
Figure 1.
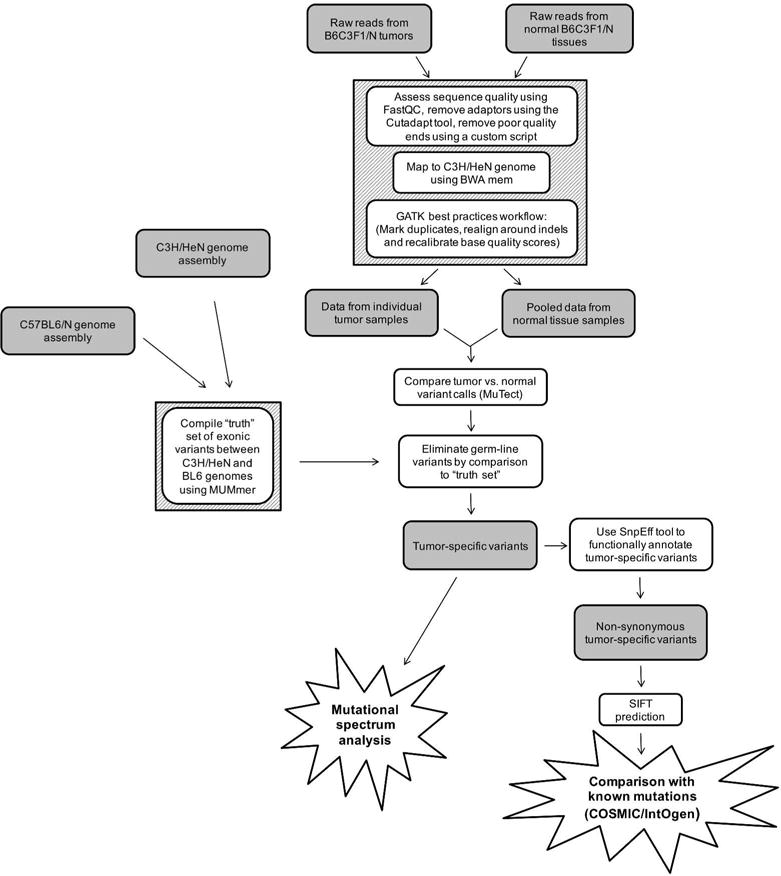
Steps used to obtain and analyze sets of tumor-specific variants. Raw reads from FF and FFPE normal tissues and individual tumor samples were filtered to remove low quality reads, mapped to the C3H genome, and subjected to a GATK best practices workflow protocol. Then, the resultant data sets for each tumor sample were compared to the pooled normal variant data set for the corresponding FF or FFPE samples. Germ-line variants were eliminated by removing the C3H versus BL6 variants in the “truth set”. The tumor-specific variants were then subject to mutational spectra analysis or filtered for non-synonymous variants and low SIFT scores prior to comparison to known mutations.