression, namely zinc finger and BTB domain-containing 20 (ZBTB20) and cyclin-dependent kinase inhibitor 2D (CDKN2D) (23). However, the regulatory mechanism between SET7/9 and these two co-factors remains unknown. The aim of the present study was to investigate the expression of SET7/9 in 68 clinical HCC samples and evaluate the association between SET7/9 expression and clinical characteristics. SET7/9-downregulated HCC cell lines were established to investigate the effects of aberrant SET7/9 expression on the behavior of HCC cells in vitro, and evidence was presented supporting the role of E2F transcription factor 1 (E2F1) as an important methylation substrate and co-regulator of SET7/9 during HCC initiation and progression.
Materials and methods
Ethics statement
This study was conducted strictly in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki) (24). The protocol was approved by the Ethics Committee of Southeast University and No. 81 Hospital of PLA (Nanjing, China). For the use of clinical materials, prior approval was obtained from No. 81 Hospital of PLA and written informed consent forms were signed by all the patients.
Tissue specimens and cell cultures
Sixty-eight HCC tissue samples from 58 male and 10 female patients aged between 26 and 87 (average age, 56 years) along with the matched adjacent healthy tissue samples from the same patient were collected between January 2015 and December 2017 at the Department of Pathology, No. 81 Hospital of PLA (Nanjing, China). None of the patients had received radiotherapy or chemotherapy prior to surgery. The medical records of the patients were reviewed to obtain the clinicopathological characteristics. The clinical stage of HCC was evaluated according to the TNM classification systems (25). All primary HCC samples were freshly frozen in liquid nitrogen (or fixed in 10% formalin) within 1 h after surgical resection. Samples for reverse transcription-quantitative polymerase chain reaction (RT-qPCR) and western blot analyses were stored at −80°C. Samples for immunohistochemical assay were embedded in paraffin, sectioned consecutively into 4-µm slices using a rotary microtome, and stained using hematoxylin and eosin.
Two human HCC cell lines (Huh7 and SK-HEP-1), as well as a normal human liver cell line (LO2), were originally purchased from the Type Culture Collection of the Chinese Academy of Science (Shanghai, China). One HCC cell line Hep3B was purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA). All the four cell lines were cultured in RPMI-1640 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS) at 37°C in a humidified atmosphere with 5% CO2. All studies using these cell lines were conducted within 6 months upon receipt or resuscitation.
Immunohistochemistry (IHC)
For IHC analysis, the tissue sections were deparaffinized and rehydrated. Endogenous peroxidase activity was inhibited with 0.3% H2O2 in methanol. For antigen retrieval, the slides were boiled in 0.01 M (pH 6.0) sodium citrate buffer for 15 min. After blocking with 5% normal goat serum, the slides were incubated with primary antibodies (rabbit anti-SET7/9, 1:50; cat. no. 2813; Cell Signaling Technology, Inc., Danvers, MA, USA; rabbit anti-E2F1, 1:1,000; cat. no. ab179445; Abcam, Cambridge, MA, USA) at 4°C overnight. Subsequently, biotinylated secondary antisera were applied and the slides were incubated with streptavidin-biotin horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (1:2,000; cat. no. 7074; Cell Signaling Technology, Inc.) for 30 min at room temperature. Finally, the visualization signal was developed by incubation in 3–3′-diaminobenzidine solution and hydrogen peroxide (Maixin, Fuzhou, China), followed by counterstaining with Mayer's haematoxylin. Images were taken (magnification, ×40) with an Olympus BX53 light microscope (Olympus Corp., Center Valley, PA, USA).
Plasmids and transfection
HCC cell lines with SET7/9 short hairpin (sh)RNA-induced silencing were generated using the pCMV-Tag5B vector. HCC cell lines with E2F1 shRNA-induced silencing were generated using the pIRES2-Zs1 vector. The following sense strand sequences of RNA duplexes for shRNA interference were designed to target the E2F1 and SET7/9 genes: shE2F1: 5′-TATAACAACGGTAGTGGATCC-3′ and shSET7/9: 5′-CACCGCTCTTCTTCCAGATCCTTATTTCAAGAGAATAAGGATCTGGAAGAAGAGCTTTTTTG-3′.
The silencing vectors of SET7/9 and E2F1, as well as the control vectors, were constructed by Invitrogen/Thermo Fisher Scientific, Inc. (Waltham, MA, USA). The vectors were transfected into HCC cells using Superfect Transfection Reagent (Qiagen Inc., Valencia, CA, USA) according to the manufacturer's instructions. Cells transfected with scrambled-shRNA vectors were used as negative controls. The silencing efficiency was confirmed using western blotting 48 h after transfection.
RNA extraction and RT-qPCR
Total RNA was extracted from HCC cells using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer's instructions. cDNA was then synthesized from 1 µg of total RNA using the RevertAid™ First Strand cDNA Synthesis kit (Fermentas, Burlington, ON, Canada). RT-qPCR was conducted on the ABI StepOne Plus Real-Time PCR System (Applied Biosystems; Thermo Fisher Scientific, Inc.) using the Platinum SYBR-Green Master Mix (Invitrogen; Thermo Fisher Scientific, Inc.). Amplification was performed in 20 µl reaction mixture containing 10 µl Supermix, 0.8 µM of each primer and 0.1–0.5 µg template cDNA. The primer sequences used for amplification are shown in Table I. Each experiment was conducted with at least three independent replicates. Relative quantification of mRNA expression was calculated with the ΔΔCq method using the expression level of GAPDH as an internal control (26).
Table I.
Primer sequences used for RT-qPCR analysis.
| Amplified genes | Primer | Oligonucleotide |
|---|---|---|
| SET7/9 | Forward (5′-3′) | CACTGGTGCCCAGGTGTACT |
| Reverse (3′-5′) | TTGGATGGCATGGCACTCATA | |
| E2F1 | Forward (5′-3′) | CATCAGTACCTGGCCGAGAG |
| Reverse (3′-5′) | TGGTGGTCAGATTCAGTGAGG | |
| GAPDH | Forward (5′-3′) | GGAGCGAGATCCCTCCAAAAT |
| Reverse (3′-5′) | GGCTGTTGTCATACTTCTCATGG |
Western blot analysis
For protein extraction, ~1×107 cells were collected and rinsed with ice-cold phosphate-buffered saline (PBS). The cells were then re-suspended and lysed in 1 ml RIPA buffer [13 PBS, 1% NP40, 0.1% sodium dodecyl sulfate (SDS), 5 mM EDTA, 0.5% sodium deoxycholate and 1 mM sodium orthovanadate] with protease inhibitors on ice. Protein concentrations were determined using the Bradford reagent (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Protein lysates were then resolved on 12% SDS polyacrylamide gel and electrotransferred to polyvinylidene fluoride membranes (ImmobilonP; Millipore, Bedford, MA, USA). After blocking with 5% milk solution in Tris-buffered saline (100 mM NaCl, 50 mM Tris and 0.1% Tween-20; pH 7.5), the membranes were immunoblotted with the following primary antibodies: SET7/9 (1:1,000; cat. no. 2813; Cell Signaling Technology, Inc.), GAPDH (1:2,000; cat. no. G9545; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany), E2F1 (1:1,000; cat. no. ab179445; Abcam), cyclin E1 (1:1,000; cat. no. 20808; Cell Signaling Technology, Inc.), cyclin A2 (1:2,000; cat. no. ab181591; Abcam) and CDK2 (1:1,000; cat. no. ab32147; Abcam) for 2 h at room temperature, followed by the addition of the respective secondary antibodies (HRP-conjugated anti-rabbit IgG, 1:5,000; cat. no. 7074; Cell Signaling Technology, Inc.; and HRP-conjugated anti-mouse IgG, 1:5,000; cat. no. 7076; Cell Signaling Technology, Inc.). Finally, signals were examined with the Pierce enhanced chemiluminescence detection system (Thermo Fisher Scientific, Inc.) according to the manufacturer's instructions.
Subcellular fractionation and co-immunoprecipitation
Subcellular fractionation was conducted using the NE-PER kit according to the manufacturer's instructions (Thermo Fisher Scientific Inc., Rockford, IL, USA). For co-immunoprecipitation, cell lysates, as well as lysates of the cytoplasm and nuclei from Huh7 cells, were cleared by centrifugation at 15,100 × g for 10 min at 4°C, followed by incubation with pre-blocked Protein A-Sepharose beads (Zymed Laboratories, Inc., San Francisco, CA, USA) and individual antibodies (SET7/9, 1:1,000; cat. no. 2813; Cell Signaling Technology, Inc.; E2F1, 1:1,000; cat. no. ab179445; Abcam; normal rabbit IgG, cat. no. A0545; Sigma-Aldrich; Merck KGaA) were applied. After incubation overnight at 4°C, complexes with Protein-A Sepharose (Zymed Laboratories, Inc.) were harvested and the beads were washed three times in lysis buffer. Bound proteins were separated with SDS/PAGE and visualized using western blotting.
Cell proliferation assay
Cells were plated at a density of 2×103 in 96-well plates containing 100 µl of culture medium and shaken on a microplate shaker for 1–5 days at 37°C in a humidified atmosphere with 5% CO2. For the 5′-deoxy-5′-methylthioadenosine (MTA)-treated group, 1 mM MTA (no. D5011, Sigma-Aldrich; Merck KGaA), an inhibitor of protein methylation, was added into each well. To determine the number of cells at each time point, 10 µl of Cell Counting Kit-8 solution (Shanghai Obio Technology Corp., Ltd., Shanghai, China) was added into each well. The plate was then incubated for another 4 h before measurement using a Microplate Autoreader (Bio-Rad Laboratories, Hercules, CA, USA). The experiment was performed with three replicates.
Wound healing assay
Cells were grown to a monolayer in 6-well plates at a density of 1.25×106 per well for 24 h to 80–90% confluence and washed twice with PBS. Then, the cells were serum-starved and cultured in PBS-free medium for 24 h. For the MTA-treated group, 1 mM MTA was added into the medium. Using a 10-µl standard pipette tip, uniform parallel scratches were created across each well. The plates were washed twice with PBS to remove the detached cells. The wound margins were evaluated by digital photography using an inverted microscope (0 h). Cells were grown in the absence or presence of MTA for 24 h. Images were captured using an image-analyzing frame-grabber card (LG-3 Scientific Frame Grabber; Scion, Frederick, MD, USA) and analyzed with the image analysis software NIH Image 1.55 (National Institutes of Health, Bethesda, MD, USA). The unfilled scratched zones were quantified by measuring the distance between the advancing margins of cells in three randomly selected microscopic fields (magnification, ×200) at each time-point. The experiment was performed with three replicates.
Migration assay
Approximately 5×104 cells were suspended in serum-free medium and seeded into the upper chamber of 8-µm-pore Transwells (Costar, Corning, NY, USA). Then, 300 µl of 10% FBS-free medium (Gibco; Thermo Fisher Scientific) was added into the lower chamber. For the MTA-treated group, 1 mM MTA was added into the medium. The cells were allowed to migrate for 24 h, after which time the cells inside the chamber were removed with a cotton swab and cells in the lower chamber were fixed with 1% paraformaldehyde and stained with hematoxylin. The number of cells migrating through the chamber was evaluated in 5 randomly selected fields (magnification, ×200) using an Olympus GX41 light microscope (Olympus Corp.). The experiment was performed with three replicates.
Statistical analysis
All statistical computations were performed using SPSS software version 20 (IBM Corp., Armonk, NY, USA). The correlations between SET7/9 and E2F1 expression and various clinicopathological parameters were evaluated with the χ2 test. The significance of difference in the expression levels of SET7/9 and E2F1 between HCC tumor tissue and paired healthy tissue was evaluated with the paired t-test. Cell proliferative and migratory rates, cell invasive ability, and the expression levels of SET7/9 and E2F1 among three different cell groups were compared using one-way analysis of variance (ANOVA) followed by Tukey's post-hoc test. A P-value of <0.05 was considered to indicate statistically significant differences.
Results
SET7/9 and E2F1 are upregulated in HCC
Expression of SET7/9 and E2F1 at the protein level was analyzed in 68 clinical samples of surgically removed HCC tissues and paired adjacent healthy tissues using IHC (Figs. 1A and 2A). Among HCC tissue samples, SET7/9 exhibited high expression in 53 cases (77.94%) and low expression in the remaining 15 cases (22.06%, Table II). In the paired healthy liver samples, SET7/9 was undetectable in 41 cases (60.29%) and weakly expressed (staining score 1; percent positivity score 0–1) in 27 (39.71%) cases. Overall, SET7/9 expression was significantly higher in HCC tumor tissues compared with that in healthy liver tissues (P<0.05; Fig. 1B). The expression level of SET7/9 protein was significantly correlated with tumor size (P<0.001) and pathological stage (P<0.001; Table II). No significant correlation was detected between SET7/9 protein expression and other clinicopathological parameters, including age, sex, lymph node metastasis and distant metastasis (Table II). In the three HCC cell lines (Huh7, Hep3B and SK-HEP-1), SET7/9 exhibited the highest expression level in Huh7 cells and the lowest expression level in SK-HEP-1 cells (Fig. 1C). However, the expression level of SET7/9 was higher in all the HCC cell lines compared with that in the normal human liver cell line LO2 (Fig. 1C).
Figure 1.

Expression of SET7/9 in clinical HCC samples and cell lines. (A) Immunostaining of SET7/9 protein in HCC tissues and the surrounding non-cancerous tissues. (a) Negative expression (0) in healthy liver tissue. (b) Weak expression (1+) in HCC tissues. (c) Moderate expression (2+) in HCC tissues. (d) Strong expression (3+) in HCC tissues. (B) Staining scores of SET7/9 protein expression in HCC tissue samples and surrounding noncancerous tissues. The staining score ranged from 0 to 3, with 0 for no staining, 1 for weak staining, 2 for moderate staining, and 3 for strong staining. *P<0.05. (C) Expression of SET7/9 and E2F1 in three human HCC cell lines Huh7, Hep3B, and SK-HEP-1, and the human healthy liver cell line LO2. HCC, hepatocellular carcinoma.
Figure 2.

Expression of E2F1 in clinical HCC samples. (A) Immunostaining of E2F1 protein in HCC tissues and the surrounding non-cancerous tissues. (a) Negative expression (0) in healthy liver tissues. (b) Weak expression (1+) in HCC tissues. (c) Moderate expression (2+) in HCC tissues. (d) Strong expression (3+) in HCC tissues. (B) Staining scores of E2F1 protein expression in HCC tissue samples and surrounding noncancerous tissues. The staining score ranged from 0 to 3, with 0 for no staining, 1 for weak staining, 2 for moderate staining, and 3 for strong staining. *P<0.05. (C) Staining scores of E2F1 protein expression in HCC tissue samples with T1, T2, T3, and T4 stage and surrounding noncancerous tissues. (D) Staining scores of E2F1 protein expression in HCC tissue samples with N0 and N1 stage and surrounding noncancerous tissues. (E) Staining scores of E2F1 protein expression in HCC tissue samples with M0 and M1 stage and surrounding noncancerous tissues. T, tumor size; N, regional lymph node metastasis; M, distant metastatsis; HCC, hepatocellular carcinoma.
Table II.
Relationship between SET7/9 expression and the clinicopathological features of the 68 HCC patients.
| SET7/9 expressionb | |||||
|---|---|---|---|---|---|
| Features | n | Low | High | χ2 | P-value |
| Age (years) | 2.138 | 0.144 | |||
| ≤56 | 34 | 5 | 29 | ||
| >56 | 34 | 10 | 24 | ||
| Sex | 3.318 | 0.069 | |||
| Male | 58 | 15 | 43 | ||
| Female | 10 | 0 | 10 | ||
| TNM staginga | |||||
| T category | 29.224 | <0.001 | |||
| 1 | 27 | 15 | 12 | ||
| 2 | 10 | 0 | 10 | ||
| 3 | 30 | 0 | 30 | ||
| 3a | 14 | 0 | 14 | ||
| 3b | 16 | 0 | 16 | ||
| 4 | 1 | 0 | 1 | ||
| N category | 3.318 | 0.069 | |||
| 0 | 58 | 15 | 43 | ||
| 1 | 10 | 0 | 10 | ||
| M category | 0.461 | 0.497 | |||
| 0 | 67 | 15 | 52 | ||
| 1 | 1 | 0 | 1 | ||
| Pathological stage | 23.229 | <0.001 | |||
| I | 27 | 15 | 12 | ||
| II | 8 | 0 | 8 | ||
| III | 23 | 0 | 23 | ||
| A | 13 | 0 | 13 | ||
| B | 10 | 0 | 10 | ||
| C | 0 | 0 | 0 | ||
| IV | 10 | 0 | 10 | ||
| A | 9 | 0 | 9 | ||
| B | 1 | 0 | 1 | ||
The TNM classification system was used to evaluate the clinical stage of the HCC samples. T, tumor size; N, regional lymph node metastasis; M, distant metastatsis.
The total immunostaining score was calculated as the sum of the percentage of positively stained tumor cells and the staining intensity. The percentage of positivity was scored from 0 to 3, with 0 for <10% positively stained cells, 1 for 10–30% stained cells, 2 for >30–50% stained cells, and 3 for >50% positively stained cells. The staining intensity was scored from 0 to 3, with 0 for no staining, 1 for weak staining, 2 for moderate staining, and 3 for strong staining. Both the percentage of positively stained cells and staining intensity were decided in a double-blinded manner. Then the score of SET7/9 expression was calculated as the product of the value of the percentage of positivity score × staining intensity score, which ranged from 0 to 9. The final expression level of SET7/9 was defined as ‘low’ (0–4) and ‘high’ (5–9). HCC, hepatocellular carcinoma.
Similar to SET7/9, E2F1 exhibited high expression in 44 cases (64.71%) and low expression in the remaining 24 cases (35.29%) of HCC tumor tissues, but was undetectable in 31 cases (45.59%) and only weakly expressed (staining score 1; percent positivity score 0–1) in 37 (54.51%) cases of the paired healthy liver samples (Table III). The expression level of E2F1 protein was significantly higher in HCC tumor tissues compared with that in healthy liver tissues (P<0.05; Fig. 2B) and was significantly correlated with tumor size (P<0.001), regional lymph node metastasis (P<0.05) and pathological stage (P<0.001; Table III; Fig. 2C and D). However, no significant relationship was detected between E2F1 protein expression and other clinicopathological parameters, including age, sex and distant metastasis (Table III; Fig. 2E). Meanwhile, markedly higher expression of E2F1 was also detected in the three HCC cell lines compared with that in the LO2 cell line (Fig. 1C).
Table III.
Relationship between E2F1 expression and the clinicopathological features of the 68 HCC patients.
| E2F1 expression | |||||
|---|---|---|---|---|---|
| Features | n | Low | High | χ2 | P-value |
| Age (years) | 2.318 | 0.128 | |||
| ≤56 | 34 | 9 | 25 | ||
| >56 | 34 | 15 | 19 | ||
| Sex | 0.544 | 0.461 | |||
| Male | 58 | 22 | 36 | ||
| Female | 10 | 2 | 8 | ||
| TNM staginga | |||||
| T category | 25.980 | <0.001 | |||
| 1 | 27 | 19 | 8 | ||
| 2 | 10 | 3 | 7 | ||
| 3 | 30 | 2 | 28 | ||
| 3a | 14 | 2 | 12 | ||
| 3b | 16 | 0 | 16 | ||
| 4 | 1 | 0 | 1 | ||
| N category | 4.711 | 0.030 | |||
| 0 | 58 | 24 | 34 | ||
| 1 | 10 | 0 | 10 | ||
| M category | 0.096 | 0.757 | |||
| 0 | 67 | 24 | 43 | ||
| 1 | 1 | 0 | 1 | ||
| Pathological stage | 27.142 | <0.001 | |||
| I | 27 | 19 | 8 | ||
| II | 8 | 3 | 5 | ||
| III | 23 | 2 | 21 | ||
| A | 13 | 2 | 11 | ||
| B | 10 | 0 | 10 | ||
| C | 0 | 0 | 0 | ||
| IV | 10 | 0 | 10 | ||
| A | 9 | 0 | 9 | ||
| B | 1 | 0 | 1 | ||
The TNM classification system was used to evaluate the clinical stage of HCC samples. T, tumor size; N, regional lymph node metastasis; M, distant metastatsis.
The total immunostaining score was calculated as the sum of the percentage of positively stained tumor cells and the staining intensity. The percentage of positivity was scored from 0 to 3, with 0 for <10% positively stained cells, 1 for 10–30% stained cells, 2 for >30–50% stained cells, and 3 for >50% positively stained cells. The staining intensity was scored from 0 to 3, with 0 for no staining, 1 for weak staining, 2 for moderate staining, and 3 for strong staining. Both the percentage of positively stained cells and staining intensity were decided in a double-blinded manner. Then the score of E2F1 expression was calculated as the product of the value of the percentage of positivity score × staining intensity score, which ranged from 0 to 9. The final expression level of E2F1 was defined as ‘low’ (0–4) and ‘high’ (5–9). HCC, hepatocellular carcinoma.
SET7/9-mediated post-translational methylation increases E2F1 abundance
In order to further elucidate the functions of SET7/9 and E2F1 and their role in HCC cells, SET7/9- and E2F1-underexpressing HCC cell lines (Huh7-SET7/9-shRNA and Huh7-E2F1-shRNA, respectively) were established and wild-type Huh7 cells transduced with scrambled-shRNA vectors were used as negative controls. The efficiency of gene silencing was confirmed using western blot analysis (Fig. 3A and C). Downregulation of SET7/9 in Huh7 cells did not affect E2F1 expression at the mRNA level, but decreased E2F1 expression at the protein level (Fig. 3A and B). Treatment with MTA, an inhibitor of protein methylation, in Huh7 cells led to the similar changes in E2F1 protein expression compared with the SET7/9-underexpressing group (Fig. 3A and B). However, no evident change in SET7/9 expression at the mRNA or protein level was observed after E2F1 downregulation or MTA treatment (Fig. 3C and D).
Figure 3.

SET7/9 directly interacts with E2F1 and regulates E2F1 abundance through post-translational methylation in HCC cells. (A) E2F1 expression at the protein level was downregulated in SET7/9-silenced cells and MTA-treated cells as compared with the control cells. (B) E2F1 showed similar mRNA expression levels in SET7/9-silenced cells, MTA-treated cells, and the control cells. (C) SET7/9 expression at the protein level did not show significant change after E2F1 downregulation and MTA treatment as compared with the control cells. (D) SET7/9 showed similar mRNA expression levels in E2F1-silenced cells, MTA-treated cells, and the control cells. (E) Co-immunoprecipitation of total cell and subcellular fractions for SET7/9 and E2F1 in the HCC cell line Huh7. Lysates were immunoprecipitated with SET7/9 antibody or control IgG and detected with E2F1 antibody on a western blotting, then immunoprecipitated with E2F1 antibody or control IgG and detected with SET7/9 antibody on a western blotting. Each error bar represents the mean ± SD of three replicate samples. HCC, hepatocellular carcinoma.
Co-immunoprecipitation analysis revealed protein-protein interaction between SET7/9 and E2F1 in HCC cells, indicating that SET7/9 was physically associated with E2F1 (Fig. 3E). Subcellular fractions derived from Huh7 HCC cells were further analyzed for the location of interaction between SET7/9 and E2F1. Direct interaction of the two proteins was detected in the cytoplasm, while no interaction was detected in the nucleus (Fig. 3E).
Silencing of SET7/9 expression suppresses the proliferation, migration and invasion of human HCC cells
To investigate whether SET7/9 plays a role in the biological behavior of HCC cells in vitro, we examined changes in cell proliferative, migratory and invasive abilities following SET7/9 downregulation. Huh7-shRNA-SET7/9 cells exhibited significantly decreased cell proliferation as compared with the control cells transduced with scrambled-shRNA vectors (P<0.05; Fig. 4A). Furthermore, a significantly lower migratory rate and slower wound closure were observed in the Huh7-shRNA-SET7/9 cells, as revealed by the Transwell migration and wound healing assays (P<0.05; Fig. 4B and C).
Figure 4.

Silencing of SET7/9 leads to changes in cellular behavior of human HCC cells. (A) SET7/9 downregulation and MTA treatment significantly reduced cell proliferation as determined by cell proliferation assay. (B) SET7/9-silenced HCC cells as well as MTA-treated cells showed decreased migration and invasion through extracellular matrix (ECM) as indicated by Transwell migration assay. Representative images (left) and quantification (right) are shown. The number of cells that migrated through the ECM after 24 h was counted in five randomly selected (×200) microscopic fields. (C) SET7/9 downregulation and MTA treatment led to reduced cell invasive ability as revealed by wound healing assay. Images were captured at 0 and 24 h. At 24 h, the extent of wound closure was 31.33% (±3.20%) for SET7/9-silenced cells, 33.00% (±5.06%) for MTA-treated cells, but was 79.50% (±3.73%) for the control cells (NC) transduced with scrambled-shRNA vectors. Each error bar represents the mean ± SD of three replicate samples. *P<0.05. HCC, hepatocellular carcinoma.
To examine whether these changes in cellular behavior are associated with the methylation activity of SET7/9, wild-type Huh7 cells were also treated with MTA, which inhibits protein methylation. The results demonstrated that treatment with MTA significantly suppressed cell proliferation, migration and invasion. The proliferative, migratory and invasive abilities of cells from the MTA group were comparable to those observed in the SET7/9-silenced group (Fig. 4).
Silencing of E2F1 expression suppresses the proliferation, migration and invasion of human HCC cells
To investigate whether E2F1 is involved in HCC initiation and metastasis as a potential target of SET7/9, changes in cell proliferative, migratory and invasive abilities were examined after E2F1 downregulation. Huh7-shRNA-E2F1 cells exhibited significantly decreased cell proliferation as compared with the control cells transduced with scrambled-shRNA vectors (P<0.05; Fig. 5A). Furthermore, a markedly lower migratory rate and slower wound closure were observed in Huh7-shRNA-E2F1 cells, as revealed by the Transwell migration and wound healing assays (P<0.05; Fig. 5B and C). Treatment of the wild-type Huh7 cells with MTA exerted similar effects. The proliferative, migratory and invasive abilities of cells from the MTA group were comparable to those observed in the E2F1-silenced group (Fig. 5).
Figure 5.

Silencing of E2F1 leads to changes in cellular behavior of human HCC cells. (A) E2F1 downregulation and MTA treatment significantly reduced cell proliferation as determined by cell proliferation assay. (B) E2F1-silenced HCC cells as well as MTA-treated cells showed decreased migration and invasion through extracellular matrix (ECM) as indicated by Transwell migration assay. Representative images (left) and quantification (right) are shown. The number of cells migrated through the ECM after 24 h was counted in five randomly selected (×200) microscopic fields. (C) E2F1 downregulation and MTA treatment led to reduced cell invasive ability as revealed by wound healing assay. At 24 h, the extent of wound closure was 33.17% (±2.79%) for E2F1-silenced cells, 37.67% (±1.86%) for MTA-treated cells, but was 95.00% (±2.61%) for the control cells (NC) transduced with scrambled-shRNA vectors. Each error bar represents the mean ± SD of three replicate samples. *P<0.05. HCC, hepatocellular carcinoma.
Silencing of SET7/9 leads to decreased expression of E2F1 downstream targets
The downstream activation of E2F1 target genes in SET7/9-silenced HCC cells was next investigated. Decreased protein abundance of E2F1, cyclin E1, cyclin A2 and CDK2 was observed in the SET7/9- silenced cells, as revealed by western blotting (Fig. 6). Treatment of wild-type Huh7 cells with MTA resulted in similar changes in the protein abundance of E2F1, cyclin E1, cyclin A2 and CDK2 as compared with those observed in SET7/9- silenced Huh7 cells (Fig. 6).
Figure 6.
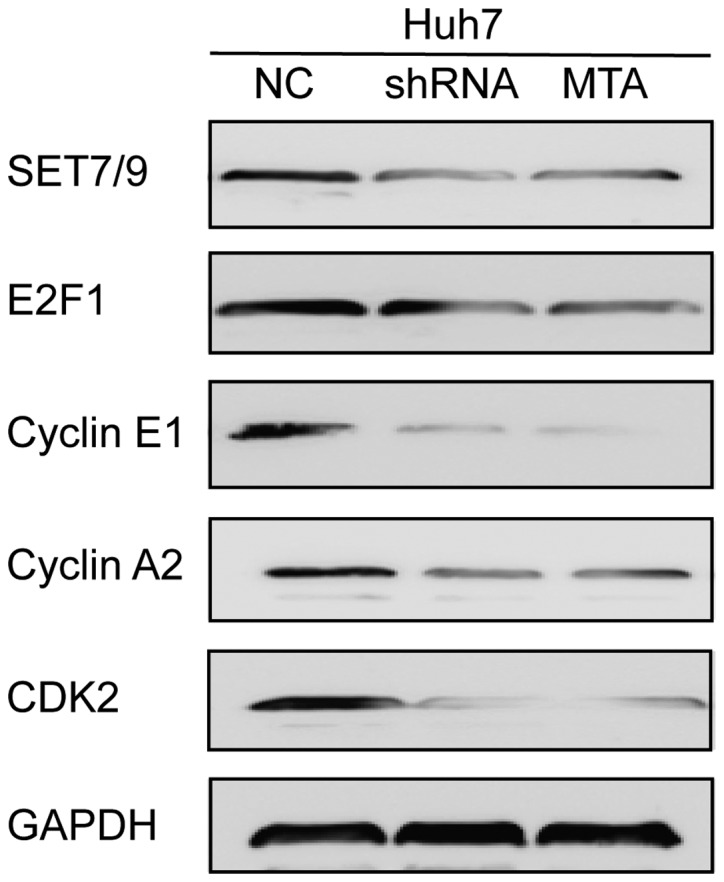
Silencing of SET7/9 leads to changes in the expression of E2F1 downstream targets. Downregulation of E2F1, cyclin E1, cyclin A2, and CDK2 was observed in both the SET7/9-silenced and the MTA-treated cells.
Discussion
SET7/9 is a methyltransferase that methylates both lysine 4 of histone 3 (H3K4) and lysine(s) of non-histone proteins. The role of SET7/9 in human cancer development has attracted attention in recent years. In breast cancer and chronic lymphocytic leukemia, SET7/9 has been implicated as a potential prognostic marker for tumor sensitivity to genotoxic therapies (17). In the present study, upregulation of SET7/9 was detected in clinical tumor samples of HCC patients, as well as in the three investigated HCC cell lines (Fig. 1). The expression level of SET7/9 was significantly correlated with tumor size (P<0.001) and pathological stage (P<0.001) (Table II), indicating a positive correlation between SET7/9 expression and HCC progression. Silencing of SET7/9 in HCC cells significantly suppressed cell proliferation, migration and invasion (Fig. 4). These results suggest an invasion-promoting effect of SET7/9 during HCC progression. Consistently, significantly upregulated expression of SET7/9 was previously detected in colon adenocarcinoma, head and neck squamous cell carcinoma, kidney carcinoma, lung adenocarcinoma, prostate cancer, pancreatic adenocarcinoma and uveal melanoma in the TCGA database (Fig. 7). In addition, in regards to breast (27), lung (28,29) and ovarian cancer (30), studies combining clinical expression analysis and in vitro functional analyses have reported an oncogenic role of SET7/9 in carcinogenesis.
Figure 7.

SET7/9 expression profiling from public databases. (A-F) Box plots from the TCGA database showing the differences in the expression level of SET7/9 in normal and tumor tissue from various types of cancer. (A) Colon adenocarcinoma (COAD), (B) head and neck squamous cell carcinoma (HNSC), (C) kidney renal clear cell carcinoma (KIRC), (D) kidney renal papillary cell carcinoma (KIRP), (E) pancreatic adenocarcinoma (PAAD), (F) uveal melanoma (UVM). (G) Summary of SET7/9 expression profiling in different cancer types in the ONCOMINE database. The expression differences in all box plots are significant with a P-value <0.05.
However, diverged expression patterns and functions of SET7/9 in different cancer types have been reported. For example, SET7/9 was shown to act as a tumor suppressor in cervical cancer (20), colon cancer (16), acute myeloid leukemia (29) and osteosarcoma (12,18,31). In the ONCOMINE database, conflicting expression profiles for SET7/9 have been documented in different studies. Significantly higher expression level of SET7/9 was detected in 6 cases of clinical cancer compared with normal tissues, whereas significantly lower SET7/9 expression was detected in two cases (P<0.05, www.oncomine.org; Fig. 7). Given the various protein substrates of SET7/9 responsible for the regulation of gene expression, cell cycle arrest, cell apoptosis, cell growth and cell viability, it was hypothesized that SET7/9-mediated lysine methylation is a ubiquitous post-translational modification that modulates proteins involved in various cellular processes (32). The final outcome may depend on the dominant target of SET7/9 in certain cancer types. Therefore, it is important to elucidate the detailed molecular mechanism underlying the carcinostasis- or carcinogenesis-related process mediated by SET7/9.
Similar to SET7/9, significant upregulation of E2F1 was observed in HCC, as revealed by our clinical analysis (Fig. 2). As one of the best characterized members of the E2F transcription factor family, E2F1 plays an integral role in the coordination of DNA replication events. Previous studies have proven that SET7/9 methylates E2F1 at lysine 185 (33). We herein demonstrated that SET7/9 physically interacts with E2F1 in the cytoplasm of HCC cells (Fig. 3). E2F1 expression at the protein level was decreased following downregulation of SET7/9, while E2F1 expression at the mRNA level remained almost unchanged (Fig. 3). Treatment with MTA, an inhibitor of protein methylation, also led to similar changes in E2F1 protein expression compared with the SET7/9-underexpressing group (Fig. 3). These results suggest that SET7/9-mediated post-translational methylation likely alters E2F1 stability and abundance in HCC cells. Furthermore, no evident change in SET7/9 expression at the mRNA or protein level was observed after silencing of E2F1 (Fig. 3). This is in accordance with the previous hypothesis that SET7/9 is located upstream of E2F1, acting as a signal transducer that assists in cell DNA damage response (31).
To examine whether E2F1 participates in the oncogenic processes downstream of SET7/9 in HCC cells, an E2F1-silenced cell line was next established. Similar to the SET7/9-silenced cells, E2F1-silenced cells exhibited decreased proliferative, migratory and invasive properties (Fig. 5). In fact, E2F1 is commonly overexpressed in HCC (34). In HCC and clear cell renal cell carcinomas, E2F1 activates transcription of the CCNE gene, the product of which, cyclin E, forms a complex with CDK2 to promote DNA replication and tumor differentiation (35,36). On the contrary, in osteosarcoma, E2F1 can induce cell apoptosis by transcriptionally activating its downstream target gene, p73 (37). It was hypothesized that when the cell apoptotic machinery malfunctions, the function of E2F1 in triggering apoptotic cell death may be reversed (38). Thus, E2F1 cannot fulfill its pro-apoptotic role, but rather upregulates the expression of its own co-factors and leads to sustained cell proliferation/migration and suppressed cell apoptosis (34,39,40). In the present study, we observed decreased expression of cyclin A2, cyclin E1 and CDK2 in SET7/9-underexpressing cells (Fig. 6). Furthermore, treatment of the wild-type HCC cells with MTA exerted similar effects (Figs. 4–6). Taken together, these results suggest that SET7/9-mediated methylation of E2F1 may be responsible for the stabilization of E2F1 protein and the activation of E2F1 downstream transcriptional targets.
In conclusion, the present study demonstrated that SET7/9 promotes HCC progression through regulation of E2F1. Previously, SET7/9 was reported to play a key role in HCC and predict poor prognosis, mainly through regulation of two different co-factors, ZBTB20 and CDKN2D, which are correlated with cell cycle arrest (23). We herein provide evidence that SET7/9 affects E2F1 stability by post-translational methylation, and silencing of SET7/9 inhibits HCC cell proliferation, migration and invasion. Of note, SET7/9-dependent methylation of E2F1 may stabilize E2F1 protein expression in colon cancer, but may interfere with the acetylation and phosphorylation of E2F1 and promote its degradation in lung cancer (28,41). Since the stability of proteins is controlled by complicated post-translational covalent modifications, it is possible that different protein-protein interactions or different types and amounts of post-translational modifiers in different cell contexts may lead to discrepant effects in the stability of the target protein (41). Therefore, to evaluate the clinical potential of SET7/9 for therapeutics and diagnostics, further studies are required to identify the co-regulators of SET7/9 involved in the modification of the E2F1 protein or in other carcinogenic processes under different genetic backgrounds.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Saskatchewan Health Research Foundation of Canada, the National Natural Science Foundation of China (grant no. 81071654), the Fundamental Research Funds for the Central Universities, the Southeast University Fundamental Research Funding Project (3224008704) and the Postgraduate Research & Practice Innovation Program of Jiangsu Province (grant no. KYCX17_0171).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
YG conducted most of the experiments and wrote the manuscript; XW performed all the immunohistochemical analyses; HL prepared the reagents and chemicals; GL collected and embedded the clinical specimens; WY designed the study and was also involved in the conception of the study; and QM helped with the purchase of some reagents for the experiments and revised the manuscript. All authors read and approved the manuscript and agree to be accountable for all aspects of the research in ensuring that the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Ethics approval and consent to participate
This study was conducted strictly in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki) (24). The protocol was approved by the Ethics Committee of Southeast University and No. 81 Hospital of PLA (Nanjing, China). For the use of clinical materials, prior approval was obtained from No. 81 Hospital of PLA and written informed consent forms were signed by all the patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 2.Bruix J, Gores GJ, Mazzaferro V. Hepatocellular carcinoma: Clinical frontiers and perspectives. Gut. 2014;63:844. doi: 10.1136/gutjnl-2013-306627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu CY, Chen SY, Peng HL, Kan PY, Chang WC, Yen CJ. Cell-free methylation markers with diagnostic and prognostic potential in hepatocellular carcinoma. Oncotarget. 2017;8:6406–6418. doi: 10.18632/oncotarget.14115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Toffanin S, Hoshida Y, Lachenmayer A, Villanueva A, Cabellos L, Minguez B, Savic R, Ward SC, Thung S, Chiang DY, et al. MicroRNA-based classification of hepatocellular carcinoma and oncogenic role of miR-517a. Gastroenterology. 2011;140(1618–1628):e16. doi: 10.1053/j.gastro.2011.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH, Aoki M, Hosono N, Kubo M, Miya F, et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. 2012;44:760–764. doi: 10.1038/ng.2291. [DOI] [PubMed] [Google Scholar]
- 6.Shen J, Wang S, Zhang YJ, Kappil M, Wu HC, Kibriya MG, Wang Q, Jasmine F, Ahsan H, Lee PH, et al. Genome-wide DNA methylation profiles in hepatocellular carcinoma. Hepatology. 2012;55:1799–1808. doi: 10.1002/hep.25569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anwar SL, Lehmann U. DNA methylation, microRNAs, and their crosstalk as potential biomarkers in hepatocellular carcinoma. World J Gastroenterol. 2014;20:7894–7913. doi: 10.3748/wjg.v20.i24.7894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cancer Genome Atlas Research Network: Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 2017;169:1327. doi: 10.1016/j.cell.2017.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang H, Cao R, Xia L, Erdjument-Bromage H, Borchers C, Tempst P, Zhang Y. Purification and functional characterization of a histone H3-lysine 4-specific methyltransferase. Mol Cell. 2001;8:1207–1217. doi: 10.1016/S1097-2765(01)00405-1. [DOI] [PubMed] [Google Scholar]
- 10.Nishioka K, Chuikov S, Sarma K, Erdjument-Bromage H, Allis CD, Tempst P, Reinberg D. Set9, a novel histone H3 methyltransferase that facilitates transcription by precluding histone tail modifications required for heterochromatin formation. Genes Dev. 2002;16:479–489. doi: 10.1101/gad.967202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ivanov GS, Ivanova T, Kurash J, Ivanov A, Chuikov S, Gizatullin F, Herrera-Medina EM, Rauscher F, III, Weinberg D, Barley NA. Methylation-acetylation interplay activates p53 in response to DNA damage. Mol Cell Biol. 2007;27:6756–6769. doi: 10.1128/MCB.00460-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kurash JK, Lei H, Shen Q, Marston WL, Granda BW, Fan H, Wall D, Li E, Gaudet F. Methylation of p53 by Set7/9 mediates p53 acetylation and activity in vivo. Mol Cell. 2008;29:392–400. doi: 10.1016/j.molcel.2007.12.025. [DOI] [PubMed] [Google Scholar]
- 13.Ea CK, Baltimore D. Regulation of NF-kappaB activity through lysine monomethylation of p65. Proc Natl Acad Sci USA. 2009;106:18972–18977. doi: 10.1073/pnas.0910439106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang J, Huang J, Dasgupta M, Sears N, Miyagi M, Wang B, Chance MR, Chen X, Du Y, Wang Y, et al. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc Natl Acad Sci USA. 2010;107:21499–21504. doi: 10.1073/pnas.1016147107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang D, Zhou J, Liu X, Lu D, Shen C, Du Y, Wei FZ, Song B, Lu X, Yu Y, et al. Methylation of SUV39H1 by SET7/9 results in heterochromatin relaxation and genome instability. Proc Natl Acad Sci USA. 2013;110:5516–5521. doi: 10.1073/pnas.1216596110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu X, Wang D, Zhao Y, Tu B, Zheng Z, Wang L, Wang H, Gu W, Roeder RG, Zhu WG. Methyltransferase Set7/9 regulates p53 activity by interacting with Sirtuin 1 (SIRT1) Proc Natl Acad Sci USA. 2011;108:1925–1930. doi: 10.1073/pnas.1019619108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lezina L, Aksenova V, Fedorova O, Malikova D, Shuvalov O, Antonov AV, Tentler D, Garabadgiu AV, Melino G, Barlev NA. KMT Set7/9 affects genotoxic stress response via the Mdm2 axis. Oncotarget. 2015;6:25843–25855. doi: 10.18632/oncotarget.4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Munro S, Khaire N, Inche A, Carr S, Thangue La NB. Lysine methylation regulates the pRb tumour suppressor protein. Oncogene. 2010;29:2357–2367. doi: 10.1038/onc.2009.511. [DOI] [PubMed] [Google Scholar]
- 19.Carr SM, Munro S, Zalmas LP, Fedorov O, Johansson C, Krojer T, Sagum CA, Bedford MT, Oppermann U, Thangue La NB. Lysine methylation-dependent binding of 53BP1 to the pRb tumor suppressor. Proc Natl Acad Sci USA. 2014;111:11341–11346. doi: 10.1073/pnas.1403737111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen C, Wang D, Liu X, Gu B, Du Y, Wei FZ, Cao LL, Song B, Lu X, Yang Q, et al. SET7/9 regulates cancer cell proliferation by influencing β-catenin stability. FASEB J. 2015;29:4313–4123. doi: 10.1096/fj.15-273540. [DOI] [PubMed] [Google Scholar]
- 21.Yang XD, Huang B, Li M, Lamb A, Kelleher NL, Chen LF. Negative regulation of NF-kappaB action by Set9-mediated lysine methylation of the RelA subunit. EMBO J. 2009;28:1055–1066. doi: 10.1038/emboj.2009.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kassner I, Andersson A, Fey M, Tomas M, Ferrando-May E, Hottiger MO. SET7/9-dependent methylation of ARTD1 at K508 stimulates poly-ADP-ribose formation after oxidative stress. Open Biol. 2013;3:120173. doi: 10.1098/rsob.120173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Y, Yang S, Hu J, Yu C, He M, Cai Z. Increased expression of SETD7 promotes cell proliferation by regulating cell cycle and indicates poor prognosis in hepatocellular carcinoma. PLoS One. 2016;11:e0154939. doi: 10.1371/journal.pone.0154939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.World Medical Association Declaration of Helsinki: Ethical principles for medical research involving human subjects. JAMA. 2013;310:2191–2194. doi: 10.1001/jama.2013.281053. [DOI] [PubMed] [Google Scholar]
- 25.Gospodarowicz MK, Brierley JD, Wittekind C, editors. TNM classification of malignant tumours. 8th edition. John Wiley & Sons; Oxford, UK: 2017. [Google Scholar]
- 26.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y, Liu J, Lin J, Zhou L, Song Y, Wei B, Luo X, Chen Z, Chen Y, Xiong J, et al. The transcription factor GATA1 and the histone methyltransferase SET7 interact to promote VEGF-mediated angiogenesis and tumor growth and predict clinical outcome of breast cancer. Oncotarget. 2016;7:9859–9875. doi: 10.18632/oncotarget.7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kontaki H, Talianidis I. Lysine methylation regulates E2F1-induced cell death. Mol Cell. 2010;39:152–160. doi: 10.1016/j.molcel.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 29.Gu Y, Wang Y, Wang X, Gao L, Yu W, Dong WF. Opposite effects of SET7/9 on apoptosis of human acute myeloid leukemia cells and lung cancer cells. J Cancer. 2017;8:2069–2078. doi: 10.7150/jca.19143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou J, Xie M, Shi Y, Luo B, Gong G, Li J, Wang J, Zhao W, Zi Y, Wu X, et al. Microrna-153 functions as a tumor suppressor by targeting set7 and zeb2 in ovarian cancer cells. Oncol Rep. 2015;34:111–120. doi: 10.3892/or.2015.3952. [DOI] [PubMed] [Google Scholar]
- 31.Lezina L, Aksenova V, Ivanova T, Purmessur N, Antonov AV, Tentler D, Fedorova O, Garabadgiu AV, Talianidis I, Melino G, et al. KMTase Set7/9 is a critical regulator of E2F1 activity upon genotoxic stress. Cell Death Differ. 2014;21:1889–1899. doi: 10.1038/cdd.2014.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rizzo PAD, Trievel RC. Substrate and product specificities of SET domain methyltransferases. Epigenetics. 2011;6:1059–1067. doi: 10.4161/epi.6.9.16069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mittenberg AG, Moiseeva TN, Barlev NA. Role of proteasomes in transcription and their regulation by covalent modifications. Front Biosci. 2008;13:7184–7192. doi: 10.2741/3220. [DOI] [PubMed] [Google Scholar]
- 34.Huynh H, Do PT, Nguyen TH, Chow P, Tan PH, Quach TH, Van T, Soo KC, Tran E. Extracellular signal-regulated kinase induces cyclin D1 and Cdk-2 expression and phosphorylation of retinoblastoma in hepatocellular carcinoma. Int J Oncol. 2004;25:1839–1847. doi: 10.3892/ijo.25.6.1839. [DOI] [PubMed] [Google Scholar]
- 35.Nauman A, Turowska O, Poplawski P, Master A, Tanski Z, Puzianowska-Kuznicka M. Elevated cyclin e level in human clear cell renal cell carcinoma: Possible causes and consequences. Acta Biochim Pol. 2007;54:595–602. [PubMed] [Google Scholar]
- 36.Wong JV, Dong P, Nevins JR, Mathey-Prevot B, You L. Network calisthenics: Control of e2f dynamics in cell cycle entry. Cell Cycle. 2011;10:3086–3094. doi: 10.4161/cc.10.18.17350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Irwin M, Marin MC, Phillips AC, Seelan RS, Smith DI, Liu W, Flores ER, Tsai KY, Jacks T, Vousden KH, et al. Role for the p53 homologue p73 in e2f-1-induced apoptosis. Nature. 2000;407:645–648. doi: 10.1038/35036614. [DOI] [PubMed] [Google Scholar]
- 38.Engelmann D, Pützer BM. The dark side of E2F1: In transit beyond apoptosis. Cancer Res. 2012;72:571–575. doi: 10.1158/0008-5472.CAN-11-2575. [DOI] [PubMed] [Google Scholar]
- 39.Wang C, Xiao Y, Hu Z, Chen Y, Liu N, Hu G. PEG10 directly regulated by E2Fs might have a role in the development of hepatocellular carcinoma. Febs Lett. 2008;582:2793. doi: 10.1016/j.febslet.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 40.Chen YL, Uen YH, Li CF, Horng KC, Chen LR, Wu WR, Tseng HY, Huang HY, Wu LC, Shiue YL. The E2F transcription factor 1 transactives stathmin 1 in hepatocellular carcinoma. Ann Surg Oncol. 2013;20:4041. doi: 10.1245/s10434-012-2519-8. [DOI] [PubMed] [Google Scholar]
- 41.Xie Q, Bai Y, Wu J, Sun Y, Wang Y, Zhang Y, Mei P, Yuan Z. Methylation-mediated regulation of E2F1 in DNA damage-induced cell death. J Recept Sig Transd. 2011;31:139. doi: 10.3109/10799893.2011.552914. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.