

Abstract
The effects of phosphorylation of a serine residue on the structural and dynamic properties of Ras-like protein, Rap, and its interactions with effector protein Ras binding domain (RBD) of Raf kinase, in the presence of GTP, are investigated via molecular dynamics simulations. The simulations show that phosphorylation significantly effects the dynamics of functional loops of Rap which participate in the stability of the complex with effector proteins. The effects of phosphorylation on Rap are significant and detailed conformational analysis suggest that the Rap protein, when phosphorylated and with GTP ligand, samples different conformational space as compared to non-phosphorylated protein. In addition, phosphorylation of SER11 opens up a new cavity in the Rap protein which can be further explored for possible drug interactions. Residue network analysis shows that the phosphorylation of Rap results in a community spanning both Rap and RBD and strongly suggests transmission of allosteric effects of local alterations in Rap to distal regions of RBD, potentially affecting the downstream signalling. Binding free energy calculations suggest that phosphorylation of SER11 residue increases the binding between Rap and Raf corroborating the network analysis results. The increased binding of the Rap-Raf complex can have cascading effects along the signalling pathways where availability of Raf can influence the oncogenic effects of Ras proteins. These simulations underscore the importance of post translational modifications like phosphorylation on the functional dynamics in proteins and can be an alternative to drug-targeting, especially in notoriously undruggable oncoproteins belonging to Ras-like GTPase family.
Introduction
Rap belongs to the family of small Ras-like GTPases, which have many roles in cellular activities like cell proliferation, apoptosis and differentiation etc.1–5. These GTPases act like molecular switches, active when GTP is bound and inactive when GDP is bound. These conformations are interconvertible by the action of the guanine nucleotide exchange factors (GEFs) and The GTPase activating proteins (GAPs). GEFs exchange GDP for GTP and GAPs catalyse the hydrolysis of GTP converting the active form into the inactive protein6. The Ras GTPases participate in many signalling pathways, including the MAPK/ERK, PI3K7–9. Many factors including their cellular location, bound ligand molecule and phosphorylation can affect how these molecules interact with downstream signalling proteins10,11, which is crucial in transmitting signal from Ras to the mitogen-activated protein kinase. Because of their role in key signalling events which are often deregulated in cancer and due to their prominent role as oncogenes, Ras family members (specifically H-, K- and N-Ras proteins) have garnered considerable attention over the years9,12–14. Considering all cancers where at least 20 tumours were counted and weighted equally, pan ras mutations were found at an incident rate of 16%14 and activating Ras mutations are associated with approximately 30% of all human cancers8. Many of these mutations render the tumour aggressive and are responsible for the death of patients. Yet there are no targeted therapies for these class of proteins as they are considered notoriously undruggable lacking specific binding pockets13.
The Rap proteins such as the Rap1A and Rap1B rose to prominence because of their high degree of identity to the Ras proteins1,4,15–19. Rap1A was identified as a suppressor of Ras activity in screening assays, a function attributed to its ability to competitively bind (in the presence of GTP) to downstream Raf without activating it and hence disrupting the signal transmission along the MAPK pathway20. Binding of Rap1 to RafB on the other hand results in activation as seen with the Ras family of proteins21. Many mutations, domain swapping experiments have indicated regions other than the RBD domain of Raf are responsible for these differences22,23. All known Ras effectors share a common Ras-binding Domain (RBD). Besides competing with Ras, Rap proteins are involved in many other crucial cellular functions such as cell adhesion, cell-cell junction formation and regulation of the actin cytoskeleton24–27.
Although not as well studied as the role of GEF proteins and GAP catalyzed changes in the nucleotide bound conformations, phosphorylation is known to regulate the functions of Ras and Rap proteins28–32. Phosphorylation is the most common reversible post translational modification(PTM) of proteins with a role in regulation of essentially all cellular functions33,34. The mechanism of how phosphorylation acts as a molecular switch that allows cells to respond instantaneously to various stimuli without the need for new protein synthesis, how phosphorylation at a remote site often influences the activity at a completely different site continue to be active areas of research. Most often phosphorylation is observed in disordered, well accessible or highly flexible regions and loops in proteins35,36. However, multiple investigators, including the author of this paper, while analyzing large scale data sets have documented that many of these phosphosites are not readily exposed to the solvent36–39. The presence of significant number of possible phosphosites in the disallowed region of phosphoconformation suggests that factors including dynamic conformational changes of proteins, binding to other proteins or regulatory factors can potentially expose these buried phosphosites to the solvent and to a kinase. Therefore understanding the role of protein dynamics in exposing such sites to solvents, interactions involved in transmitting the effects of phosphorylation to other functional sites is essential. Inherent loop dynamics of proteins are known to play a critical role in functioning of the protein40–43. Local mutations and PTMs impact the local loop dynamics and global structure and function of the protein. However it is not trivial to obtain phosphoproteins in amounts large enough for experimental investigations even when the kinase involved is known. MD simulations are invaluable tools successfully employed on multiple occasions to understand the effect of phosphorylation on the structure, dynamics, allosteric effect, conformational stabilization and map the electrostatic interactions in the proteins and thereby deduce the effect on functions44–48.
In this paper, we study the effects of phosphorylation of a single residue, SER11, identified as a possible phosphosite in Rap1A (Supplemental Table 1 of 37 and Table S1 of 49) (see Fig. 1), on the conformational dynamics of the Rap1A and its interactions with the effector protein kinase c-Raf1. It is to be noted that this particular residue, serine, occurs only in Rap GTPases and is replaced by alanine in Ras GTPases50. Phosphorylation at the same site has been observed in tumor samples of lung cancer patients as well (see Supplemental table of 51) and SER11 phosphosite in RAP1 carries motifs for many kinases, some with high and others with moderate scores: for example putative sites CK1, Aurora and ATM kinases, are predicted by KinasePhos2 a webserver for phosphosite predictions52. In addition to SER11 residue, SER39, SER179 and SER180 are other possible phosphosites in Rap1 that are either predicted or experimentally determined. Proximity of SER11 to the nucleotide ligand, which alters the activity of GTPases such as Rap1, renders the investigation of effects of phosphorylation particularly interesting. We also explore the effects of such phosphorylation on the dynamics of functional loops such as Switch I and Switch II loops to characterize the allosteric pathways within Rap-Raf complex and subsequently gain some insight into possible mechanisms through which Rap may affect the downstream MAPK signalling pathway.
Figure 1.

Rap-Raf protein complex (crystal structure, PDB ID 1C1Y) showing the location of important functional loops like P-loop, Switch I, Switch II and RBD loop regions. The phosphosite SER11 is shown in green.
Results
Effects of phosphorylation on structure and dynamics of Rap
The stability of the structures monitored via the root mean squared deviation of the GTP-bound Rap protein with and without the SER11 phosphorylated is shown in Fig. S1. The mobility of various parts of the protein and the effect of phosphorylation on the mobility of the Rap protein is measured through the root mean squared fluctuations (RMSF) of each residue, averaged over last 50 ns, 350–400 ns (Fig. 2). In the GTP bound form, the effects of SER11 phosphorylation are centered around the Switch I and Switch II loop regions. Proteins belonging to Ras superfamily are known to inhabit multiple conformational states and the two important loops that determine such conformational flexibility are Switch I (residues 25–40) and Switch II (residues 57–74) loops. The mobility of the loop region containing residues 80–85, which is spatially proximal to the phosphosite, is increased in the case of phosphorylated SER11. This is due to the acquired favourable interactions between the phosphorylated SER11 and the Switch II loop (discussed later) and consequently disrupt the interaction between the phosphosite and the loop containing residues 80–85. To understand the effects of complex formation on the structure of Rap, we also simulated the Rap protein alone (not in complex with Raf), with GTP ligand. The RMSF plots for all the four cases of Rap protein with or without complex formation with RBD of Raf protein and with and without phosphorylation of SER11 is shown in Fig. S2. The results suggest that the loop with residues 138–141 is mobile in all cases except the case of Rap-Raf complex with SER11 phosphorylated. This is due to favourable electrostatic interactions between ASN140 and ASP108. Results (Fig. S2) suggest that the mobility of both switch loops is very high when Rap is not in complex with RBD of Raf but undergoes reduction of mobility when SER11 is phosphorylated. The Solvent accessible surface area (SASA) of SER11 residue calculated for the molecular dynamics trajectory of Raf-Rap complex with GTP (measured using VMD keeping the probe radius to 1.4 Å) indicates that the dynamical nature of the P-loop allows the buried phosphosite SER11 to be exposed to water for a significant time on the present simulation timescale, suggesting a strong possibility of a kinase phosphorylating the SER11 residue and lends validity to our simulation studies involving phosphorylated Rap protein (Fig. S3). In the following sections we describe how phosphorylation at SER11 influences the dynamics that affects a) Switich I loop interaction with nucleotide and RBD domain and b) Switch II loop interaction with the nucleotide.
Figure 2.
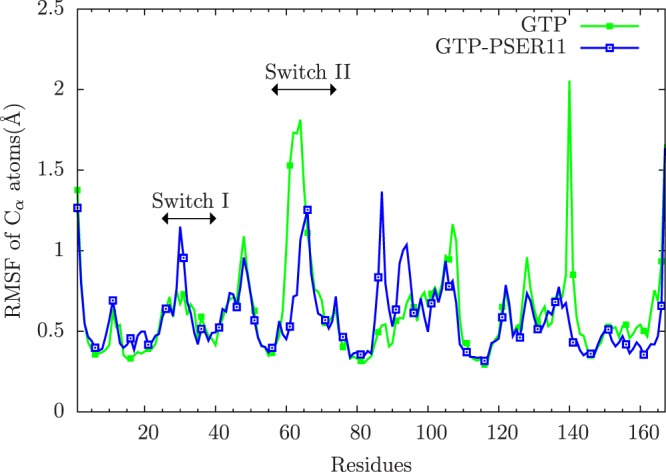
The RMSF of residues of Rap Cα atoms averaged over 350 to 400 ns of simulation for both unphosphorylated (green) and phosphorylated (blue). The relevant Switch I and Switch II loops are indicated in the figure.
Switch II loop interacts primarily with the GDP/GTP exchange factors (known as GEFs), which accelerate the release of the previously bound GDP to the proteins and to be replaced by GTP. The conformation of Switch II loop undergoes profound changes when GDP is exchanged with GTP ligand6,53. The GEF proteins are known to make extensive contacts with residues in the Switch II loop inducing local conformational changes near the nucleotide binding site, which results in the release of the bound nucleotide. Thus the mobility of the Switch II loop plays an important role in binding the GEF proteins leading to the GTP-bound conformations of G-proteins and consequently affect their ability to bind to downstream effectors. From the Fig. 2, it can be seen that the Switch II loop is most mobile in the GDP bound form of Rap, whose mobility reduces in the GTP bound form. This can be understood as the conformational stability that the loop acquires upon its interaction with the GEF proteins. Phosphorylation of the SER11 residue further reduces the mobility of this important functional loop in the GTP bound form of the protein. To understand this difference in the dynamics of the Switch II loop, its interactions with residues in the nearby P-loop (to which SER11 residue belongs) were investigated. In the Rap protein, as with the Ras proteins, the nucleotide pocket is flanked by primarily three loops: Switch I, Switch II and P-loop. As can be seen in Fig. 3, the conformation of Switch II loop is drastically altered when SER11 is phosphorylated in Rap-GTP protein. There are two strong electrostatic interactions which underlie such a significant conformational change. The Switch II loop contains several polar and charge residues including ARG68, which is positively charged. On phosphorylation of the SER11, the pocket region close to this residue acquires more negative charge compared to the unphosphorylated Rap. The simulations strongly suggest there is a phosphorylation induced change in conformation of Arg 68, which forms a stable salt bridge with the phosphate group of the SER11. This strong interaction results in pulling of the Switch II region into the nucleotide binding pocket which results in formation of another stable electrostatic interaction between the main chain carbonyl oxygen and the amide nitrogen atoms belonging to GLY 60 (Switch II) and GLY 12(P-loop) respectively. The distance between the two residues in the Rap-GTP protein with and without phosphorylation throughout the simulation timescale are shown in Fig. 4(a) and the difference is almost of the order of 7 Å. The GLY60 residue also forms stable favourable interactions with the oxygen atoms of the GTP ligand when SER11 is phosphorylated (Fig. 4b). The resulting favourable electrostatic interaction locks the Switch II loop into a conformation that reduces drastically its mobility.
Figure 3.
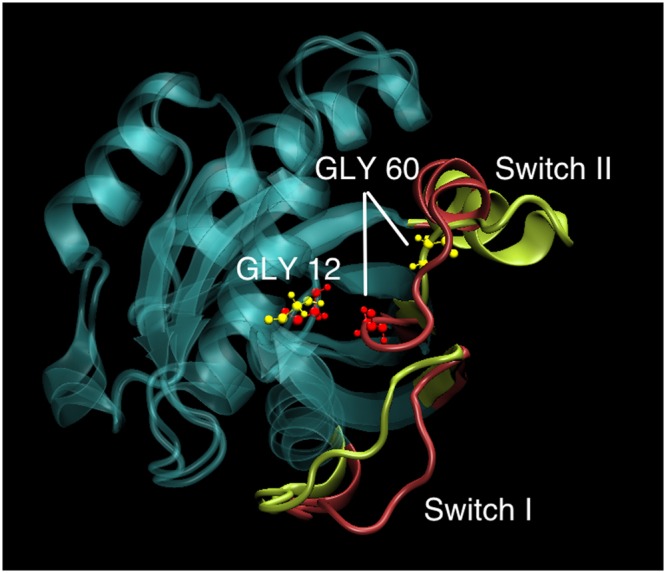
Overlapped snapshots of GTP bound Rap protein showing the conformation of functional Switch I and Switch II loops with (red) and without (yellow) phosphorylation towards the end of the simulation. The positions of residues GLY 12 and GLY60 with (red) and without(yellow) phosphorylation is also marked.
Figure 4.
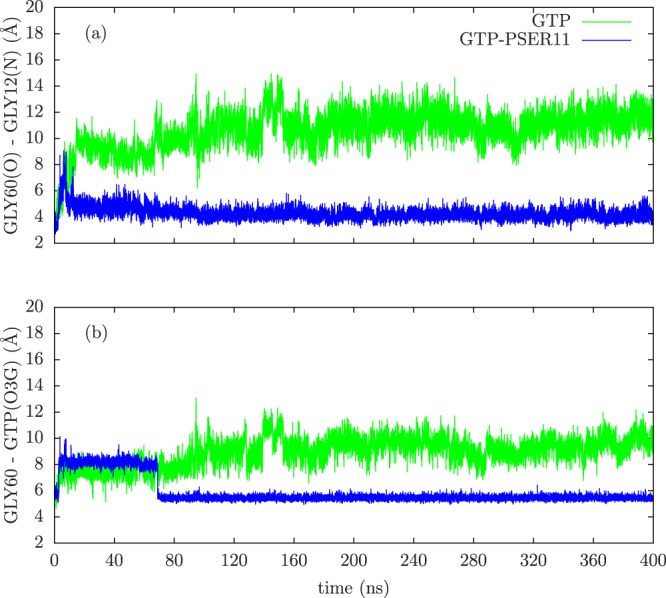
The time evolution of (a) distance between GLY60(O) and GLY12(N) atoms, (b) distance between GLY60(center of mass) and GTP(O3G) atoms for both unphosphorylated (green) and phosphorylated (blue) cases.
Switch I loop of Rap interacts directly with the effector protein Raf kinase, and the strength of the interaction strongly depends on the bound nucleotide. Compared to the inactive state of GDP bound Ras, RBD binds to active GTP bound Ras, almost 1000 times more strongly54. This mode of interaction is conserved in Ras superfamily of proteins10,55,56. Hence, the dynamic mobility of the Switch I loop is of crucial importance in the interaction between proteins in Ras superfamily and their effectors. The RMSF plot in Fig. 2 shows that the Switch I loop has slightly increased mobility when SER11 is phosphorylated, compared to the unphosphorylated Rap-GTP protein (in the GTP bound forms). The increased mobility is largely due to the movement of Glu30 of Switch I loop region (Fig. 5(a)). In the unphosphorylated Rap-GTP case, the GTP ligand forms several favourable interactions with residues lining the nucleotide pocket. This includes a strong and persistent hydrogen bond between the the oxygen molecules attached to the γ-phosphate atom and the hydroxyl group of TYR32, which has been observed in many crystal structures of Ras super family including Rap57,58. The other favourable electrostatic interaction of GTP ligand with Switch I loop is between GLU 30 and hydroxyl groups attached to ribose moieties of GTP ligand (see Fig. 5). These interactions reduce the mobility of the Switch I loop and participate in the stability of the complex formation with RBD loop of Raf kinase. With the inward movement of the Switch II loop into the nucleotide pocket region, as mentioned above, the position of GTP ligand changes and the interaction between GLU30 and GTP ligand is broken (see Fig. 5(b)), resulting in increased mobility of the residue GLU30 (see Fig. 2). It is to be noted that the position of TYR32 in the crystal structure used in the present simulations (1C1Y) is in the conformation in which the residue is located within the active site59,60. This conformation of TYR32 is expected to play a crucial role for catalysis of Rap proteins61, independent of GAP proteins, and that this conformation of TYR32 is preserved even when nucleotide pocket is significantly perturbed when SER11 is phosphorylated. This alteration of dynamics of Switch I loop by phosphorylation of SER11, which is located spatially and sequentially away from Switch I, clearly shows that allosteric mechanism is involved in communication.
Figure 5.

Snapshots of nucleotide pocket of Rap-GTP with and without phosphorylation of SER11. The functional loops Switch I and Switch II loops are shown in red and yellow colour respectively. The relevant residues in the two loops are also shown and the hydrogen bonds between GTP and residues in Switch I loop, when relevant, are shown in white dashed lines.
The most significant effect of phosphorylation of SER11 is in the position of THR61 residue on the Switch II loop, with respect the bound ligand GTP as shown in Fig. 6. In Ras proteins, the residue 61 (which is GLN) plays a very crucial role, along with the GAP proteins, in the GTP hydrolysis. The residue 61 is one of the most mutated site found in human tumours which impairs or abolishes the hydrolysis of GTP can lend the Ras protein to be in perpetually ON state14. In Rap proteins, this important residue is replaced by a threonine and experimental studies have shown that THR61, unlike GLN61 in Ras, plays a predominant role in binding of GAP proteins and does not participate in GTP hydrolysis59,62. The solved structure of Rap1 in complex with Rap1GAP has shown that the conformation of THR61 is away from the active site59. In the Rap-RBD structure used in the present simulation also, the Switch II is in a disordered state and THR61 is pointed away from the nucleotide. The simulations with the unphosphorylated Rap-RBD complex shows that the Switch II loop remains mobile and the THR61 moves away from the nucleotide during the course of the simulation. However, the phosphorylation of SER11 residue brings the THR61 into the nucleotide active site by forming a stable bond with GTP (as seen in Fig. 6). This conformation and location of THR61 inside the active site can have profound effect on the ability of the GAP proteins to hydrolyse the GTP ligand and can potentially affect Rap’s interaction with Raf.
Figure 6.
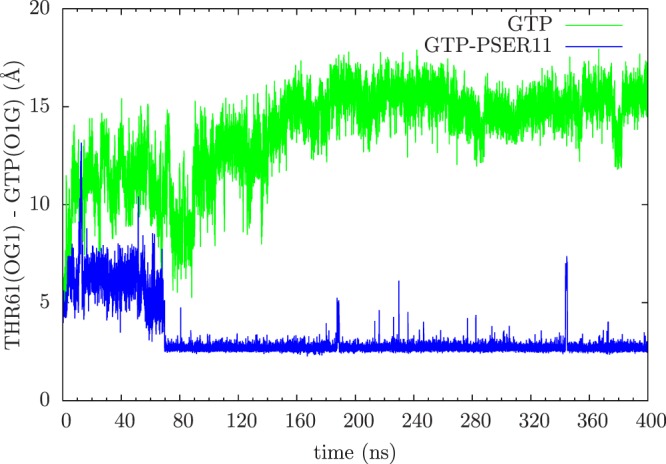
The time evolution of distance between the catalytic residue THR61 and GTP ligand without (green) and with (blue) phosphorylation.
Conformational sampling of Rap: Effects of phosphorylation
Having found that there is a reciprocal relationship in the dynamics of the switch loops that has a strong influence on nucleotide binding and Raf interaction the role of phophorylation on the conformation of different forms of the protein and effect on Rap activity is further explored in this section. Covariance analysis, using the cross correlation matrix as defined in the Methods section, is a very useful tool in getting insights into the relative correlated motions of different parts of the protein. The cross correlation matrix is computed by measuring the positional deviations of individual residues from an averaged structure and it is further averaged over equilibrium trajectory time scale. For all the simulations considered in this study, the cross correlation matrix is constructed over last 100 ns of simulation time in a run of 400 ns. We would like to emphasise that in our simulations, all rotations and translations were removed before performing the cross correlation analysis, as is the norm. The presence of hinges and possible large scale movement about the hinges can potentially complicate the positional cross correlation measurements, but no such global changes have been observed in all our simulations. It is very evident from the results (shown in Fig. 7) that the phosphorylation significantly alters the correlation between various functional loops. In the unphosphorylated Rap, the Switch II loop is anticorrelated with both Switch I and P-loop (shaded in blue 1and 2 respectively in Fig. 7), which disappears in the case of phosphorylated Rap protein. In addition, a strongly positively correlated motion (region 4 in Fig. 7) appears between Switch II and P-loop region. These results are consistent with the observations made in the previous section: the attractive interactions between the GLY60 in Switch II loop and GLY12 in P-loop triggers the observed positively correlated motion between the two loops. The conformational change in Switch II loop also removes strong anti-correlation between Switch II and helix 4 (residues 75 to 100) in Rap (region 3 in Fig. 7), further suggesting an overall increase in the correlated motion between different parts of the Rap protein when SER11 is phosphorylated.
Figure 7.

Cross-correlation plots of the Rap complex for 300 to 400 ns trajectory data of GTP- and GTP-PSER11 cases.
To understand the change in configurational space explored by the Rap protein when SER11 is phosphorylated, PCA analysis is employed. As has been described earlier, the two major regions which experience considerable changes after phosphorylation are localized regions in Switch I and Switch II loops. It has been long proposed that the proteins involved in the complex formation undergo changes in conformational entropy to compensate for the loss of translational entropy, due to complex formation, and it would be interesting to see if the phosphorylation can affect such conformational sampling. The mobility of the Switch II loop is considerably reduced when SER11 is phosphorylated and the loop’s configuration also changes such that the residues on the loop are pulled significantly towards the nucleotide binding site. The Switch I experiences an increase in its mobility on phosphorylation, but the change is much less compared to the reduction of mobility of Switch II loop. From these results, it can be expected that the Rap molecule acquires an overall tighter configuration on phosphorylation which can be verified through monitoring the subspace defined by the two largest principal components (PC1 and PC2) of the projected MD trajectory. Towards this we have analyzed the trajectory data generated over the last 25 ns of MD simulations (375–400 ns) by fitting the coordinates of all the frames of Cα atoms. The results clearly show that the Rap protein occupies a different conformational space compared to the unphosphorylated form and that the over all conformational density is smaller, strongly indicating a tighter conformation (see Fig. 8). The first two principal component vectors PC1 and PC2 captured nearly 83% of the information content from the last 25 ns data of MD trajectories of Rap domain Cα atoms for the GTP liganded forms with and without SER11 phosphorylation.
Figure 8.
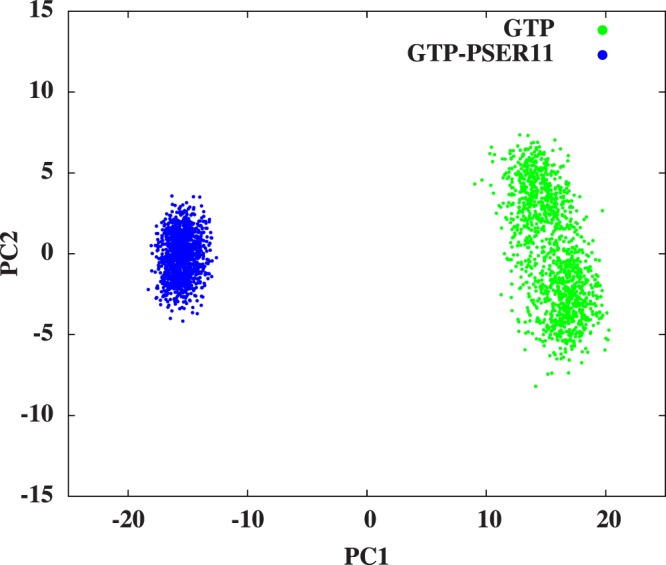
Conformer plots of Rap and Raf domains respectively(375 to 400 ns simulation data). The plot shows conformational space sampled by Rap protein in terms of PC1(80.13%) and PC2(2.36%).
The conformational changes in Rap, observed both in terms of interactions between various loop regions via covariance analysis and overall conformational flexibility of the protein, prompted us to look for phosphorylation induced changes in the distribution of pockets within the protein. The results are shown in Fig. 9. The largest pocket in the Rap protein, by volume, is identified (averaged over last 5 ns of simulation) and the results are shown for Rap-GTP with and without phosphorylation and the original crystal structure are also shown for comparison. The pocket location remains more or less the same for all the forms of Rap protein, except when Rap is phosphorylated at SER11 and with ligand GTP (shown in dark grey in fig Fig. 9). As can be seen from Table S2, the residues lining the pocket are very similar in all the three forms of Rap-PSER11. Most of the residues that line the nucleotide pocket, not surprisingly, belong to Switch I, Switch II and P-loop (as seen from the colouring of the residues in Table S2). However in the case of GTP-PSER11, the residues are predominantly only from P-loop and Switch II and the Switch I loop residues are not part of the residues lining the nucleotide pocket, which can be rationalised in terms of increased mobility of Switch I loop. The pocket volume is also changed and is much smaller for the phosphorylated case. As discussed earlier, the change in the pocket location can be rationalised in terms significant perturbation to the nucleotide binding pocket of the Rap protein upon phosphorylation. The results clearly show that the location of the largest pocket is near the nucleotide in all the cases except when the protein is phosphorylated in the presence of GTP (GTP-PSER11 case). The observation that the largest pocket size in the case of phosphorylated Rap protein, in the presence of GTP, is smaller than all the other case, suggests a more compact structure when phosphorylated and in the presence of GTP which is consistent with the PCA results in Fig. 8.
Figure 9.
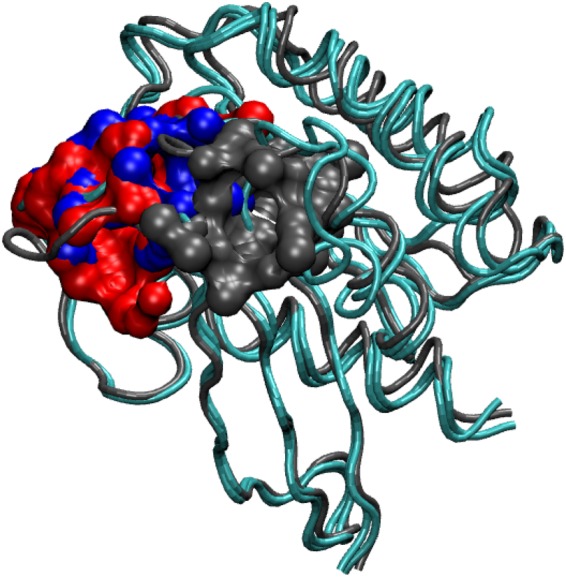
Largest cavity location of Rap domain in original crystal structure and last 5 ns averaged structures of GTP (red; volume: 605.96 Å3), Crystal structure(blue; volume: 521.01 Å3) and GTP-PSER11(black; 278.01 Å3) cases.
Effects of phosphorylation on complex and interactions at the interface
In this section we describe in depth the effect of phosphorylation on the interaction between Rap and Raf. Studies have shown that one of the functions of Rap proteins is to bind to the Raf effector via the RBD domain effectively trapping the Raf protein in an inactive complex24. The interaction between Ras and Raf is essential for activation of Raf kinase domain (which is located in the C-terminal half of the Raf protein), which plays a crucial signalling role in the MAPK pathway and Rap proteins can interefere with this mechanism by making Raf unavailable to Ras proteins. Proteins in Ras superfamily, including Rap protein, interact with effector proteins like Raf via Switch I loop10,56. Experimental studies63 have shown that single mutation of a conserved residue like THR 35 can significantly alter the dynamics of the Switch I loop and consequently affect the interaction of Ras superfamily proteins with the effector proteins like Raf. Switch I region (also called effector loop) is identical between Ras and Rap proteins. As seen in previous section, phosphorylation of SER11 residue leads to considerable changes in the mobility of the two main functional loops: Switch I and II of Rap protein which liganded with GTP. RMSF of Rap protein in the presence of GTP ligand shows that, the mobility of the Switch II and Switch I loop decreases and increases respectively (Fig. 2). Protein-protein binding exploits the inherent flexibility of the proteins to undergo conformational changes and form a complex64,65. In this section we explore the changes to the interface of Rap with Raf when SER11 of Rap is phosphorylated.
Unlike the majority of protein-protein interfaces which have more hydrophobic contacts66, the crystal structure of Rap-Raf suggests that there are many polar interactions between the Switch I loop of Rap and RBD of Raf57. The Switch I loop is not in close contact with the nucleotide binding pocket region in Rap protein but residue TYR32 and its conformational changes when GDP is exchanged for GTP, plays a crucial role in binding of the Switch I loop with the RBD of Raf protein16. This conformational change in Tyr 32 presumably facilitates the formation of a polar contact between residues ASP38 of Rap and ARG89 of Raf. As shown in previous section, the phosphorylation of SER11 significantly perturbs and reorganizes the nucleotide binding region and the surrounding loop conformations (See Figs 3 and 5). Due to breaking of strong polar interaction between GLU 30 and hydroxyl groups attached to ribose moieties of GTP ligand on phosphorylation of SER11, the Switch I acquires additional mobility and the loop moves away significantly compared to the unphosphorylated Rap-GTP case (See Fig. 3). This movement of Switch I loop, on phosphorylation, results in introduction of additional interactions between RBD of Raf protein and Switch I loop. Figure 10(a) shows the evolution of distance between TYR32 of Rap and LYS84 of Raf and in the case of phosphorylated Rap-GTP protein, the movement of the Switch I loop decreases the distance between Switch I loop and LYS84 of Rap by more than 6 Å. The consequences of such movement can be seen in the polar interactions between the RBD of Raf and residues of Rap at their interface (Fig. 10(b) and in Fig. S4). The time evolution of distance profiles suggest that few polar interactions like SER39-ARG89, GLU37-ARG59, ASP33-ARG84 remain unchanged with phosphorylation of SER11. However phosphorylation leads to changes in other polar interactions at the interface with disruptions in ASP33-ARG73 and GLU37-ARG67 interactions and enhancement in GLU54-ARG67 interaction. The polar interactions involving ASP38 of Rap are required interactions for effector binding to the Rap protein16 show positive enhancement upon phosphorylation. As can be seen in Figs 10(b) and S4(c), in phosphorylated Rap case, the ASP38 residue forms long-surviving ionic interactions with both ARG89 and THR68 residues of RBD of Raf protein suggesting increased binding between Rap and Raf with phosphorylation. Allosteric network analysis confirms this enhanced interaction (Fig. 11).
Figure 10.
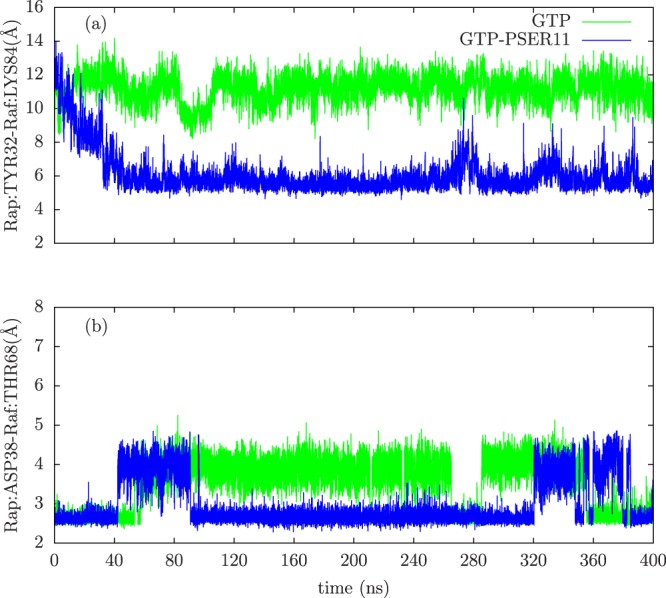
The time evolution of (a) distance between Rap:TYR32(CA) and Raf:LYS84(CA) atoms, (b) distance between Rap:ASP38(OD1) and Raf:THR68(OG1) atoms.
Figure 11.

The communities detected in (a) GTP- and (b) GTP-PSER11 cases. The yellow community represents the L4 loop.
From the allosteric network analysis, we see that the number of detected communities remain the same (i.e 10 communities). However, the organizationof the communities at the interface is drastically different when Rap is phosphorylated. The most striking aspect of the network analysis is that the GTP-PSER11 case has atleast 3 communities in common to the interface between Rap and Raf proteins (cyan, purple and red communities in Fig. 11(b)), which is absent in the unphosphorylated case. New community (shown in purple in Fig. 11(b)) that connects the Switch II loop with the L4 loop of the Raf protein (which is closer to the Cysteine Rich Domain part of Raf), via the protein-protein interface emerges in the phosphorylated form. In a recent combined experimental and simulation study on a Ras-Raf complex, a similar result was obtained due to mutation of GLN61LEU58. The mutation resulted in altering the allosteric pathways in which a single community network was found to form between the interface of Ras-Raf complex and the distant L4 loop of Raf. The present observations of global effects of phosphorylation of a single residue SER11 in Rap protein reiterates the fact that such changes can be allosterically communicated to spatially distant regions in the complex and suggest how a local, single residue phosphorylation can have global effects.
Effects of phosphorylation on binding energy of the complex
In the previous sections, we described how the phosphorylation of SER11 affects the dynamics of functional Switch I and Switch II loops and consequently how interacting communities spanning the complex interface emerges. In this section we look at the effect of phosphorylation on the binding free energy of the Rap-Raf complex, in particular to gain insight into the emergence of community across the complex interface. To do this, we used the standard MM-GBSA technique, as described in the Methods section. The results of the MM-GBSA calculations are shown in Table 1.
Table 1.
Free energy contribution of GTP liganded simulations.
| Contribution | GTP-SER11 system (Kcal/mol) | GTP-PSER11 system (Kcal/mol) |
|---|---|---|
| ΔGbind | −62.88 ± 8.59 | −68.67 ± 8.35 |
| ΔEelec | −498.66 ± 109.20 | −520.18 ± 61.72 |
| ΔEvdW | −45.88 ± 5.69 | −48.48 ± 5.92 |
| ΔGsolv | 481.66 ± 104.23 | 500.00 ± 57.16 |
| ΔGsolv−np | −8.95 ± 0.54 | −10.01 ± 0.51 |
| ΔGsolv−polar | 490.62 ± 104.17 | 510.02 ± 57.19 |
The binding energy values show that ΔGbind of the complex is lower when the SER11 is phosphorylated suggesting an increased binding of Rap-Raf complex. It can be seen that the contribution to the increased binding energy of the complex has main contribution from the and ΔEelec and ΔGsolv−polar terms. The binding free energy results and its contributions are consistent with increased interactions at the interface as seen in formation of long-surviving ionic interactions between ASP38 residue of Rap with both ARG89 and THR68 residues of RBD of Raf protein in Fig. 10 and also the emergent community at the complex interface shown in Fig. 11(b). The predominant contribution of electrostatic interactions to the binding free energy, seen here, is consistent with earlier work on thermodynamic analysis of Ras/Effector Complex Interfaces67. The entropy calculations were performed to understand any possible effects of phosphorylation of SER11, using the quasi-harmonic approach, and are shown in Table S2 of Supplementary Information. The calculations clearly suggest that effects of phosphorylation on the overall complex is minimal but, the Rap protein is less dynamic when SER11 is phosphorylated. This result is consistent with our PCA results, which suggests that the conformational sampling of the Rap protein is smaller when SER11 is phosphorylated.
Discussion
The focus of the present paper is to understand the effects of phosphorylation of a single residue SER11 in Rap on the interactions between Rap and RBD of Raf and speculate on how network of new interactions, that emerge as a consequence of phosphorylation, are used to communicate the changes at the interface to the distal region of Raf. The SER11 phosphosite is present in the P-loop (see Fig. 1), close to the Switch I region and is relatively buried. This phosphorylation was identified earlier in an independent experimental study49. The mobility of the Rap protein, and in particular those of various functional loops, as seen from the simulations here suggest that the dynamic nature of the loop regions can potentially expose this buried phosphosite. Indeed solvent accessible surface area (SASA) measurements of SER11 residue in a 400 ns long simulation (when unphosphorylated) and with GTP ligand revealed that for a significant time on the simulation time scale, the SASA value of SER11 is greater than that of its crystal structure value [See Fig. S3]. This also corroborates the observation in Fig. 2 that the P-loop region is marginally dynamic. These two observations together lend credence to our assumption that though the phosphosite is buried in the crystal structure, the inherent protein dynamics can potentially expose the site to solvent and other relevant kinases which can phosphorylate the SER11 residue. Further simulations of the SER11 Phosphorylated GTP bound Raf Rap complex helped us to understand the effects of the phosphorylation on the possible global conformational changes in Rap protein, its interface with RBD of Raf and possible allosteric effects transmitted to spatially far away locations.
All of the observed changes upon phosphorylation converge on one unifying theme: SER11 phosphorylation stabilizes the GTP bound Rap structure, likely to prevent GTP hydrolysis by pulling and closing the Switch II loop over the active site and establishes an allosteric network that can potentially transmit these changes to the distal L4 loop of Raf. Furthermore the PCA results indicated that the conformational space occupied by this SER11 GTP bound Rap is dramatically different from the unphosphorylated form and reflects a tighter conformation of the protein. The simulations also reveal that there is significant alteration in the pocket location and its size when the SER11 residue in Rap is phosphorylated. This new pocket is flanked predominantly by residues in Switch II loop (and from P-loop), and is different from the unphosphorylated form in which the largest pocket is surrounded by Switch I loop residues. The allosteric network analysis suggests that there is an increase in communities across the interface with phosphorylation. More significantly a single community spanning residues in the Switch II loop all the way to distal L4 loop emerges on phosphorylation. The net effect of SER11 phosphorylation is an allosteric relay of signals from Switch II region in Rap to L4 loop in RBD of Raf kinase which could result in constitutive activation of Rap and consequently that of Raf kinase potentially affecting the downstream signalling. A recent work on Hsp90 family of proteins also suggested that such buried post translational modification sites can play an important role in allosteric conformational changes and can potentially act as mediators of global dynamics in the Hsp90 structures68. The binding free energy calculations concur with the other results and show that the binding of Rap, with GTP ligand, with RBD of Raf is stronger when SER11 is phosphorylated, though the difference may not be very strong given the possible errors in such calculations. This plausible increased binding has its origin in favourable electrostatic interactions between residues of the two proteins at the interface due to cascading effects of phosphorylation of SER11 and can have important consequences in the downstream MAPK signaling pathways. One of the functions of Rap proteins is to competitively bind to Raf, without activating it, and disrupt the Ras-Raf binding20. Increased binding of Rap to RBD, due to phosphorylation of SER11 residue, can potentially make Raf even less available to Ras. In addition, such phosphorylation is also seen to induce possible long-range allosteric communication between Rap (via Switch II) and L4 loop of RBD of Raf which connects RBD to the cystein rich domain (CRD) of Raf. The entropy calculations suggest that the phosphorylation of SER11 does not change the overall entropy of the complex significantly but when considered individually, the Rap protein has lower entropy inits phosphorylated state. This result is quite consistent with our PCA results and together they suggest that the Rap protein has a ‘tighter’ conformational sampling when phosphorylated. This possibly due to increase in favourable electrostatic attractions within the Rap protein, which are are cascadng effects of phosphorylation of SER11. Whether such increased binding of phosphorylated Rap wth RBD of Raf can trigger conformational changes in the CRD of Raf, further affecting the binding of Raf to Ras, since it has been suggested that CRD of Raf also binds to Ras23,69–72, is open to speculation.
There is a significant parallel between these changes observed in the present MD simulations with those of Ras protein GTP bound crystal structure when GLN61 is mutated to a LEU. This mutation is a well known oncogenic mutation that prevents GTP hydrolysis locking Ras in a constitutively active form58. The authors predicted that the extended long range allosteric effect transmitted across the interface to the L4 loop is responsible for the kinase activity of Raf. The similarity between the two observations strongly suggests that the phosphorylation at SER11 mimics the oncogenic mutation in Ras which when extrapolated to function suggests that Rap may be constitutively activated by such phoshorylation. This SER11 phosphorylation in Rap was observed in Hela cells when the EGFR is activated. This phosphorylation increases upon nacodazole treatment49. Other highthroughput studies have detected the same phosphoryation in tumors51. Although this phosphorylation does not seem to occur at high enough occupancy, it is nevertheless detected with high confidence. These observations suggests a likely scenario. It is possible that this phosphorylation of Rap happens in normal cells during ligand binding to receptors. It is also possible that aberrant signalling due to some over active kinase often seen in cancers may phosphorylate Rap and activate it. Consequently this may lead to disruption of Rap-Mapk signaling or it may independently activate other RBD domain containing effector proteins. Such a possibility is supported by the significant parallel between the observations reported here and those of Ras protein GTP.
In summary we believe that this case study is an example as to how integrating tools that can probe dynamics can yield wealth of biological information hidden in crystal structures and highthroughput studies. They can provide probable mechanism by which single site PTM or point mutations affect functions of a protein. In addition these results reveal new binding pockets in proteins not evident in static crystal structures but evolve due to dynamic changes in proteins. Such dynamic pockets may be trapped by small molecules to inhibit the functions of the protein thus expanding the repertoire of druggable genome space. Arguably one may target the kinase responsible for the phosphorylation of such proteins thus providing alternative strategies to inhibit the functions of notoriously un druggale and elusive protein such as the Ras GTPases.
Methods
MD Simulations
The effects of phosphorylation on the dynamics of complex Rap-Raf are studied using the available complex structure of Ras-related protein Rap1A (referred to as Rap henceforth) liganded with a GTP-analogue molecule, Mg2+ ion and RBD (Ras binding Domain) region of Raf protein variant c-Raf1 (referred to as Raf henceforth) downloaded from protein data bank with PDB id 1C1Y with a resolution of 2.2 Å57. All-atom classical MD simulations were done for the systems listed in Table S1, using CHARMM3673,74 forcefield with the aid of NAMD75 software. The visualization was done using software VMD76 and analysis of data using Tcl scripting language which is embedded with VMD, Matlab and Grcarma77 software. The protonation states for all ionizable residues were determined using PDB2PQR server78 before solvating them. Each system was solvated in a water box (using TIP3P79 water model) and overall charge neutrality was achieved through the addition of appropriate counter ions. The systems were then subjected to energy minimization runs using the conjugate gradient method for 5000 steps, followed by MD simulation runs in NPT ensemble. The GTP analogue in the crystal structure,GppNHp was replaced by GTP and GDP molecules for the present simulations. The simulations of Rap-Raf complex with GTP and GDP ligands with and without SER 11 phosphorylated were done for 400 ns with a time step of 2 fs. The Nosé-Hoover-Langevin piston with a decay period of 100 fs and a damping time of 50 fs was used to maintain a constant pressure of 1 atm80,81. Berendsen thermostat82 was used to control temperature at 298 K. A cut-off distance of 12 Å was used to compute all short-range van der Waals (VDW) interactions and the long-range electrostatics interactions was treated with the Particle Mesh Ewald(PME) method83,84.
Covariance Analysis
Covariance Analysis is done on Rap-Raf complex Cα atoms of liganded cases to analyze the coupling between residues. The covariance matrix constructed from the displacements with respect to the average structure can give information regarding correlated/anti-correlated movements between various regions of the complex. The covariance matrix is constructed for the last 50 ns of the 400 ns long simulations. The covariance matrix is defined as following:
| 1 |
where 〈⋅〉 stands for the averaged values, Δri and Δrj are the ith and jth atom’s displacements with respect to the corresponding averaged structure atoms.
Principal Component Analysis
Molecular dynamics data is inherently high dimensional in nature and Principal component analysis (PCA) is therefore used to reduce the data into linear dimension for analyzing MD trajectory data85–88. The aim of this technique is to transform the coordinate system of the data such that the most variance is captured in least number of coordinates(principal axes). This representation is found by computing the eigen value decomposition on the covariance matrix of the data. The resulting coordinate vectors are the principal components. The largest eigen value corresponds to the most significant eigen vector (or first principal component) and the second largest eigen value corresponds to second principal component(PC) axis and so on. The nth eigen value represents the variance corresponding to the respective principal axis and the information content captured by nth PC is measured by the following expression:
| 2 |
We apply PCA to M × 3N dimensional MD trajectory where M is the number of frames (which are the time snapshots from MD trajectory) and N is the number of Cα atoms (3N because of coordinates). In most of the cases we can capture about 70% of the information in the first very few (like 3 to 10) principal components. PCA was done on the cartesian coordinate data of Cα atoms from MD trajectory files (for Rap and Raf chains under different conditions) produced from simulations. This approach enables us to deconvolute the information available in MD trajectories and visualize how the proteins sample the conformational space.
Pocket Identification
We used CASTp server89 for pocket identification in the Rap domain. The largest cavities of the last 5 ns time averaged structure of Rap domain for 4 different cases (as in Table S2) were computed. These calculations can give information about the flexibilty of the protein cavities and provides the necessary insights into comparative analysis of different forms of the protein as well as how each simulation is different from the original starting structures.
Community Network Analysis
Community network analysis is a unique tool to identify possible signaling pathways between distant yet functionally relevant protein sites based on topology of communities that are derived from the long molecular dynamics trajectories90–94. The community network analysis on the MD trajectory data is done using NetworkView90,95 plugin in VMD. A network is a set of nodes connected using edges. Each Cα atom of amino acid in the protein is represented as a node here. Edges connect pairs of nodes if the corresponding residues are in contact, and 2 nonconsecutive monomers are said to be in contact if any Cα atoms from the 2 monomers are within 4.5 Å of each other for at least 75% of the frames (corresponding to the last 50 ns MD simulation trajectory of GTP- and GTP-PSER11 case) analyzed. The edges are weighted using correlation matrix(Cij) data between the Cα atoms using the relation:
| 3 |
These weights in the form of correlation matrices are calculated using Carma software96. The correlations in the residue motion is used as a measure for information transfer between the two residues in contact. The suboptimal path is defined as the length of a path Dij between 2 distant nodes i and j is the sum of edge weights between the consecutive nodes (k,l) along the path:
| 4 |
The shortest distance between all pair of nodes is found using Floyd-Warshall algorithm97. The average of all shortest paths, known as critical path length (CPL), is a measure of the network size. The community detection analysis was done using software “gncommunities”90.
MM-GBSA for Binding free energy measurements
MM-GBSA(Molecular Mechanics-Generalised Born Surface Area)98–101 calculations were done to estimate the binding free energy of Rap-Raf complex. MM-GBSA method has been used in many studies to calculate the binding free energy of ligands with biomolecules as well as between biomolecules102–104. This method is computationally cheaper than other free energy methods such as steered molecular dynamics105, free energy perturbation and metadynamics. In MM-GBSA method, the free energy of binding of either two proteins or a ligand a protein is obtained by calculating the sum of difference between the gas-phase molecular mechanical, the solvation free energy and the entropy terms. In the present work, we follow the single-trajectory method of simulating the entire complex and then decomposing the required trajectories out of it, which has been used in earlier works and has also been shown to yield more accurate results of free energy of binding than three separate simulations involving the complex and the two proteins99,106,107. In this work, the MM-GBSA analysis was performed on three subsets: the Rap-Raf complex, only Rap and only Raf for both GTP-SER11 and GTP-PSER11 systems and all the GBSA calculations were performed on these three trajectories for the last 100 ns (300–400 ns or 5000 frames) of simulations using NAMD75 software. In all the cases, water and counterions were removed for the calculations and equivalent implicit solvent model parameters were used in the NAMD. The free energy was calculated using the equation below:
| 5 |
where HMM is the sum of the bonded, electrostatic and Lennard-Jones energy terms, Gsolv is the sum of polar and non-polar solvation energies,T is the temperature and Sconf is the configurational entropy. Gsolv is included in the electrostatic measurements done via NAMD script for GBSA calculations. In the single trajectory method, employed here, the bonded energy contribution to the change in HMM will be zero. Regarding the inclusion of entropic term in the free energy of binding, we would like to note that the usual method of computing the entropy via normal modes108 is computationally very expensive, especially for protein-protein complexes and other methods may have convergence issues and has been omitted in many earlier works, which we follow in this work as well98,99,101,109–118. The binding free energy for complex formation is then calculated as:
| 6 |
where , and are the free energies corresponding to Rap-Raf complex, only Rap and only Raf trajectories. In addition to the binding free energy measurements, we also analysed the change in entropy of the individual as well as the complex using quasi harmonic approach implemented in the Wordom program23,32,49,51,99,101,119,120.
Electronic supplementary material
Acknowledgements
The simulations were carried out on the supercomputing machines Annapurna and Nandadevi at The Institute of Mathematical Sciences. S.V would like to acknowledge useful discussions with Dr Sneha Menon and Dr Neelanjana Sengupta regarding the manuscript.
Author Contributions
S.V. and P.V. conceived the simulations. D.T. conducted the simulations. D.T., S.V. and P.V. analysed the results. All authors reviewed the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Prasanna Venkatraman, Email: vprasanna@actrec.gov.in.
Satyavani Vemparala, Email: vani@imsc.res.in.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-31234-7.
References
- 1.Frische E, Zwartkruis F. Rap1, a mercenary among the Ras-like GTPases. Dev Biol. 2010;340:1–9. doi: 10.1016/j.ydbio.2009.12.043. [DOI] [PubMed] [Google Scholar]
- 2.Bourne HR, Sanders DA, McCormick F. The GTPase superfamily: a conserved switch for diverse cell functions. Nature. 1990;348:125–132. doi: 10.1038/348125a0. [DOI] [PubMed] [Google Scholar]
- 3.Cox AD, Der CJ. Ras history: The saga continues. Small GTPases. 2010;1:2–27. doi: 10.4161/sgtp.1.1.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raaijmakers JH, Bos JL. Specificity in Ras and Rap signaling. J Biol Chem. 2009;284:10995–10999. doi: 10.1074/jbc.R800061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Altschuler DL, Peterson SN, Ostrowski MC, Lapetina EG. Cyclic AMP-dependent Activation of Rap1b. J Biol Chem. 1995;270:10373–10376. doi: 10.1074/jbc.270.18.10373. [DOI] [PubMed] [Google Scholar]
- 6.Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007;129:865–877. doi: 10.1016/j.cell.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 7.Alanazi FG, et al. Ras Signaling Pathway, Historical View. J Cell Sci Ther. 2014;5:1. doi: 10.4172/2157-7013.1000e117. [DOI] [Google Scholar]
- 8.Lu S, Jang H, Gu S, Zhang J, Nussinov R. Drugging Ras GTPase: a comprehensive mechanistic and signalling structural view. Chem Soc Rev. 2016;45:4929–4952. doi: 10.1039/C5CS00911A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 10.Lu S, et al. Ras Conformational Ensembles, Allostery, and Signaling. Chem Rev. 2016;116:6607–6665. doi: 10.1021/acs.chemrev.5b00542. [DOI] [PubMed] [Google Scholar]
- 11.Molina JR, Adjei AA. The Ras/Raf/MAPK pathway. J Thorac Oncol. 2006;1:7–9. doi: 10.1016/S1556-0864(15)31506-9. [DOI] [PubMed] [Google Scholar]
- 12.Castellano E, Santos E. Functional Specificity of Ras Isoforms: So Similar but So Different. Genes Cancer. 2011;2:216–231. doi: 10.1177/1947601911408081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov. 2014;13:828. doi: 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72:2457–2467. doi: 10.1158/0008-5472.CAN-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cook SJ, Rubinfeld B, Albert I, McCormick F. RapV12 antagonizes Ras-dependent activation of ERK1 and ERK2 by LPA and EGF in Rat-1 fibroblasts. EMBO J. 1993;12:3475. doi: 10.1002/j.1460-2075.1993.tb06022.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sprang SR. How Ras works: structure of a Rap–Raf complex. Structure. 1995;3:641–643. doi: 10.1016/S0969-2126(01)00198-8. [DOI] [PubMed] [Google Scholar]
- 17.Bos JL. Ras-like GTPases. BBA-REV CANCER. 1997;1333:M19–M31. doi: 10.1016/s0304-419x(97)00015-2. [DOI] [PubMed] [Google Scholar]
- 18.Rodriguez-Viciana P, Sabatier C, McCormick F. Signaling specificity by Ras family GTPases is determined by the full spectrum of effectors they regulate. Mol Cell Biol. 2004;24:4943–4954. doi: 10.1128/MCB.24.11.4943-4954.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boussiotis VA, Freeman GJ, Berezovskaya A, Barber DL, Nadler LM. Maintenance of human T cell anergy: blocking of IL-2 gene transcription by activated Rap1. Science. 1997;278:124–128. doi: 10.1126/science.278.5335.124. [DOI] [PubMed] [Google Scholar]
- 20.Kitayama H, Sugimoto Y, Matsuzaki T, Ikawa Y, Noda M. A ras-related gene with transformation suppressor activity. Cell. 1989;56:77–84. doi: 10.1016/0092-8674(89)90985-9. [DOI] [PubMed] [Google Scholar]
- 21.Ohtsuka T, Shimizu K, Yamamori B, Kuroda S, Takai Y. Activation of brain B-Raf protein kinase by Rap1B small GTP-binding protein. J Biol Chem. 1996;271:1258–1261. doi: 10.1074/jbc.271.3.1258. [DOI] [PubMed] [Google Scholar]
- 22.Hu C-D, et al. Coassociation of Rap1A and Ha-Ras with Raf-1 N-terminal Region Interferes with Ras-dependent Activation of Raf-1. J Biol Chem. 1997;272:11702–11705. doi: 10.1074/jbc.272.18.11702. [DOI] [PubMed] [Google Scholar]
- 23.Okada T, et al. The strength of interaction at the raf cysteine-rich domain is a critical determinant of response of raf to ras family small gtpases. Molecular and cellular biology. 1999;19:6057–6064. doi: 10.1128/MCB.19.9.6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bos JL, de Rooij J, Reedquist KA. Rap1 signalling: adhering to new models. Nat Rev Mol Cell Bio. 2001;2:369–377. doi: 10.1038/35073073. [DOI] [PubMed] [Google Scholar]
- 25.Kooistra MR, Dubé N, Bos JL. Rap1: a key regulator in cell-cell junction formation. J Cell Sci. 2007;120:17–22. doi: 10.1242/jcs.03306. [DOI] [PubMed] [Google Scholar]
- 26.Bos JL. Linking Rap to cell adhesion. Curr Opin Cell Biol. 2005;17:123–128. doi: 10.1016/j.ceb.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 27.Gloerich M, Bos JL. Regulating Rap small G-proteins in time and space. Trends Cell Biol. 2011;21:615–623. doi: 10.1016/j.tcb.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 28.Ting PY, et al. Tyrosine phosphorylation of RAS by ABL allosterically enhances effector binding. FASEB J. 2015;29:3750–3761. doi: 10.1096/fj.15-271510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frémin C, et al. ERK1/2-induced phosphorylation of R-Ras GTPases stimulates their oncogenic potential. Oncogene. 2016;35:5692–5698. doi: 10.1038/onc.2016.122. [DOI] [PubMed] [Google Scholar]
- 30.Bunda S, et al. Src promotes GTPase activity of Ras via tyrosine 32 phosphorylation. Proc Natl Acad Sci USA. 2014;111:E3785–E3794. doi: 10.1073/pnas.1406559111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang X, et al. Cyclase-associated protein 1 (cap1) is a prenyl-binding partner of rap1 gtpase. Journal of Biological Chemistry. 2018;293:7659–7673. doi: 10.1074/jbc.RA118.001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sahyoun N, McDonald OB, Farrell F, Lapetina EG. Phosphorylation of a ras-related gtp-binding protein, rap-1b, by a neuronal ca2+/calmodulin-dependent protein kinase, cam kinase gr. Proceedings of the National Academy of Sciences USA. 1991;88:2643–2647. doi: 10.1073/pnas.88.7.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Khoury, G. A., Baliban, R. C. & Floudas, C. A. Proteome-wide post-translational modification statistics: frequency analysis and curation of the swiss-prot database. SCI REP-UK 1 (2011). [DOI] [PMC free article] [PubMed]
- 34.Graves JD, Krebs EG. Protein Phosphorylation and Signal Transduction. Pharmacol Therapeut. 1999;82:111–121. doi: 10.1016/S0163-7258(98)00056-4. [DOI] [PubMed] [Google Scholar]
- 35.Iakoucheva LM, et al. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004;32:1037–1049. doi: 10.1093/nar/gkh253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiménez JL, Hegemann B, Hutchins JR, Peters J-M, Durbin R. A systematic comparative and structural analysis of protein phosphorylation sites based on the mtcPTM database. Genome Biol. 2007;8:R90. doi: 10.1186/gb-2007-8-5-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Somavarapu AK, Balakrishnan S, Gautam AKS, Palmer DS, Venkatraman P. Structural interrogation of phosphoproteome identified by mass spectrometry reveals allowed and disallowed regions of phosphoconformation. BMC Struct Biol. 2014;14:9. doi: 10.1186/1472-6807-14-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karasev D, et al. Spatial features of proteins related to their phosphorylation and associated structural changes. Proteins. 2018;86:13–20. doi: 10.1002/prot.25397. [DOI] [PubMed] [Google Scholar]
- 39.Johnson JR, et al. Prediction of functionally important phospho-regulatory events in xenopus laevis oocytes. Plos Comput Biol. 2015;11:e1004362. doi: 10.1371/journal.pcbi.1004362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karshikoff A, Nilsson L, Ladenstein R. Rigidity versus flexibility: the dilemma of understanding protein thermal stability. FEBS J. 2015;282:3899–3917. doi: 10.1111/febs.13343. [DOI] [PubMed] [Google Scholar]
- 41.Matsunaga S, et al. Structural transition of solvated h-ras/gtp revealed by molecular dynamics simulation and local network entropy. J Mol Graph Model. 2017;77:51–63. doi: 10.1016/j.jmgm.2017.07.028. [DOI] [PubMed] [Google Scholar]
- 42.Lukman S, Lane DP, Verma CS. Mapping the structural and dynamical features of multiple p53 dna binding domains: Insights into loop 1 intrinsic dynamics. PloS One. 2013;8:e80221. doi: 10.1371/journal.pone.0080221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vemparala S, Mehrotra S, Balaram H. Role of loop dynamics in thermal stability of mesophilic and thermophilic adenylosuccinate synthetase: a molecular dynamics and normal mode analysis study. BBA. 2011;1814:630. doi: 10.1016/j.bbapap.2011.03.012. [DOI] [PubMed] [Google Scholar]
- 44.Espinoza-Fonseca LM, Kast D, Thomas DD. Molecular dynamics simulations reveal a disorder-to-order transition on phosphorylation of smooth muscle myosin. Biophys J. 2007;93:2083–2090. doi: 10.1529/biophysj.106.095802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lyons AJ, Gandhi NS, Mancera RL. Molecular dynamics simulation of the phosphorylation-induced conformational changes of a tau peptide fragment. Proteins. 2014;82:1907–1923. doi: 10.1002/prot.24544. [DOI] [PubMed] [Google Scholar]
- 46.Yonezawa Y. Molecular Dynamics Study of the Phosphorylation Effect on the Conformational States of the C-Terminal Domain of RNA Polymerase II. J Phys Chem B. 2014;118:4471–4478. doi: 10.1021/jp4101976. [DOI] [PubMed] [Google Scholar]
- 47.Bueren-Calabuig JA, Michel J. Impact of Ser17 Phosphorylation on the Conformational Dynamics of the Oncoprotein MDM2. Biochemistry-US. 2016;55:2500–2509. doi: 10.1021/acs.biochem.6b00127. [DOI] [PubMed] [Google Scholar]
- 48.Smart JL, McCammon JA. Phosphorylation stabilizes the N-termini of α-helices. Biopolymers. 1999;49:225–233. doi: 10.1002/(SICI)1097-0282(199903)49:3<225::AID-BIP4>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 49.Sharma K, et al. Ultradeep human phosphoproteome reveals a distinct regulatory nature of tyr and ser/thr-based signaling. Cell reports. 2014;8:1583–1594. doi: 10.1016/j.celrep.2014.07.036. [DOI] [PubMed] [Google Scholar]
- 50.Noguchi H, et al. The structure and conformational switching of Rap1B. Biochem Bioph Res Co. 2015;462:46–51. doi: 10.1016/j.bbrc.2015.04.103. [DOI] [PubMed] [Google Scholar]
- 51.Schweppe DK, Rigas JR, Gerber SA. Quantitative phosphoproteomic profiling of human non-small cell lung cancer tumors. Journal of proteomics. 2013;91:286–296. doi: 10.1016/j.jprot.2013.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wong Y-H, et al. Kinasephos 2.0: a web server for identifying protein kinase-specific phosphorylation sites based on sequences and coupling patterns. Nucleic acids research. 2007;35:W588–W594. doi: 10.1093/nar/gkm322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cŕechet J-B, Bernardi A, Parmeggiani A. Distal Switch II Region of Ras2p Is Required for Interaction with Guanine Nucleotide Exchange Factor. J Biol Chem. 1996;271:17234–17240. doi: 10.1074/jbc.271.29.17234. [DOI] [PubMed] [Google Scholar]
- 54.Herrmann C, Horn G, Spaargaren M, Wittinghofer A. Differential interaction of the ras family GTP-binding proteins H-Ras, Rap1A, and R-Ras with the putative effector molecules Raf kinase and Ral-guanine nucleotide exchange factor. J Biol Chem. 1996;271:6794–6800. doi: 10.1074/jbc.271.12.6794. [DOI] [PubMed] [Google Scholar]
- 55.Mott HR, Owen D. Structures of Ras superfamily effector complexes: What have we learnt in two decades? Crit Rev Biochem Mol. 2015;50:85–133. doi: 10.3109/10409238.2014.999191. [DOI] [PubMed] [Google Scholar]
- 56.Simanshu DK, Nissley DV, McCormick F. Ras proteins and their regulators in human disease. Cell. 2017;170:17–33. doi: 10.1016/j.cell.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nassar N, et al. The 2.2A° crystal structure of the Ras-binding domain of the serine/threonine kinase c-Raf1 in complex with RaplA and a GTP analogue. Nature. 1995;375:554. doi: 10.1038/375554a0. [DOI] [PubMed] [Google Scholar]
- 58.Fetics SK, et al. Allosteric effects of the oncogenic RasQ61L mutant on Raf-RBD. Structure. 2015;23:505–516. doi: 10.1016/j.str.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scrima A, Thomas C, Deaconescu D, Wittinghofer A. The Rap-RapGAP complex: GTP hydrolysis without catalytic glutamine and arginine residues. EMBO J. 2008;27:1145–1153. doi: 10.1038/emboj.2008.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Buhrman G, Holzapfel G, Fetics S, Mattos C. Allosteric modulation of Ras positions Q61 for a direct role in catalysis. Proc Natl Acad Sci USA. 2010;107:4931–4936. doi: 10.1073/pnas.0912226107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cherfils J, et al. Crystal structures of the small G protein Rap2A in complex with its substrate GTP, with GDP and with GTPγs. EMBO J. 1997;16:5582–5591. doi: 10.1093/emboj/16.18.5582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chakrabarti PP, et al. Insight into catalysis of a unique GTPase reaction by a combined biochemical and FTIR approach. J Mol Biol. 2007;367:983–995. doi: 10.1016/j.jmb.2006.11.022. [DOI] [PubMed] [Google Scholar]
- 63.Spoerner M, Herrmann C, Vetter IR, Kalbitzer HR, Wittinghofer A. Dynamic properties of the Ras switch I region and its importance for binding to effectors. Proc Natl Acad Sci USA. 2001;98:4944–4949. doi: 10.1073/pnas.081441398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Atwell S, Ultsch M, De Vos AM, Wells JA. Structural plasticity in a remodeled protein-protein interface. Science. 1997;278:1125–1128. doi: 10.1126/science.278.5340.1125. [DOI] [PubMed] [Google Scholar]
- 65.Gu Y, Li D-W, Brüschweiler R. Statistical database analysis of the role of loop dynamics for protein–protein complex formation and allostery. Bioinformatics. 2017;33:1814–1819. doi: 10.1093/bioinformatics/btx070. [DOI] [PubMed] [Google Scholar]
- 66.Yan C, Wu F, Jernigan RL, Dobbs D, Honavar V. Characterization of protein–protein interfaces. PROTEIN J. 2008;27:59–70. doi: 10.1007/s10930-007-9108-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kiel C, Serrano L, Herrmann C. A detailed thermodynamic analysis of ras/effector complex interfaces. Journal of molecular biology. 2004;340:1039–1058. doi: 10.1016/j.jmb.2004.05.050. [DOI] [PubMed] [Google Scholar]
- 68.Stetz G, Tse A, Verkhivker GM. Dissecting Structure-Encoded Determinants of Allosteric Cross-Talk between Post-Translational Modification Sites in the Hsp90 Chaperones. Scientific reports. 2018;8:6899. doi: 10.1038/s41598-018-25329-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Luo Z, Diaz B, Marshall MS, Avruch J. An intact raf zinc finger is required for optimal binding to processed ras and for ras-dependent raf activation in situ. Molecular and cellular biology. 1997;17:46–53. doi: 10.1128/MCB.17.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mott HR, et al. The solution structure of the raf-1 cysteine-rich domain: a novel ras and phospholipid binding site. Proceedings of the National Academy of Sciences. 1996;93:8312–8317. doi: 10.1073/pnas.93.16.8312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li S, Jang H, Zhang J, Nussinov R. Raf-1 cysteine-rich domain increases the affinity of k-ras/raf at the membrane, promoting mapk signaling. Structure. 2018;26:513–525. doi: 10.1016/j.str.2018.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li Z-L, Prakash P, Buck M. A tug of war maintains a dynamic protein–membrane complex: Molecular dynamics simulations of c-raf rbd-crd bound to k-ras4b at an anionic membrane. ACS central science. 2018;4:298–305. doi: 10.1021/acscentsci.7b00593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.MacKerell AD, Jr., et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 74.Best RB, et al. Optimization of the additive charmm all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ1 and χ2 dihedral angles. J Chem Theory Comput. 2012;8:3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Phillips JC, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Humphrey W, Dalke A, Schulten K. VMD – Visual Molecular Dynamics. J Mol Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 77.Koukos PI, Glykos NM. Grcarma: A fully automated task-oriented interface for the analysis of molecular dynamics trajectories. J Comput Chem. 2013;34:2310–2312. doi: 10.1002/jcc.23381. [DOI] [PubMed] [Google Scholar]
- 78.Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- 80.Martyna GJ, Tobias DJ, Klein ML. Constant pressure molecular dynamics algorithms. J Chem Phys. 1994;101:4177–4189. doi: 10.1063/1.467468. [DOI] [Google Scholar]
- 81.Feller SE, Zhang Y, Pastor RW, Brooks BR. Constant pressure molecular dynamics simulation: the langevin piston method. J Chem Phys. 1995;103:4613–4621. doi: 10.1063/1.470648. [DOI] [Google Scholar]
- 82.Berendsen HJ, Postma JV, van Gunsteren WF, DiNola A, Haak J. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–3690. doi: 10.1063/1.448118. [DOI] [Google Scholar]
- 83.Darden T, York D, Pedersen L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. doi: 10.1063/1.464397. [DOI] [Google Scholar]
- 84.Essmann U, et al. A smooth particle mesh ewald method. J Chem Phys. 1995;103:8577–8593. doi: 10.1063/1.470117. [DOI] [Google Scholar]
- 85.Balsera MA, Wriggers W, Oono Y, Schulten K. Principal component analysis and long time protein dynamics. J Phys Chem-US. 1996;100:2567–2572. doi: 10.1021/jp9536920. [DOI] [Google Scholar]
- 86.Kapoor A, Travesset A. Mechanism of the Exchange Reaction in HRAS from Multiscale Modeling. PloS One. 2014;9:e108846. doi: 10.1371/journal.pone.0108846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kapoor A, Travesset A. Differential dynamics of RAS isoforms in GDP- and GTP-bound states. Proteins. 2015;83:1091–1106. doi: 10.1002/prot.24805. [DOI] [PubMed] [Google Scholar]
- 88.Grant BJ, Gorfe AA, McCammon JA. Ras conformational switching: simulating nucleotide-dependent conformational transitions with accelerated molecular dynamics. PLoS Comput Biol. 2009;5:e1000325. doi: 10.1371/journal.pcbi.1000325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dundas J, et al. CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006;34:W116–W118. doi: 10.1093/nar/gkl282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sethi A, Eargle J, Black AA, Luthey-Schulten Z. Dynamical networks in trna: protein complexes. Proc Natl Acad Sci USA. 2009;106:6620–6625. doi: 10.1073/pnas.0810961106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Girvan M, Newman ME. Community structure in social and biological networks. Proc Natl Acad Sci USA. 2002;99:7821–7826. doi: 10.1073/pnas.122653799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bhattacharyya M, Ghosh A, Hansia P, Vishveshwara S. Allostery and conformational free energy changes in human tryptophanyl-trna synthetase from essential dynamics and structure networks. Proteins. 2010;78:506–517. doi: 10.1002/prot.22573. [DOI] [PubMed] [Google Scholar]
- 93.Ghosh A, Vishveshwara S. Variations in clique and community patterns in protein structures during allosteric communication: Investigation of dynamically equilibrated structures of methionyl trna synthetase complexes. Biochemistry. 2008;47:11398–11407. doi: 10.1021/bi8007559. [DOI] [PubMed] [Google Scholar]
- 94.Burendahl S, Nilsson L. Computational studies of lxr molecular interactions reveal an allosteric communication pathway. Proteins. 2012;80:294–306. doi: 10.1002/prot.23209. [DOI] [PubMed] [Google Scholar]
- 95.Eargle J, Luthey-Schulten Z. NetworkView: 3D display and analysis of protein_RNA interaction networks. Bioinformatics. 2012;28:3000–3001. doi: 10.1093/bioinformatics/bts546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Glykos NM. Software news and updates carma: A molecular dynamics analysis program. J Comput Chem. 2006;27:1765–1768. doi: 10.1002/jcc.20482. [DOI] [PubMed] [Google Scholar]
- 97.Floyd RW. Algorithm 97: Shortest path. Commun Acm. 1962;5:345. doi: 10.1145/367766.368168. [DOI] [Google Scholar]
- 98.Hou T, Wang J, Li Y, Wang W. Assessing the performance of the mm/pbsa and mm/gbsa methods. 1. the accuracy of binding free energy calculations based on molecular dynamics simulations. Journal of chemical information and modelling. 2010;51:69–82. doi: 10.1021/ci100275a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Genheden S, Ryde U. The mm/pbsa and mm/gbsa methods to estimate ligand-binding affinities. Expert opinion on drug discovery. 2015;10:449–461. doi: 10.1517/17460441.2015.1032936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kollman PA, et al. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Accounts of chemical research. 2000;33:889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 101.Vergara-Jaque A, Comer J, Monsalve L, Gonzalez-Nilo FD, Sandoval C. Computationally efficient methodology for atomic-level characterization of dendrimer–drug complexes: a comparison of amine-and acetyl-terminated pamam. The Journal of Physical Chemistry B. 2013;117:6801–6813. doi: 10.1021/jp4000363. [DOI] [PubMed] [Google Scholar]
- 102.Gohlke H, Case DA. Converging free energy estimates: Mm-pb (gb) sa studies on the protein–protein complex ras–raf. Journal of computational chemistry. 2004;25:238–250. doi: 10.1002/jcc.10379. [DOI] [PubMed] [Google Scholar]
- 103.Hu G, Cao Z, Xu S, Wang W, Wang J. Revealing the binding modes and the unbinding of 14-3-3σ proteins and inhibitors by computational methods. Scientific reports. 2015;5:16481. doi: 10.1038/srep16481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Menon S, Sengupta N. Influence of hyperglycemic conditions on self-association of the alzheimer?s amyloid β (aβ1–42) peptide. Acs Omega. 2017;2:2134–2147. doi: 10.1021/acsomega.7b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cuendet MA, Michielin O. Protein-protein interaction investigated by steered molecular dynamics: the tcr-pmhc complex. Biophysical journal. 2008;95:3575–3590. doi: 10.1529/biophysj.108.131383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Špacková N, et al. Molecular dynamics simulations and thermodynamics analysis of dna- drug complexes. minor groove binding between 4 ?, 6-diamidino-2-phenylindole and dna duplexes in solution. Journal of the American Chemical Society. 2003;125:1759–1769. doi: 10.1021/ja025660d. [DOI] [PubMed] [Google Scholar]
- 107.Lepšĭk M, Křìž Z, Havlas Z. Efficiency of a second-generation hiv-1 protease inhibitor studied by molecular dynamics and absolute binding free energy calculations. Proteins: Structure, Function, and Bioinformatics. 2004;57:279–293. doi: 10.1002/prot.20192. [DOI] [PubMed] [Google Scholar]
- 108.Genheden S, Kuhn O, Mikulskis P, Hoffmann D, Ryde U. The normal-mode entropy in the mm/gbsa method: effect of system truncation, buffer region, and dielectric constant. Journal of chemical information and modeling. 2012;52:2079–2088. doi: 10.1021/ci3001919. [DOI] [PubMed] [Google Scholar]
- 109.Foloppe N, Hubbard R. Towards predictive ligand design with free-energy based computational methods? Current medicinal chemistry. 2006;13:3583–3608. doi: 10.2174/092986706779026165. [DOI] [PubMed] [Google Scholar]
- 110.Wang J, Hou T, Xu X. Recent advances in free energy calculations with a combination of molecular mechanics and continuum models. Current Computer-Aided Drug Design. 2006;2:287–306. doi: 10.2174/157340906778226454. [DOI] [Google Scholar]
- 111.Homeyer N, Gohlke H. Free energy calculations by the molecular mechanics poisson- boltzmann surface area method. Molecular Informatics. 2012;31:114–122. doi: 10.1002/minf.201100135. [DOI] [PubMed] [Google Scholar]
- 112.Yang T, et al. Virtual screening using molecular simulations. Proteins: Structure, Function, and Bioinformatics. 2011;79:1940–1951. doi: 10.1002/prot.23018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Das P, Chacko AR, Belfort G. Alzheimer?s protective cross-interaction between wild-type and a2t variants alters aβ42 dimer structure. ACS chemical neuroscience. 2016;8:606–618. doi: 10.1021/acschemneuro.6b00357. [DOI] [PubMed] [Google Scholar]
- 114.Gorham RD, Jr., Rodriguez W, Morikis D. Molecular analysis of the interaction between staphylococcal virulence factor sbi-iv and complement c3d. Biophysical journal. 2014;106:1164–1173. doi: 10.1016/j.bpj.2014.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhang L, et al. Probing immobilization mechanism of alpha-chymotrypsin onto carbon nanotube in organic media by molecular dynamics simulation. Scientific reports. 2015;5:9297. doi: 10.1038/srep09297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chen F, et al. Assessing the performance of the mm/pbsa and mm/gbsa methods. 6. capability to predict protein–protein binding free energies and re-rank binding poses generated by protein–protein docking. Physical Chemistry Chemical Physics. 2016;18:22129–22139. doi: 10.1039/C6CP03670H. [DOI] [PubMed] [Google Scholar]
- 117.Tuffery P, Derreumaux P. Flexibility and binding affinity in protein–ligand, protein–protein and multi-component protein interactions: limitations of current computational approaches. Journal of The Royal Society Interface. 2012;9:20–33. doi: 10.1098/rsif.2011.0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sun H, Li Y, Tian S, Xu L, Hou T. Assessing the performance of mm/pbsa and mm/gbsa methods. 4. accuracies of mm/pbsa and mm/gbsa methodologies evaluated by various simulation protocols using pdbbind data set. Physical Chemistry Chemical Physics. 2014;16:16719–16729. doi: 10.1039/C4CP01388C. [DOI] [PubMed] [Google Scholar]
- 119.Zhang L, Borthakur S, Buck M. Dissociation of a dynamic protein complex studied by all-atom molecular simulations. Biophysical journal. 2016;110:877–886. doi: 10.1016/j.bpj.2015.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Seeber M, Cecchini M, Rao F, Settanni G, Caflisch A. Wordom: a program for efficient analysis of molecular dynamics simulations. Bioinformatics. 2007;23:2625–2627. doi: 10.1093/bioinformatics/btm378. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.