

Abstract
Key points
The ClC‐3 2Cl−/1H+ exchanger modulates endosome pH and Cl− concentration. We investigated the relationships between ClC‐3‐mediated ion transport (steady‐state transport current, I SS), gating charge (Q) and cytoplasmic alkalization.
ClC‐3 transport is functionally unidirectional. ClC‐5 and ClC‐3 display indistinguishable exchange ratios, but ClC‐3 cycling is less “efficient”, as reflected by a large Q/I SS. An M531A mutation predicted to increase water‐wire stability and cytoplasmic proton supply improves efficiency.
Protonation (pH 5.0) of the outer glutamate gate (Gluext; E224) reduces Q, inhibits transport, and weakens coupling.
Removal of the central tyrosine anion gate (Y572S) greatly increases uncoupled anion current. Tyrosine –OH removal (Y572F) alters anion selectivity and impairs coupling.
E224 and Y572 act as anion barriers, and contribute to gating. The Y572 side chain and –OH regulate Q movement kinetics and voltage dependence. E224 and Y572 interact to create a “closed” inner gate conformation that maintains coupling during cycling.
Abstract
We utilized plasma membrane‐localized ClC‐3 to investigate relationships between steady‐state transport current (I SS), gating charge (Q) movement, and cytoplasmic alkalization rate. ClC‐3 exhibited lower transport efficiency than ClC‐5, as reflected by a larger Q/I SS ratio, but an indistinguishable Cl−/H+ coupling ratio. External SCN− reduced H+ transport rate and uncoupled anion/H+ exchange by 80–90%. Removal of the external gating glutamate (“Gluext”) (E224A mutation) reduced Q and abolished H+ transport. We hypothesized that Methionine 531 (M531) impedes “water wire” H+ transfer from the cytoplasm to E224. Accordingly, an M531A mutation decreased the Q/I SS ratio by 50% and enhanced H+ transport. External protons (pH 5.0) inhibited I SS and markedly reduced Q while shifting the Q–voltage (V) relationship positively. The Cl−/H+ coupling ratio at pH 5.0 was significantly increased, consistent with externally protonated Gluext adopting an outward/open position. Internal “anion gate” removal (Y572S) dramatically increased I SS and impaired coupling, without slowing H+ transport rate. Loss of both gates (Y572S/E224A) resulted in a large “open pore” conductance. Y572F (removing only the phenolic hydroxide) and Y572S shortened Q duration similarly, resulting in faster Q kinetics at all voltages. These data reveal a complex relationship between Q and ion transport. Q/I SS must be assessed together with coupling ratio to properly interpret efficiency. Coupling and transport rate are influenced by the anion, internal proton supply and external protons. Y572 regulates H+ coupling as well as anion selectivity, and interacts directly with E224. Disruption of this “closed gate” conformation by internal protons may represent a critical step in the ClC‐3 transport cycle.
Keywords: ClC‐3, gating current, pH dependence, proton transport, chloride‐proton exchange
Key points
The ClC‐3 2Cl−/1H+ exchanger modulates endosome pH and Cl− concentration. We investigated the relationships between ClC‐3‐mediated ion transport (steady‐state transport current, I SS), gating charge (Q) and cytoplasmic alkalization.
ClC‐3 transport is functionally unidirectional. ClC‐5 and ClC‐3 display indistinguishable exchange ratios, but ClC‐3 cycling is less “efficient”, as reflected by a large Q/I SS. An M531A mutation predicted to increase water‐wire stability and cytoplasmic proton supply improves efficiency.
Protonation (pH 5.0) of the outer glutamate gate (Gluext; E224) reduces Q, inhibits transport, and weakens coupling.
Removal of the central tyrosine anion gate (Y572S) greatly increases uncoupled anion current. Tyrosine –OH removal (Y572F) alters anion selectivity and impairs coupling.
E224 and Y572 act as anion barriers, and contribute to gating. The Y572 side chain and –OH regulate Q movement kinetics and voltage dependence. E224 and Y572 interact to create a “closed” inner gate conformation that maintains coupling during cycling.
Introduction
The ClC‐3 Cl−/H+ antiporter localizes primarily to intracellular vesicles, including endosomes (Hara‐Chikuma et al. 2005b; Jentsch, 2007; Miller et al. 2007; Stauber & Jentsch, 2010), lysosomes (Stauber & Jentsch, 2010), Golgi (Gentzsch et al. 2003) and secretory vesicles (Barg et al. 2001; Stobrawa et al. 2001; Salazar et al. 2004; Moreland et al. 2006, 2007; Maritzen et al. 2008; Deriy et al. 2009; Riazanski et al. 2011). The functional contribution of ClC proteins to endosome biology remains controversial (Pusch & Zifarelli, 2015). ClC antiporters (ClC‐3 to ‐7) have been broadly proposed to support vesicular acidification, primarily by contributing a “shunt” Cl− conductance that provides charge compensation for vacuolar ATPase (V‐ATPase)‐mediated H+ transport (Gunther et al. 2003; Hara‐Chikuma et al. 2005a, b ; Jentsch, 2007; Stauber et al. 2012). This proposed role requires ClC‐mediated exchange of Cl− into, and H+ out of, endosomes (Novarino et al. 2010). However, this requirement is at odds with physiologically recorded ClC‐3, ‐4 and ‐5 transport currents, which are essentially completely rectifying and have a polarity corresponding to vesicular H+ influx and Cl− efflux (Li et al. 2000, 2002; Alekov & Fahlke, 2009; Smith & Lippiat, 2010a ; Guzman et al. 2013; Pusch & Zifarelli, 2015). An alternative proposal is that the physiologically favoured polarity of ClC‐3 exchange could provide charge compensation for electron flow into endosomes via NADPH oxidases (Miller et al. 2007; Lamb et al. 2009). ClC exchangers may support different functions depending upon subtype, and vesicle‐specific biochemistry.
Until recently, defining the biophysical properties of ClC‐3 currents posed a significant challenge compared to ClC family relatives. Heterologously expressed ClC‐5 is trafficked to the plasma membrane (PM), and reliably produces robust whole‐cell exchange currents (Steinmeyer et al. 1995; Scheel et al. 2005). In contrast, ClC‐3 is cycled rapidly through the PM and incorporated into endocytic vesicles, with only 5–10% of the total protein residing in the PM at steady state (Zhao et al. 2007; Okada et al. 2014). Initial analyses of heterologously expressed ClC‐3 suggested an association between ClC‐3 and the volume‐activated anion current (Duan et al. 1997, 2001). Subsequent plasmid‐mediated ClC‐3 expression yielded small, rapidly activating and highly rectifying currents (Li et al. 2000, 2002). Conversely, adenoviral ClC‐3 overexpression produced slowly activating, pH‐dependent currents (Matsuda et al. 2008, 2010). These disparate results raised the possibility that endosomal ClC‐3 overexpression alters membrane trafficking of various anion channels, or modulates reactive oxygen production (Matsuda et al. 2010) which can in turn activate volume‐activated channels (Varela et al. 2004). The recent identification of the LRRC8 (leucine rich repeat containing 8) protein family as the mediators of volume‐activated currents (Jentsch et al. 2016) lends needed clarity to this controversy.
The ClC‐3 N‐terminal sequence strongly influences retention of the protein in the PM. Disruption of a clathrin‐binding dileucine “retention” sequence in ClC‐3a (ClC‐313–19A; LLDLLDE mutated to AAAAAAA) greatly enhances plasma membrane localization and transport currents (Zhao et al. 2007; Guzman et al. 2013, 2015). ClC‐313–19A expression resulted in larger currents that displayed rapid activation and strong outward rectification characteristic of wild type ClC‐3, and provided the first confirmation that ClC‐3 is a Cl−/H+ exchanger (Guzman et al. 2013). In voltage‐clamp recordings, ClC‐3 and ‐5 currents exhibit characteristic transient “On” and “Off” charge movements at the onset and termination of depolarizing steps. These “gating” currents are proposed to result from incomplete transport cycles. Q Off is linked to the efficiency of internal proton transfer to the external gating glutamate (Gluext), and is magnified by loss‐of‐function mutations to the internal “proton glutamate” (Gluin), or by transport‐inhibiting anions (Grieschat & Alekov, 2012, 2014; Zifarelli et al. 2012; Alekov, 2015). Compared to its close relatives, ClC‐313–19A mediates slower transport, characterized by smaller steady‐state currents and a much larger charge movement component (Guzman et al. 2013, 2015), consistent with a lower efficiency of proton transfer from Gluin to Gluext. It was speculated that ClC‐3 gating capacitance could support endosome acidification by contributing to charge‐neutralization of the V‐ATPase thereby (Guzman et al. 2013).
We took a novel approach of substituting the cytoplasmic N‐terminus (‐ter) of ClC‐5 (M1–A46), which lacks a dileucine internalization sequence, onto the “core” ClC‐3 protein (M60–C‐ter). The resulting “ClC‐5/3” protein is localized to the PM of cells of the human embryonic kidney cell line HEK 293, permitting electrophysiological and optical assays of ion transport. We characterized ClC‐5/3 transport efficiency and Cl−/H+ coupling relative to ClC‐5 by quantifying steady state current (I SS), gating charge (Q), and the rate of transport‐mediated cytoplasmic alkalization. We then quantified the influence of external H+, an uncoupling anion (SCN−), and selective ClC‐3 mutations on these variables. The fundamental properties of ClC‐3 are similar to, but qualitatively differ from those of ClC‐5. The large gating transients of ClC‐3 provide a unique opportunity to assess the impact of targeted interventions on gating. The data demonstrate that ClC‐3 gating and Cl−/H+ coupling ratio are influenced by extracellular protons. The transported anion also impacts coupling and the rate of outward proton movement. Our results support a conformational interaction between Gluext (E224) and the inner tyrosine gate (Y572) that effectively obstructs the anion pathway and prevents uncoupled anion passage.
Methods
Expression of ClC‐3 and ClC‐5 plasmids
HEK 293T cells obtained from the American Tissue Culture Collection were maintained at 37 °C in 5% CO2 in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% fetal bovine serum (FBS, Hyclone, GE Healthcare Life Sciences, Marlboro, MA, USA) and 1.25% penicillin–streptomycin (25 U/ml, Life Technologies, Thermo Fisher Scientific, Waltham, MA, USA). The ClC‐3a (“short”) N‐terminal isoform of ClC‐3 (Guzman et al. 2013, 2015) (rat sequence NM_053363 in pEGFP‐N1, a gift from S. A. Weinman, University of Kansas) was subcloned into pcDNA3.1. Rat ClC‐3b (“long” N‐terminal isoform; NM_053363) was created by adding the 58 amino acid N‐terminus extension onto rat ClC‐3a. This full‐length rat sequence differs from murine ClC‐3 by one amino acid (V668I), and from human ClC‐3 by two amino acids (V668I and S605N). Short human ClC‐5 (NP_001121370.1) in pFrog (a gift from A. Accardi, Cornell University) was subcloned into pEYFP‐N1. Mutant and chimeric constructs were created using the Stratagene (La Jolla, CA, USA) QuikChange Lightning kit. Plasmids were transfected using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer's instructions. Numbering of wild‐type and mutated amino acids is according to the sequence of ClC‐3a. ClC‐3a and ClC‐3b served as controls for ClC‐5/3.
Immunostaining, fluorescence imaging and analysis
Cells were grown on glass‐bottom dishes or coverslips coated with 25 μg/ml polyethylenimine (PEI) in order to improve adherence. For immunostaining, cells were fixed using 3.7% formaldehyde in PBS for 10 min at room temperature, washed 3× with PBS, then blocked and permeabilized with 1% bovine serum albumin (BSA) and 0.5% Triton X‐100. Cells were incubated with a polyclonal rabbit anti‐ClC‐3 (Matsuda et al. 2008) at 1:400 in PBS + 1% BSA (1 h at 37 °C), washed 3× with PBS, and incubated with an anti‐rabbit secondary antibody conjugated to Cy3 (1:300; 1 h at 37 °C). Nuclei were stained with 0.5 μM ToPro3‐iodide for 1 min at room temperature prior to mounting using Prolong Gold anti‐fade reagent. Live and fixed cell imaging of yellow fluorescent protein (YFP)‐ and green fluorescent protein (GFP)‐fusion proteins and fluorescent antibody staining was performed on a Zeiss 510 confocal microscope (Vanderbilt Cell Imaging Shared Resource) at 16–24 h post‐transfection. Images of single confocal slices (0.6 μm) and Z‐series projections were constructed using ImageJ software. YFP and GFP fluorescence images were also acquired immediately prior to all whole cell recordings on an Olympus IX71 microscope (60× objective), equipped with a Hamamatsu C10600 digital camera and Till Photonics Oligochrome and image acquisition/analysis hardware and software (Hunt Optics & Imaging, Inc., Pittsburgh, PA, USA).
Electrophysiology
Cells were seeded into 6‐ or 12‐well plates for ≥1–2 h, and transfected overnight with YFP‐ or GFP‐fusion constructs of ClC‐3, ClC‐5 or ClC‐5/3 (DMEM + 1.5–2.5 μg/μl cDNA, 2.5% Lipofectamine 2000, +10% FBS). Whole‐cell patch clamp recordings were made from freshly trypsinized and dissociated cells. Most recordings were performed on the 2nd day (∼40 h) post‐transfection, with a minority performed on the 1st day (16–20 h). Bath recording saline (300 mOsm/kg, pH 7.4) contained (in mM): 130 NaCl, 1.8 MgCl2, 1.8 CaCl2, 10 HEPES, 1–2 NaOH. Pipette saline (290 mOsm/kg, pH 7.2) contained (in mM): 120 mM CsCl, 4 TEACl, 2 MgCl2, 5 Na2ATP, 10 HEPES, 1–2 CsOH, 1.186 CaCl2, 5 EGTA (estimated free Ca2+ of 61 μM using WEBMAXC (http://www.stanford.edu/~cpatton/webmaxc/webmaxcS.htm)). Phloretin (Cayman Chemical, Ann Arbor, MI, USA), which was previously demonstrated to effectively block endogenous acid‐activated currents (I Cl,acid; Matsuda et al. 2008), was added to external salines (60–350 μM) in some experiments. Osmolality was measured (Micro‐Osmette osmometer, Precision Systems, Natick, MA, USA) and adjusted by addition of 1 M mannitol. All recordings were made at room temperature (∼22 °C). Liquid junction potentials were minimized by using a 3 M CsCl agar bridge bath electrode. Voltage values in current–voltage (I–V) and Q–V plots were corrected for a 5 mV liquid junction potential measured between the pipette and control bath salines.
Currents were filtered at 10 kHz and sampled at 100 kHz using a Molecular Devices Axopatch 200B amplifier driven with a pCLAMP 10 interface (Molecular Devices, San Jose, CA, USA). Pipette resistances were typically 2.5–3.5 MΩ. Fast pipette capacitance was compensated after seal formation. Cell capacitance (C m) and series resistance (R S) were measured in whole‐cell configuration using the Clampex 10 membrane test utility while applying a +20 to +40 mV step from a holding potential (V H) of −40 mV. R S averaged 6.5 ± 0.1 MΩ. The RC transient was cancelled using the amplifier C m and R S controls, and 65–70% R S compensation (75% prediction) was typically applied. I–V and Q–V relationships were recorded by applying 20 ms test pulses (−100 to +190 mV, in 10 or 20 mV increments) from a V H of −40 mV. Leak subtraction (P/4 for test pulses ≤+50 mV; P/6 for test pulses ≥+60 mV) was applied to subtract linear capacitance and endogenous leak currents. I SS was averaged over the last 1 ms of each pulse except where otherwise indicated, and current densities are expressed as pA/pF. Cell capacitance averaged 19 ± 1 pF among the genotype groups. Leak‐subtracted current traces are displayed, except for E224A mutant genotypes, which lack rectification (Figs 2 and 6). Zero‐current levels are indicated in all sets of traces by a dashed line.
Figure 2. The non‐halide thiocyanate (SCN−) increases anion transport rate in ClC‐5/3 wild‐type and E224A Gluext mutant.
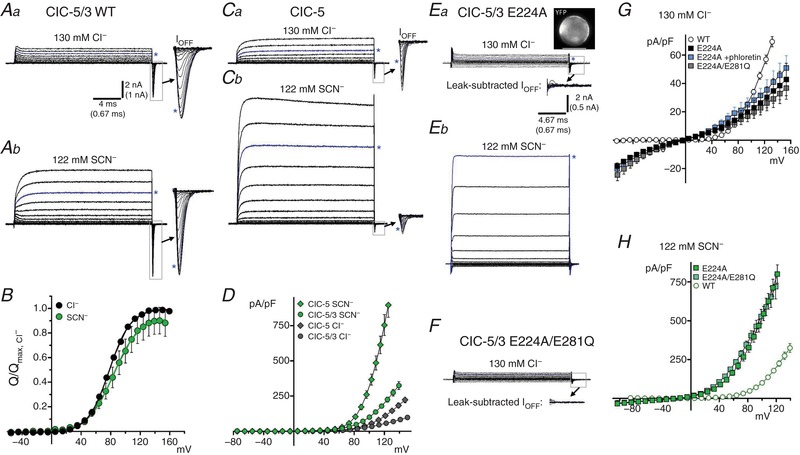
A, ClC‐5/3 steady‐state (I SS) transport and transient gating currents in normal (a, 130 mM Cl−, pH 7.35) and SCN−‐containing (b, 122 mM SCN−/8 mM Cl−; pH 7.35) external saline. I SS traces are displayed up to +140 mV depolarization. I Off transients (boxed regions) are shown at expanded scale to the right (arrows; displayed to +160 mV). Asterisks (blue traces) indicate currents at +115 mV. SCN− greatly increases I SS, consistent with increased anion “slippage”. B, Q Off–V relationships for ClC‐5/3 in Cl− and SCN−‐containing saline, normalized to same‐cell values in Cl−. Continuous lines show fits to Boltzmann functions. Relative Q max is not significantly altered in SCN−, while V 1/2 of Q–V is shifted by +6 mV (see Table 1). C, ClC‐5 I SS and transient gating currents in Cl− (a) and SCN−‐containing external saline (b), showing a large increase in SCN− current magnitude. I SS traces are displayed up to +140 mV as in A; I Off transients expanded to the right (arrows) are displayed to +180 mV (scale as in A). Asterisks (blue traces) indicate current trace recorded at +120 mV. I Off in SCN− (b) is likely obscured due to the extremely large I SS magnitude (see Methods). D, I SS–V relationships compared for ClC‐5/3 and ClC‐5 in external Cl− and SCN−. SCN− produces a larger‐fold increase in ClC‐5 currents. Untransfected HEK cells exhibit a significant endogenous SCN− conductance (29 pA/pF at +115 mV; see Table 1). E, unsubtracted currents in external Cl− (a) and SCN− (b) in an E224A mutant‐expressing cell; dashed line indicates zero‐current level. Image (top right) shows YFP protein expression in the recorded cell. E224A abolishes voltage‐dependent transient charge movements and results in a loss of I SS rectification and decreased current amplitudes at depolarized voltages. Lower inset in Ea shows leak‐subtracted currents at a magnified scale from the indicated boxed region. E224A transport is dramatically increased in external SCN− (b; non‐subtracted traces are displayed up to +120 mV). F, E22A/E281Q double mutant currents are essentially indistinguishable from E224A; scale of currents and magnified inset as in E, top. Dashed line indicates zero‐current level. G, I SS–V relationships in external Cl− for E224A single and E224A/E281Q double mutants (filled and shaded symbols), compared to wild‐type ClC‐5/3 (WT, open symbols). The Cl− channel blocker phloretin (100–150 μM) produces no significant block of E224A currents. H, I SS–V relationships in external SCN− for E224A mutants (shaded symbols) compared to wild‐type. E224A mutation impairs maximal Cl− transport, but greatly facilitates SCN− transport. [Color figure can be viewed at http://wileyonlinelibrary.com]
Figure 6. Y572 internal “anion gate” regulates anion conductance and Cl−/H+ coupling.
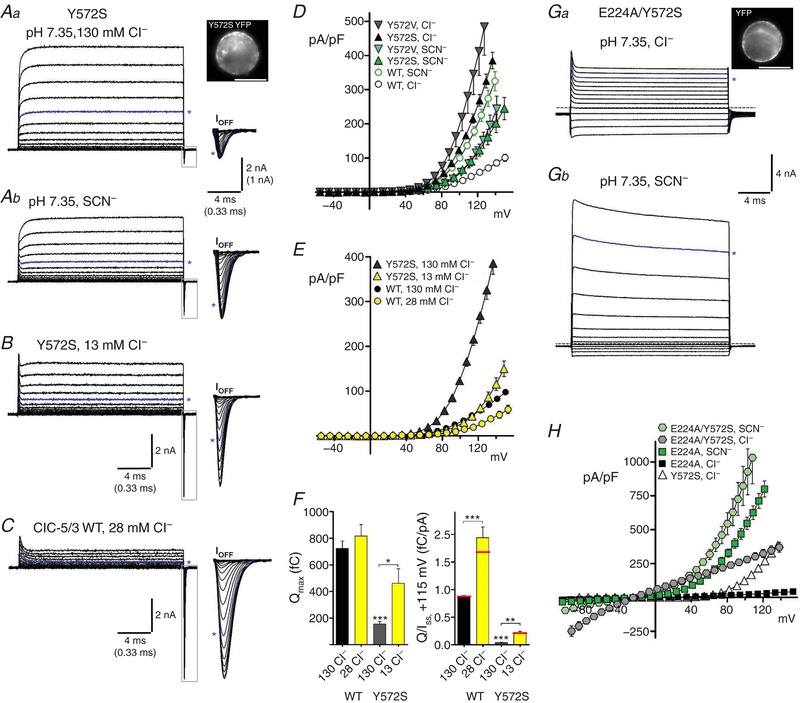
A, transport currents recorded from a Y572S‐expressing cell (YFP expression is shown at top right). Steady‐state (I SS) currents are displayed to a voltage of +160 mV; Off‐gating transients (boxed regions, expanded at right) are displayed to a voltage of +180 mV. Asterisks indicate traces at +120 mV applied potential. Y572S exhibits large I SS and small gating transients in normal Cl−‐containing external saline (Aa, pH 7.35). In external SCN− (Ab, 122 mM SCN−/8 mM Cl−), I SS amplitude is decreased while the gating component is increased. Both changes were reversed upon return to control 130 mM Cl− (not shown). B, in 13 mM external Cl− (Cl− replaced with MeSO4 −), Y572S I SS is decreased and gating currents are markedly increased. C, in 28 mM external Cl−, ClC‐5/3 wild‐type I SS is reduced relative to gating charge, compared to normal Cl−. D, I SS–V relationships for Y572S and Y572V mutants in external Cl− and SCN−. ClC‐5/3 wild‐type I–V plots (open circles) are included for comparison. Mutants display greatly elevated I SS in Cl−, while SCN− I SS is reduced to below wild‐type levels. E, I SS–V relationships for Y572S and wild‐type in normal 130 mM Cl− and lowered Cl− conditions. F, left: quantified Q max for ClC‐5/3 wild‐type and Y572S in normal and low Cl− salines. Y572S Q max is increased by ∼3‐fold in 13 mM Cl− (*** P < 0.0001 vs. WT in 130 mM Cl−; * P < 0.05 vs. 130 mM control; n = 3). Right: Q/I SS ratios (fC/pA, +115 mV) for wild‐type and Y572S are increased in reduced external Cl− saline (*** P < 0.0001 vs. WT in 130 mM Cl−, n = 5; ** P < 0.01 vs. 130 mM control; n = 3). Red lines indicate mean ratios after correction of Q and I to a corrected voltage of +115 mV. G, E224A/Y572S “double gate” mutant exhibits large non‐rectifying currents in normal external Cl− (a; non‐subtracted current traces are displayed to a voltage of +140 mV), and extraordinarily large currents in external SCN− (b; currents are displayed to a voltage of +130 mV). Asterisks indicate traces at +120 mV; dashed line indicates zero‐current level. Image (a, top right) illustrates YFP fusion protein expression. H, I SS–V relationships for Y572S and E224A single and double mutants in external Cl− or SCN−. [Color figure can be viewed at http://wileyonlinelibrary.com]
Gating charge (Q) was measured as the area under the inward “I Off” transient current within a manually determined window (typically 0.5–1.5 ms duration), using PClamp10 Clampfit Statistics functions. A Boltzmann equation was fitted to individual Q–V plots (Alekov, 2015) using Clampfit 10 curve fitting, and to averaged normalized Q–V plots using Graphpad Prism (v.5; Graphpad Software, La Jolla, CA, USA). Maximal Q Off (Q max) and V 1/2 of the Q–V relationship were determined from the fit parameters. In a minority of recordings, Q Off was too small for reliable measurement, or partly occluded by a large I SS and poorly described by a Boltzmann relationship. Such examples occurred primarily for genotypes with large I SS and comparatively small Q Off (ClC‐5, and ClC‐5/3 Y572S and Y572V mutants), and were excluded from quantification of average Q max and Q/I. Charge movement kinetics were assessed by measuring I Off half‐width (duration at 50% of peak amplitude) and 90–10% decay time. We estimated a clamp time constant of ∼40 μs, based on average cell capacitance (19 pF) and uncompensated RS of 2.2 MΩ, corresponding to a clamp bandwidth of ∼4kHz (Axopatch 200B “Theory and Operation”, 1997–1999; Molecular Devices, San Jose, CA, USA). Additional gating current recordings were made using up to 80% R S compensation (80–85% prediction). Transient current kinetics were sensitive to compensation level, but measured Q was largely independent of the level of R S compensation (0–80%; see Fig. 8 J).
Figure 8. Y572 regulates the rate and voltage dependence of gating charge movement.
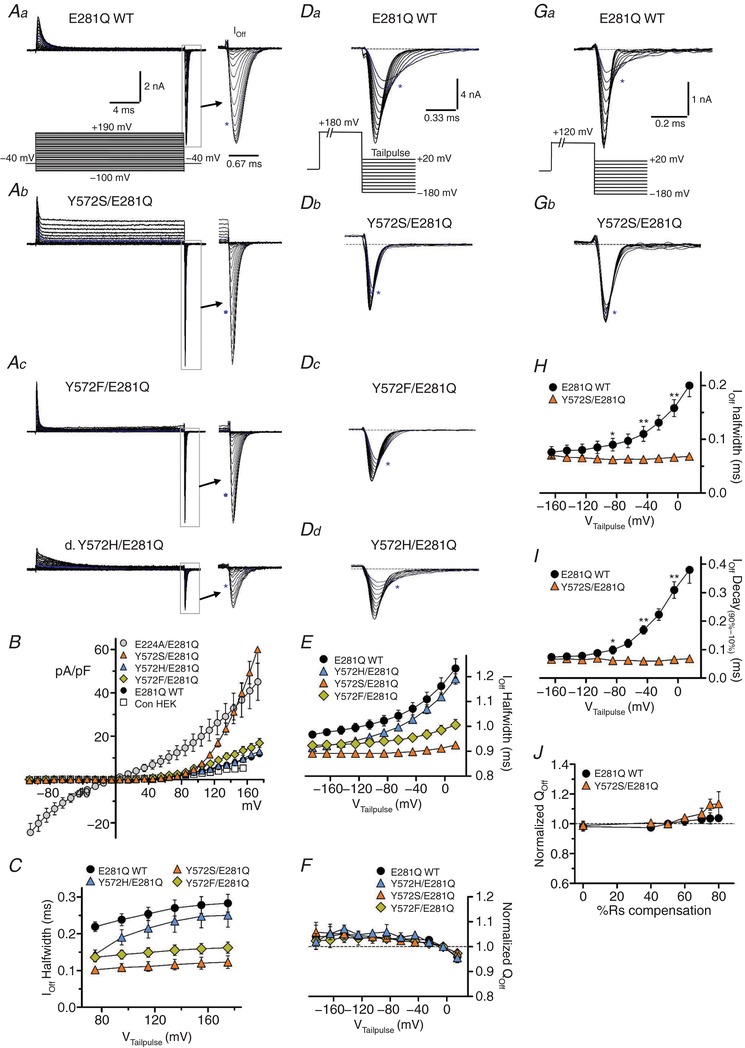
All genotypes shown are in the E281Q mutant background. A, steady‐state and transient gating currents, recorded with the illustrated voltage pulse protocol (a, lower: 20 ms test pulses from −100 to +190 mV, −40 mV V H). Off‐gating transients (boxed areas) are shown with expanded time course (arrows); asterisks indicate trace corresponding to +120 mV applied test pulse). Aa, wild‐type Y572 (E281Q WT) displays minimal I SS with large gating transients. Ab, Y572S displays a significant outwardly rectifying I SS at strongly depolarized voltages (>+100 mV), and large gating transients with more rapid kinetics. Ac, Y572F restricts I SS, but also results in faster transient kinetics. Ad, histidine substitution (Y572H) does not alter I SS, and partly restores normal transient durations. B, I SS–V relationships for E281Q mutant combinations (E224A, wild‐type, Y572S, Y572F and Y572H). Open squares show basal current level in non‐expressing control cells. C, Off‐transient half‐width (ms) plotted against depolarizing test pulse voltage. Y572S and Y572F transient durations are significantly shorter over the entire voltage range. Y572H duration approaches wild‐type values with increasing voltage. D, Off‐gating currents at increasingly negative tail pulse voltages (+20 to −180 mV) following a maximally activating depolarization (+180 mV; protocol is illustrated in a. Dotted lines show zero‐current level; asterisks indicate traces recorded at 0 mV tail pulse potential. Da, for E281Q (wild‐type Y572), the rate of charge movement is strongly dependent on repolarization voltage. Db, Y572S charge movement is faster at all voltages, and nearly voltage independent. Dc, Y572F also displays faster charge movement with only weak voltage dependence. Dd, Y572H displays intermediate kinetics with clear voltage dependence. E, Off‐gating transient half‐width plotted against tail pulse voltage. Wild‐type charge movement is slower at all voltages and strongly voltage dependent. Y572S and Y572F exhibit faster charge movement, with minimal voltage dependence, while Y572H displays intermediate rates, but pronounced voltage dependence. Y572S and Y572F mutants showed similarly increased I Off decay rate compared to E281Q WT (not shown). F, normalized Q Off plotted against tail pulse voltage. Charge recovery upon repolarization is essentially complete at all negative voltages. G, Off‐gating currents recorded in E281Q‐ (a) and Y572S/E281Q‐ (b) expressing cells. To reduce maximal I Off amplitude, cells were selected with lower protein expression level, and gating was activated with a +120 mV depolarizing test pulse (protocol illustrated in Ga; note difference in scale compared to D). R S compensation for these recordings was increased to 70–75% (80–85% prediction), resulting in faster gating kinetics. As in D, E281Q WT charge movement kinetics (Ga) are strongly voltage dependent, while Y572S charge movement kinetics are voltage independent (Gb). H, Off‐transient half‐width plotted against tail pulse voltage. I, Off‐transient 90–10% decay time plotted against tail pulse voltage. Both kinetic parameters are strongly voltage dependent in E281Q WT, but display no voltage dependence in Y572S. J, normalized Q Off (+120 mV activation, −160 mV tail pulse) is plotted against R S compensation level (0–80%). Gating Q (area) measurements in this study are relatively insensitive to compensation level. [Color figure can be viewed at http://wileyonlinelibrary.com]
Correction for estimated voltage errors (ΔV m) resulting from residual uncompensated R S (1.7–2.5 MΩ) is applied to average I–V and Q–V relationships for each genotype and experimental condition. For most recordings in external Cl−, ΔV m did not exceed 5% of the command potential (3 mV at +115 mV; Table 1). Increased I SS levels in external SCN− increased average ΔV m to ∼6 mV for wild‐type ClC‐5/3 and ∼13 mV for ClC‐5 (Table 1). Y572S/V and E224A/Y572S mutant combinations exhibited substantially larger I SS and voltage errors, noted in Table 1. Mean current densities are reported at +115 mV. Corrected I SS current (pA) and Q magnitude at +115 mV were determined by interpolating the averaged, voltage‐corrected I–V and Q–V relationships, respectively. Q/I ratios (Table 1) are corrected to +115 mV.
Table 1.
Summary of ClC‐3 ion transport and gating charge
| Anion, pH | I SS (pA/pF)a | ΔV m a , b | I SS (pA)c | Q max (fC)d | Q/I SS c | V 1/2 of Q–V | |
|---|---|---|---|---|---|---|---|
| Control HEK | Cl−, pH 7.35e | 3.4 ± 0.5 (22) | <0.5 mV | n.d. | |||
| SCN−, pH 7.35d | 29.1 ± 1.9 (4) | <0.5 mV | n.d. | ||||
| ClC‐5 WT | Cl−, pH 7.35e | 80.0 ± 8.4 | 2.8 mV | 1573 (11) | 204 ± 25 (9) | 0.05 ± 0.01 * | 126 ± 4 mV |
| SCN−, pH 7.35d | 367 ± 39 (9) | 13 mV§ | 11,482 (9) | 68 ± 19% (4) | n.m. | n.m. | |
| ClC‐3a WT | Cl−, pH 7.35e | 5.0 ± 0.8 | <0.5 mV | 111 (8) | n.d. | n.d. | n.d. |
| ClC‐3b WT | Cl−, pH 7.35 | 12.9 ± 4.3 (9) | <0.5 mV | 159 (9) | 109 ± 31 (8) | 0.69 ± 0.16 (8) | 82 ± 3 mV (7) |
| ClC‐3/5 | Cl−, pH 7.35 | 4.0 ± 0.6 (6) | <0.5 mV | n.d. | n.d. | n.d. | |
| ClC‐5/3 WT | Cl−, pH 7.35e | 45.4 ± 2.4 | 1.7 mV | 800 (28) | 755 ± 76 | 0.86 ± 0.03 | 78 ± 1 mV |
| 28 mM Cl− | 19.1 ± 3.3 | 0.6 mV | 298 (5) | 817 ± 77 | 1.67 ± 0.17† | 107 ± 1 mV† | |
| SCN−, pH 7.35f | 136 ± 9 (6) | 6.7 mV | 2865 (6) | 94 ± 7% | 0.18 ± 0.02 | 84 ± 5 mV† | |
| Cl−, pH 6.0 | 36.1 ± 3.4† | 1.8 mV | 832 (7) | 74 ± 3% | n.m. | 89 ± 4 mV† | |
| Cl−, pH 5.0g | 25.6 ± 3.2† | 1.5 mV | 468 (12) | 40 ± 3% | n.m. | 107 ± 2 mV† | |
| E224A | Cl−, pH 7.35e | 26.2 ± 6.3 | 1.1 mV | 442 (5) | n.d. | n.d. | |
| SCN−, pH 7.35f | 467 ± 40 | 20 mV§ | 11,228 (5) | n.d. | n.d. | ||
| M531A | Cl−, pH 7.35e | 67.2 ± 6.4 * | 2.6 mV | 1250 (17) | 504 ± 39 * | 0.35 ± 0.03 * | 79 ± 1 mV * |
| SCN−, pH 7.35f | 150 ± 10 | 4.7 mV | 2628 (2) | 128 ± 24% | 0.14 ± 0.02 | 103 ± 1 mV† | |
| Cl−, pH 6.0 | 53.5 ± 10 (8) | 2.2 mV | 1067 (8) | 83 ± 5% (8) | n.m. | 97 ± 4 mV | |
| Cl−, pH 5.0g | 30.5 ± 5.0 (8) | 1.3 mV | 596 (8) | 34 ± 2% (8) | n.m. | 112 ± 5 mV† | |
| E281Q | Cl−, pH 7.35e | 4.5 ± 0.6 | <0.5 mV | 97 (23) | 1663 ± 144* | 25.6 ± 9.4 | 88 ± 1 mV * |
| SCN−, pH 7.35f | 24 ± 3 | <1 mV | 387 (3) | ||||
| Cl−, pH 6.0 | 3.3 ± 1.6 (7) | <0.5 mV | 98 ± 3% (8) | n.m. | 106 ± 3 mV† | ||
| Cl−, pH 5.0g | n.m. | <0.5 mV | 87 ± 4% (12) | n.m. | 131 ± 3 mV† | ||
| E224A/E281Q | Cl−, pH 7.35e | 22.8 ± 5.0 | 1.1 mV | 446 (5) | n.d. | n.d. | |
| SCN−, pH 7.35f | 464 ± 38 | 23 mV§ | 12,960 (5) | n.d. | n.d. | ||
| E224A/Y572S | Cl−, pH 7.35e | 279 ± 26 | 13 mV§ | 5128 (6) | n.d. | n.d. | |
| SCN−, pH 7.35f | 629 ± 100 | 29 mV§ | 18,328 (4) | n.d. | n.d. | ||
| Y572S | Cl−, pH 7.35e | 166 ± 11 | 8.0 mV § | 4892 (28) | 198 ± 20 (15) * | 0.04 ± 0.005 * | 72 ± 3 mV |
| 13 mM Cl− | 43.8 ± 5.8 | 1.9 mV | 1068 (3) | 462 ± 110† | 0.22 ± 0.02† | 114 ± 4 mV† | |
| SCN−, pH 7.35f | 93 ± 13 | 4.5 mV | 2432 (13) | 214 ± 31% (8)† | 0.09 ± 0.02 | 80 ± 5 mV | |
| Y572V | Cl−, pH 7.35e | 220 ± 40 | 13 mV§ | 8328 (7) | 208 ± 37 (5)* | 0.03 ± 0.02* | 74 ± 2 mV |
| SCN−, pH 7.35f | 83 ± 13 | 4.8 mV | 2500 (7) | 83 ± 14% (5) | 0.06 ± 0.01 (5) | 87 ± 5 mV | |
| Y572F | Cl−, pH 7.35e | 55.4 ± 5.5 * | 2.0 mV | 949 (10) | 1162 ± 161 * | 0.88 ± 0.05 * | 90 ± 2 mV * |
| SCN−, pH 7.35f | 69 ± 5† | 2.5 mV | 1197 (8) | 72 ± 2% (5)† | 0.44 ± 0.04 (5) | 83 ± 4 mV | |
| Y572S/E281Q | Cl−, pH 7.35e | 11.4 ± 1.4‡ | <1 mV | 210 (11) | 959 ± 108‡ | 1.77 ± 0.34‡ | 115 ± 1 mV‡ |
| Y572F/E281Q | Cl−, pH 7.35 | 7.3 ± 1.1 | <1 mV | 154 (13) | 1384 ± 141 | 11.0 ± 2.2‡ | 103 ± 2 mV‡ |
| Y572H/E281Q | Cl−, pH 7.35 | 4.8 ± 0.9 | <1 mV | 86 (7) | 844 ± 70‡ | 3.62 ± 0.58‡ | 131 ± 2 mV‡ |
Values are means ± SEM; numbers in parentheses indicate number of experiments or observations. aValues at +115 mV applied potential. I SS (pA/pF): steady‐state transport current density. bΔV m: average R S‐associated voltage error at steady state. c I SS (pA): average current magnitude at a corrected voltage of +115 mV; Q/I SS: average ratio at a corrected voltage of +115 mV. d Q max determined in control saline (pH 7.35, 130 mM Cl−); %Q is normalized to control Q max value. e130 mM Cl− control saline at pH 7.35. f122 mM SCN−/8 mM Cl− saline at pH 7.35. gExternal saline included 100–300 μM phloretin to block endogenous I Cl,acid. Symbols: *differs significantly (P ≤ 0.05) from value in ClC‐5/3 wild‐type; †differs significantly (P ≤ 0.05) from value in control saline; ‡differs significantly (P ≤ 0.05) from E281Q “wild‐type”; §greater than 5% average steady‐state voltage error at +115 mV applied potential. n.d., undetected/not present; n.m., not measured/uninterpretable.
Cellular pH recordings
Simultaneous recordings of whole‐cell current and associated intracellular pH changes were made using bath and pipette solutions with pH adjusted to 7.0, and the pipette contained a reduced concentration of HEPES (1 mM) and 100–150 μM 2′,7′‑bis‑(2‑carboxyethyl)‑5‑(and‑6)‑carboxyfluorescein (BCECF, free acid; Life Technologies) (Alekov & Fahlke, 2009). One to two minutes after establishing whole‐cell access, a brief control I–V was recorded (20–50 ms pulses, −80 to +160 mV potentials) to determine leak‐subtracted current density. Cellular BCECF was sequentially excited at 445 and 494 nm (50 ms, 4–5 Hz), and emitted fluorescence collected at 537 nm (Alekov & Fahlke, 2009), while applying 3.2 s depolarizing pulses (0, +40, +80, +120, +160 mV potentials). The final two pulses were preceded by ∼20 s rest intervals to allow intracellular pH re‐equilibration. BCECF fluorescence was calibrated in whole‐cell configuration by imaging the recorded cell in a series of pH standard buffers containing (in mM): 150 KCl, 10 HEPES, 10 μM nigericin (pH 6.5, 7.0, 7.5, 8.0). The ratio of integrated cellular fluorescence at the two excitation wavelengths (abbreviated here as F 490/F 440) was background‐corrected and measured offline. For each cell, F 490/F 440 values measured in the standard buffers was plotted vs. buffer pH, and the slope of this relationship was determined by a linear regression (Microsoft Excel 2010). Changes in cellular pH vs. time during depolarizing pulses were calculated as Slope × ΔF 490/F 440, relative to prepulse baseline level. The initial rate of pH change (ΔpH/s) was determined by linear fits to the first 1.0–2.5 s of each response. Statistical comparison was made between ΔpH/s values measured at +120 mV applied potential. Average ΔV m values in pH experiments are indicated in Table 2. Relative coupling ratios were determined from the slope of the relationship between alkalization rate and current density (ΔpH/s vs. I SS), which is independent of voltage.
Table 2.
ClC‐3 proton coupling and turnover
| Anion, pH | ΔpH/s (+115 mV)a | ΔV m (mV)b | Relative I SS c | Sloped | Couplinge | H+ turnoverf | |
|---|---|---|---|---|---|---|---|
| ClC‐5 | Cl−, pH 7.35g | 0.197 ± 0.060 (4) * | 2.7 mV | 1.76 | 0.264 ± 0.010‡ | 2.0i | 1.97 |
| SCN−, pH 7.35h | 0.076 ± 0.004 (4) | 11.0 mV § | 14.40 | 0.021 ± 0.002 | ∼18 | 0.89 | |
| ClC‐5/3 WT | Cl−, pH 7.35 | 0.117 ± 0.012 (17) | 2.9 mV | 1.00 | 0.281 ± 0.013 | 2.0 j | 1.00 |
| pH 5.0, 130 mM Cl− | 0.034 ± 0.007 (7) * | <1.5 mV | 0.59 | 0.170 ± 0.018 † | 3.3 | 0.36 | |
| 28 mM Cl−, pH 7.35 | 0.030 ± 0.005 (5) † | <1.0 mV | 0.37 | 0.245 ± 0.015¶ | 2.2 | 0.34 | |
| SCN−, pH 7.35 | 0.044 ± 0.007 (6) | 12.7 mV § | 3.58 | 0.038 ± 0.005 | ∼15 | 0.48 | |
| M531A | Cl−, pH 7.35 | 0.227 ± 0.051 (8) * | 2.1 mV | 1.56 | 0.289 ± 0.008‡ | 1.94 | 1.61 |
| SCN−, pH 7.35 | 0.103 ± 0.030 (2) | 4.8 mV | 3.29 | 0.066 ± 0.004 | ∼8.8 | 0.75 | |
| Y572S | Cl−, pH 7.35 | 0.139 ± 0.017 (8)‡ | 8.9 mV § | 6.12 | 0.070 ± 0.008 † | 8.0 | 1.53 |
| SCN−, pH 7.35 | 0.048 ± 0.006 (4) | 6.7 mV § | 3.04 | 0.033 ± 0.005 | ∼17 | 0.36 | |
| Y572F | Cl−, pH 7.35 | 0.151 ± 0.031 (4)‡ | 2.7 mV | 1.19 | 0.206 ± 0.012 † | 2.7 | 0.88 |
| SCN−, pH 7.35 | 0.013 ± 0.002 (3) | 3.1 mV | 1.50 | 0.020 ± 0.002 | ∼28 | 0.11 | |
| E224A | Cl−, pH 7.35 | −0.001 ± 0.001 (4) | 1.2 mV | 0.55 | 0.0025 ± 0.0024 | >220 | <0.01 |
Values are means ± SEM; numbers in parentheses indicate number of experiments or observations. aCellular alkalization rate at +115 mV applied potential. bAverage steady‐state R S‐associated voltage error at +115 mV; cMean current magnitude at a corrected voltage of +115 mV, relative to ClC‐5/3 wild‐type. dΔpH/s per 100 pA/pF conductance. eEstimated anion:H+ coupling ratio assuming a ratio of 2.0 for ClC‐5/3 wild‐type at pH 7.35 in external Cl−. fRelative H+ transport efficiency vs. ClC‐5/3 wild‐type in control saline. g130 mM Cl− control saline at pH 7.35. h122 mM SCN−/8 mM Cl− saline at pH 7.35. iValue of 2.0 determined by Zifarelli & Pusch (2009). jThe relationship for ClC‐5/3 displays a slope equal to that of ClC‐5 (Fig. 3 I). Symbols: * P ≤ 0.03 and † P ≤ 0.001, respectively, vs. wild‐type ClC‐5/3, control saline. All values in external SCN− differ significantly (P ≤ 0.01) from corresponding values in external Cl−. ‡Not significant (P ≥ 0.05) vs. wild‐type ClC‐5/3, control saline; ¶not significant (P ≥ 0.05) vs. value in 130 mM Cl−; §voltage errors exceeding 5% of applied potential.
Data analysis and statistics
Statistical calculations and comparisons were made with Excel (2010) and Graphpad Prism 5 analysis software. Statistical comparisons were made using one‐way ANOVA with either Tukey, Kruskal–Wallis non‐parametric, or Wilcoxon matched pairs methodology. The Mann–Whitney non‐parametric test was used for two‐way comparisons. Quantified results are expressed and shown graphically as mean ± SEM.
Results
The ClC‐5/3 protein is localized to the plasma membrane
Mammalian isoforms of ClC‐3 (NM_001829) and ClC‐5 (BC130429.1) are highly conserved within their core channel domains (helices B–R, 89% similar, 74% identical) and cytoplasmic C‐ter (89% similar, 79% identical; identical length). The two proteins differ primarily within their cytoplasmic N‐ter sequences (Fig. 1 A). The ClC‐3a N‐ter is 13 amino acids longer and contains a dileucine cluster (LLDLLD; residues 13–18) implicated in clathrin binding (Zhao et al. 2007). Mutation of this motif (ClC‐3a13–19A) results in protein retention within the plasma membrane (PM), permitting detailed biophysical analysis (Guzman et al. 2013, 2015). Though ClC‐5 is also proposed to function primarily in endosomes, ClC‐5 lacks a dileucine motif and has an appreciable level of PM localization when heterologously expressed in cells (Steinmeyer et al. 1995; Guzman et al. 2013; Alekov, 2015). We replaced the ClC‐3a N‐ter (amino acids 1–59) with the N‐ter of ClC‐5 (amino acids 1–46), predicting that this substitution would effectively localize ClC‐3 to the PM. We compared the properties of the resulting “ClC‐5/3” chimera to wild‐type ClC‐5, and examined the consequence of mutations within the ClC‐3 anion–proton exchange pathway.
Figure 1. Functional expression and ion transport features of ClC‐5/3 compared to ClC‐3 and ClC‐5.
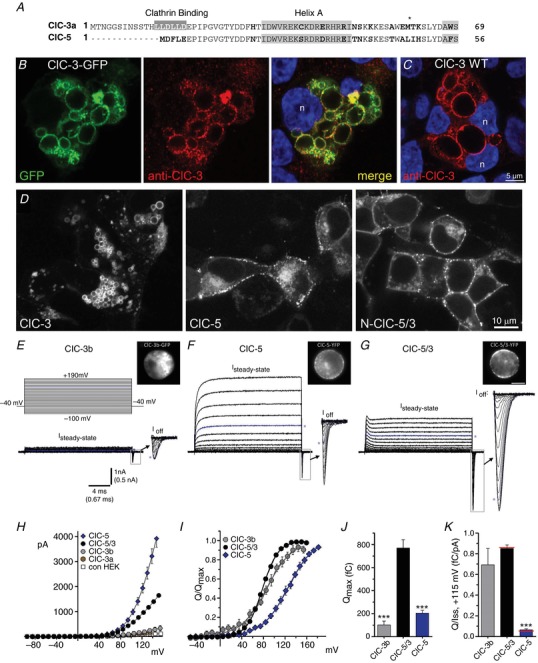
A, alignment of N‐terminal amino acid sequences of wild‐type ClC‐3a (“short” ClC‐3; top) and ClC‐5 transporters. Non‐conserved residues are indicated in bold. The clathrin binding sequence (LLDLLD, shaded) promotes ClC‐3a removal from the plasma membrane. B, HEK 293T cells transfected with wild‐type ClC‐3a‐GFP fusion construct (GFP, left) are stained with anti‐ClC‐3 (red, centre). DAPI co‐staining (blue, right) shows individual cell nuclei (n). ClC‐3‐GFP localizes strongly to abnormally large cytoplasmic vesicles, and co‐localizes with anti‐GFP vesicular labelling (merged panel, right). C, cells transfected with wild‐type (WT) non‐fusion ClC‐3a and co‐stained with anti‐ClC‐3 (red) and DAPI (blue) exhibit a similar pattern of protein localization to enlarged vesicles, confirming that the C‐terminus GFP tag does not cause the vesicle phenotype or alter ClC‐3 trafficking. D, typical YFP‐fusion protein expression patterns and vesicular morphology in cells transfected with wild‐type ClC‐3a (left), wild‐type ClC‐5 (centre), and the ClC‐5/3 chimera (right). ClC‐3a localizes primarily to enlarged vesicles, with little detectable plasma membrane localization. ClC‐3b overexpression features are entirely similar (not shown). In contrast, ClC‐5 and ClC‐5/3 show similar localization to both the plasma membrane and vesicular compartments. E–G, leak‐subtracted whole‐cell ion currents recorded from cells expressing wild‐type ClC‐3b (E), ClC‐5 (F) and ClC‐5/3 (G). Images show GFP‐ and YFP‐fusion protein expression and distribution in the recorded cells. Voltage protocol is depicted in E (top) (−40 mV V H; 20 ms test pulses ranging from −100 to +190 mV). Steady‐state (I SS) and transient Off‐gating current components are illustrated; “I Off” transients (boxed regions) are shown at magnified scale (arrows) at right. Currents are displayed up to maximum depolarizations of +160 mV, except for ClC‐5 I Off (+180 mV maximum). Asterisks (blue traces) indicate currents recorded at +120 mV applied potential. ClC‐5/3 is distinguished by larger and more prolonged gating currents compared to ClC‐5. ClC‐3a‐expressing cells did not exhibit significant I SS or transients. H, mean I SS (pA)–V relationships for ClC‐5, ClC‐5/3, ClC‐3a, ClC‐3b and non‐transfected HEK controls. I, normalized Off‐transient gating charge (Q/Q max) plotted vs. test pulse voltage for ClC‐3b, ClC‐5/3 and ClC‐5. Continuous lines show fits to Boltzmann functions. J, quantified Q max measured for the three genotypes (*** P < 0.0005 vs. ClC‐5/3, one‐way ANOVA). K, ratio of Q Off to transport current (Q/I SS, +115 mV; fC/pA) for the three genotypes (*** P < 0.0005 vs. ClC‐5/3, one‐way ANOVA). Red lines indicate mean ratios after correction of Q and I values to a corrected voltage of +115 mV. [Color figure can be viewed at http://wileyonlinelibrary.com]
C‐terminal YFP and GFP fusion proteins for ClC‐3, ClC‐5 and ClC‐5/3 expressed in HEK cells were imaged by confocal microscopy. Wild‐type ClC‐3a and 3b isoforms localize primarily to the membranes of enlarged intracellular vesicles, as previously reported (Li et al. 2002), while PM expression was visually low or undetectable (Fig. 1 B–D). ClC‐3‐GFP cellular localization was confirmed by anti‐ClC‐3 immunostaining (Fig. 1 B). Overexpression of ClC‐3a without a GFP tag also resulted in enlarged vesicles and a similar pattern of vesicular protein expression (Fig. 1 C). To confirm the requirement for the N‐ter in protein trafficking, we expressed an N‐ter deletion mutant, ΔN‐ClC‐3a‐GFP (ΔM1–E59). This protein accumulated solely within the endoplasmic reticulum (ER), as demonstrated by fluorescence co‐localization with an ER marker (data not shown). Overexpressed ClC‐5 prominently localized to the cell surface, exhibiting a non‐homogeneous distribution characterized by weaker smooth fluorescence punctuated by brighter fluorescence aggregates (Fig. 1 D, centre; Fig. 1 F inset). ClC‐5 exhibited variable intracellular distribution and expression level, but its overexpression did not produce conspicuously enlarged vesicles, as observed with ClC‐3. Overexpression of ClC‐5/3 likewise resulted in pronounced PM localization and distribution closely resembling that of ClC‐5 (Fig. 1 D, right; Fig. 1 G inset).
ClC‐5/3 displays prominent gating currents and completely rectifying ion transport
In ClC‐3a‐expressing cells, whole‐cell steady‐state ion currents (I SS) were not significantly larger than endogenous background current level (Fig. 1 H and Table 1), consistent with findings from other studies (Guzman et al. 2013, 2015). In contrast, ClC‐3b‐expressing cells exhibited significant I SS (12.9 ± 4.3 pA/pF, +115 mV; P < 0.01 vs. ClC‐3a, one‐way ANOVA; Table 1). ClC‐3b currents were accompanied by small transient “Off” gating currents evident upon repolarization after depolarizing steps (I Off; inset, Fig. 1 E), consistent with a low level of functional PM expression. Similar but smaller transient gating currents were previously recorded in cells expressing wild‐type ClC‐3a (Guzman et al. 2013).
ClC‐5‐expressing cells displayed large, strongly outwardly rectifying I SS (80 ± 8 pA/pF, +115 mV; Table 1) with rapid transient Off gating currents (Fig. 1 F and H), consistent with other reports (Guzman et al. 2013; Alekov, 2015). ClC‐5/3‐expressing cells also displayed rapidly activating and strongly outwardly rectifying I SS, with prominent On and Off gating transients associated with positive depolarizing pulses (Fig. 1 G). ClC‐5/3 I SS density was ∼60% that of ClC‐5 at +115 mV (45 ± 2 pA/pF, Fig. 1 H and Table 1). ClC‐5/3 expression and biophysical features qualitatively resemble those described for ClC‐3a13–19A and ClC‐3b13–19A dileucine mutants, as well as for the ClC‐3c N‐terminal splice variant, which is targeted to recycling endosomes and exhibits PM localization (Guzman et al. 2013, 2015). ClC‐5 N‐ter substitution therefore effectively targets functional ClC‐3 to the PM, and C‐ter GFP and YFP fusion tags do not disrupt ion current expression. As an additional test, we replaced the N‐ter of ClC‐5 with ClC‐3a M1–E59. This “ClC‐3/5” chimera failed to localize to the PM, but was predominantly localized to large intracellular vesicles as described for wild‐type ClC‐3a (data not shown). ClC‐3/5 expression predictably failed to produce significant I SS (n = 6; Table 1). These results confirm that the N‐ter sequence is a fundamental determinant of ClC‐3 and ClC‐5 membrane trafficking. The N‐ter is required for ClC‐3 trafficking into cytoplasmic vesicles, and overexpression of either ClC‐3 or ClC‐3/5 causes enlargement and expansion of this vesicular compartment.
Voltage‐dependent gating charge (Q) mobilization was assessed by measuring the area under I Off transients, and fitting the Q Off–V relationship to a Boltzmann equation (Fig. 1 I). ClC‐5/3 exhibited far larger maximal Q Off (Q max; 755 ± 76 fC; n = 27) than ClC‐3b or ClC‐5 (Fig. 1 J and Table 1), but the half‐maximal activation voltage of Q–V (V 1/2) was comparable for ClC‐5/3 (78 ± 1 mV) and ClC‐3b (82 ± 3 mV) (Table 1). ClC‐5 V 1/2 was shifted by about +50 mV (126 ± 4 mV; Table 1) relative to ClC‐3. Interpreting gating charge movements and Q–V relationships to reflect the voltage‐dependent transport activation process (Smith & Lippiat, 2010b ; Orhan et al. 2011; Zifarelli et al. 2012), ClC‐3 transport is 80–90% maximally activated at +115 mV, while ClC‐5 is less than 50% activated at the same voltage (Fig. 1 I).
Our results are consistent with previous studies demonstrating significantly different gating and conductance for ClC‐3 and ClC‐5 (Guzman et al. 2013, 2015; Alekov, 2015), with ClC‐3 displaying an inherently lower efficiency of steady‐state ion transport than ClC‐5. Since gating charge movements are assumed to result from incomplete (non‐exchanging) transport cycles, we compared the ratio of gating Q Off to transport conductance (Q/I; fC/pA) at +115 mV, as a relative index of transport efficiency (Grieschat & Alekov, 2012; Guzman et al. 2013; Alekov, 2015). Q/I ratio is comparable for ClC‐5/3 (0.86 ± 0.03; n = 27) and ClC‐3b (0.69 ± 0.16; n = 8), but less than 10% as large for ClC‐5 (0.05 ± 0.01; n = 9; Fig. 1 K and Table 1). We would therefore predict the Q/I ratio to inversely reflect the rate or probability of a completed exchange cycle, that is, the efficiency of proton transfer from the cytoplasm to the inner “proton gate” (Gluin; E281 of ClC‐5/3), to the outer “gating glutamate” (Gluext; E224) and into the extracellular milieu. We test this assumption below by combining measurements of Q Off and I SS with optical quantification of cytoplasmic alkalization, and assessing the impact of altered ionic conditions or mutations predicted to impact anion or proton pathways in ClC‐5/3. Ion transport and gating charge parameters under different ionic and pH conditions are compiled in Table 1.
External SCN− increases ClC‐5/3 and ClC‐5 anion transport
The non‐halide thiocyanate (SCN−) anion reduces anion/H+ exchange coupling in ClC‐4 and ClC‐5 (Alekov & Fahlke, 2009; Orhan et al. 2011; Grieschat & Alekov, 2012). A “slippage” mode of exchange is proposed to occur in external SCN−, whereby multiple (10–20) anions are conducted for each cycle of proton transport (Alekov & Fahlke, 2009). We assayed ClC‐5/3 and ClC‐5 transport currents following the substitution of SCN− for external Cl− (122 mM SCN−, 8 mM Cl−). In SCN−, ClC‐5/3 I SS increased by 3‐ to 4‐fold at +115 mV (Fig. 2 A and D, and Table 1). This relative increase is comparable to that previously reported for ClC‐4 in external SCN− (Alekov & Fahlke, 2009). In contrast, SCN− increased ClC‐5 I SS to a greater degree in same‐cell recordings (Fig. 2 C and D, and Table 1). The large size of SCN− currents in some cases generated appreciable (>5%) voltage errors (ΔV m), particularly for ClC‐5 (Table 1). After applying voltage correction to I–V relationships, we estimated ClC‐5 I SS to be increased by 7‐ to 8‐fold in SCN− (+115 mV; Fig. 2 D and Table 1). The observed increase for ClC‐5 in SCN− is nevertheless smaller than the 10‐ to 15‐fold increase previously reported (Grieschat & Alekov, 2012). ClC‐5/3 Q max typically underwent little change in external SCN− (94 ± 7% of Q max in external Cl−; Fig. 2 B), but V 1/2 of Q–V shifted by +6 mV (Table 1). Assuming that Q Off largely reflects unprotonated (incompletely cycled) Gluext (Grieschat & Alekov, 2012; Alekov, 2015), these data suggest that the efficiency of ClC‐3 Gluext protonation is not significantly altered when SCN− is the transported anion. For ClC‐5, the large amplitude of SCN− ion currents more clearly hindered the ability to resolve the Q movements, precluding reliable measurement (Fig. 2 C).
Transport rectification and gating charge movements are abolished by loss of the external gating glutamate E224
To confirm that in ClC‐5/3 E224 functions as Gluext and assess its contribution to gating Q, we characterized currents for an E224A mutation predicted to abolish proton transport. E224A‐YFP localized to the PM similarly to the wild‐type protein (Fig. 2 E, inset). E224A‐expressing cells exhibited significant inward currents immediately after establishing whole‐cell configuration. Unsubtracted I–V relationships showed comparable inward and outward currents (20–30 pA/pF) at −100 mV and +115 mV (Table 1) that reversed near 0 mV, consistent with a non‐rectifying Cl− conductance (Fig. 2 E and G). Transient charge‐movement components were undetectable in leak‐subtracted traces (Fig. 2 E, inset). An E224A/E281Q double mutant displayed indistinguishable behaviour, with non‐rectifying I SS and an absence of Q transients (Fig. 2 F and G). The anion channel blocker phloretin (120–250 μM) had no inhibitory effect on the E224 mutant currents (Fig. 2 G). E224 gate removal thus readily permits Cl− efflux regardless of the presence of E281Q, demonstrating that Gluext represents an external barrier to anions. E224A I SS is nevertheless reduced to < 40% of wild‐type transport at +115 mV, consistent with the finding that Cl− transport rate is reduced by severalfold in the homologous ClC‐ec1 E148A mutant (Accardi & Miller, 2004). It was of interest to examine transport behaviour of the “slippery” SCN− anion in the non‐coupled E224A mutant. Unexpectedly, SCN− dramatically increased outward I SS by 20‐fold or more (+115 mV; Fig. 2 E and H, and Table 1). Though these I SS levels are severely underestimated (>20 mV ΔV m error; Table 1), they greatly exceed the magnitude of wild‐type SCN− I SS (Fig. 2 H). This result suggests that Gluext also functions to limit the influx rate of uncoupling non‐halide anions. This suggestion is further supported by results below.
An M531A mutation predicted to facilitate “water wire” proton transfer increases transport efficiency
The formation of a “water‐wire” along the hydrophobic central proton pathway is proposed to facilitate proton transfer from Gluin to Gluext in the bacterial ClC‐ec1 (Han et al. 2014). H+ transport is inhibited by bulk‐increasing side‐chain substitutions at ClC‐ec1 A404, the most highly hydrated residue lining the proton pathway (Han et al. 2014). Interestingly, both ClC‐3 (M531 in ClC‐3a) and ClC‐5 have methionine at the analogous position. We made a ClC‐5/3 M531A mutant, predicting that reversion to the bacterial residue would increase the efficiency of water‐wire proton transfer. Consistent with this prediction, M531A significantly increased I SS while decreasing gating Q max in external Cl− (P < 0.02 vs. wild‐type; Fig. 3 A–C), resulting in a 50% reduction in Q/I (+115 mV; P < 0.0001 vs. wild‐type; Fig. 3 C). V 1/2 of Q–V was unchanged relative to wild‐type (79 ± 1 mV; Table 1). External SCN− substitution altered M531A transport in a manner resembling wild‐type, increasing I SS by over 2‐fold (Fig. 3 B). In contrast, V 1/2 of Q–V was much more strongly shifted, increasing by over +25 mV (Table 1).
Figure 3. ClC‐5/3 coupled H+/Cl− exchange.
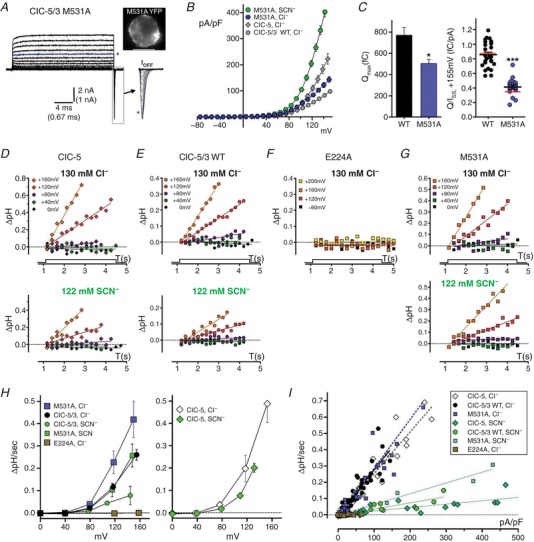
A–C, the M531A “water‐wire” mutation increases transport rate. A, steady‐state and transient gating currents in a M531A‐expressing cell (inset, top right), recorded in Cl−‐containing external saline. Currents are displayed up to maximum depolarizations of +160 mV; asterisk (blue trace) indicates current recorded at +120 mV applied potential. I Off gating transients (boxed region) are shown at expanded time scale to right. B, quantified I SS–V plots for M531A in external Cl− (n = 17) and SCN− (n = 2) compared to wild‐type ClC‐5/3 and ClC‐5 I SS in external Cl− (grey symbols). C, M531A Q max (left panel) and Q/I SS ratio (+115 mV, right panel) are significantly decreased compared to wild‐type (* P < 0.02; *** P < 0.0001, Mann–Whitney test vs. wild‐type; see also Table 1). Red lines in Q/I SS plot indicate mean ratios after correction of Q and I to a corrected voltage of +115 mV. D–G, whole‐cell pH changes recorded during depolarizing voltage pulses (0, +40, +80, +115 and +160 mV) in BCECF‐loaded cells expressing ClC‐5 (D), ClC‐5/3 wild‐type (E), ClC‐5/3 E224A (F) and ClC‐5/3 M531A (G). pH changes are plotted relative to pre‐pulse baseline (dotted line); lines are fitted to the data by linear regression. Top row: responses in 130 mM external Cl−. Bottom row: same‐cell responses in external SCN−. H, rates of pH change (ΔpH/s) plotted vs. corrected voltage for ClC‐5/3 (left) and ClC‐5 (right), in Cl−‐ and SCN−‐containing saline. ClC‐5/3 H+ transport is increased by M531A (P ≤ 0.02, Mann–Whitney test) and completely abolished by E224A. Wild‐type and M531A H+ transport is substantially reduced in external SCN−. ClC‐5 H+ transport rate is significantly greater than for ClC‐5/3 at voltages >+120 mV (P ≤ 0.03, Mann–Whitney test), and reduced in external SCN−. I, recorded rates of pH change (ΔpH/s) are plotted against corresponding I SS density (pA/pF) for the experimental groups illustrated in D–G. Lines are fitted to each data group by linear regression. In external Cl−, wild‐type ClC‐5/3 and ClC‐5 exhibit the same slope (P > 0.30), and M531A slope is unchanged from wild‐type (see Table 2). ClC‐5/3 and ClC‐5 display significantly different slopes in SCN− (P < 0.002). Slopes in SCN− indicate anion:H+ coupling ratios of ∼15:1 for ClC‐5/3 and ∼18:1 for ClC‐5. [Color figure can be viewed at http://wileyonlinelibrary.com]
Proton transport rate is increased by M531A and decreased in external SCN−
Fluorescence‐based measurements of intracellular pH changes have demonstrated Cl−/H+ exchange in mammalian cells expressing ClC‐4 and ClC‐5 (Alekov & Fahlke, 2009; Grieschat & Alekov, 2012), and provided the first demonstration that ClC‐3 is likewise a Cl−/H+ exchanger (Guzman et al. 2013). We combined whole‐cell current recordings and BCECF ratiometric fluorescence measurements to assess relative Cl−/H+ exchange capacity in ClC‐5/3‐ and ClC‐5‐expressing cells. In BCECF‐loaded cells, intracellular alkalization resulting from proton export was readily evident during depolarizing voltage pulses (+80 to +160 mV) in Cl−‐containing external saline (Fig. 3 D–G, top panels). Alkalization rate (ΔpH/s) increased with depolarization, with ClC‐5 predictably displaying greater relative ΔpH/s (Fig. 3 H). In external SCN−, ΔpH/s for ClC‐5/3 was significantly decreased (lower panels, Fig. 3 D–G), while for ClC‐5 the decrease was less pronounced (Fig. 3 H and Table 2). ΔpH/s was linearly correlated with I SS density (Fig. 3 I). ClC‐5/3 and ClC‐5 exhibited indistinguishable normalized slopes (ΔpH/s vs. pA/pF) in external Cl− (P > 0.30; Fig. 3 I and Table 2), confirming that ClC‐3 possesses a 2Cl−:1H+ coupling stoichiometry, as previously established for ClC‐5 (Zifarelli & Pusch, 2009). In external SCN−, slopes were greatly reduced, suggesting SCN−/H+ coupling ratios of ∼15:1 (ClC‐5/3) and ∼18:1 (ClC‐5) (Table 2). No pH increases were detected during depolarizing steps (+115 to +195 mV) for the ClC‐5/3 E224A mutant (Fig. 3 F, H and I and Table 1), confirming that E224A is functionally non‐coupled.
An increased efficiency of proton transport in the M531A mutant was confirmed by increased alkalization rates relative to ClC‐5/3 wild‐type (P < 0.02; Fig. 3 H and Table 2). M531A normalized slope (ΔpH/s vs. pA/pF) was unaltered, indicating an unaltered 2:1 coupling stoichiometry (Fig. 3 I). As observed for wild‐type, M531A alkalization rate was significantly reduced in external SCN− (Fig. 3 H and Table 2), with a decreased slope indicating an anion:H+ coupling ratio of ∼9:1 (Fig. 3 I and Table 2).
ClC‐3‐mediated ion transport is distinguishable from an acid‐activated endogenous current
The extracellular face of PM‐localized ClC proteins is normally exposed to an acidic environment within the endosomal vesicle pathway. A key aim of this work was to assess the influence of external protons and internal proton supply on ClC‐5/3 activation and transport. This analysis is complicated by an endogenous anion conductance(s) activated by external protons, which is present in many cell types. Previously, we described an acid‐activated outwardly rectifying, phloretin‐sensitive Cl− current with slow kinetics (I Cl,acid) in HEK and vascular smooth muscle cells (Matsuda et al. 2010). In cells overexpressing wild‐type human ClC‐3 in an adenovirus vector (Ad‐ClC‐3), slowly activating phloretin‐sensitive currents were observed at pH 7.35, and I Cl,acid was increased (Matsuda et al. 2008, 2010). These and other supporting data led us to conclude that I Cl,acid represents an uncoupled ClC‐3 current (Matsuda et al. 2010). This interpretation is incompatible with the rapid activation kinetics and gating transients demonstrated by PM‐targeted ClC‐3.
To resolve this issue, we implemented high‐resolution leak‐subtracted recording methods to record I Cl,acid. Non‐transfected cells in control pH 7.35 and pH 6.0 external saline exhibited insignificant endogenous basal I SS, and no detectable transient charge movements (Fig. 4 Aa). In pH 5.0 saline, depolarizing voltages activated an outwardly rectifying current (74 ± 7 pA/pF, +115 mV; n = 22; Fig. 4 Ab), with no evidence of gating transients (insets, Fig. 4 A). This acid‐activated current displayed much slower activation and deactivation kinetics compared to ClC‐5/3‐mediated currents, and was progressively inhibited by increasing concentrations of external phloretin (60–250 μM) at voltages ≤+115 mV (Fig. 4 Ac). At 150–250 μM, phloretin blocked ∼90% of I Cl,acid at +115 mV, with a voltage‐dependent relief of blockade evident with stronger depolarization (Fig. 4 B). In contrast, ClC‐5/3 wild‐type (Fig. 4 B) and E224A mutant transport (Fig. 2 G) were insensitive to 100–150 μM phloretin, in agreement with previous studies of heterologously expressed ClC‐3 currents (Okada et al. 2014). I Cl,acid moreover displayed less‐pronounced outward rectification, with current appearing at +10 to +20 mV, in contrast to ClC‐5/3 currents which require depolarizations of +50 or greater for significant activation (Fig. 4 B and D). We conclude that ClC‐3‐ and ClC‐5/3‐mediated currents have no identity with endogenous I Cl,acid, nor with currents we previously attributed to Ad‐ClC‐3. We speculate that adenovirus‐based ClC‐3 overexpression results in altered expression and/or trafficking of unidentified protein(s) responsible for I Cl,acid.
Figure 4. External protons inhibit ClC‐5/3 transport and decrease gating charge.
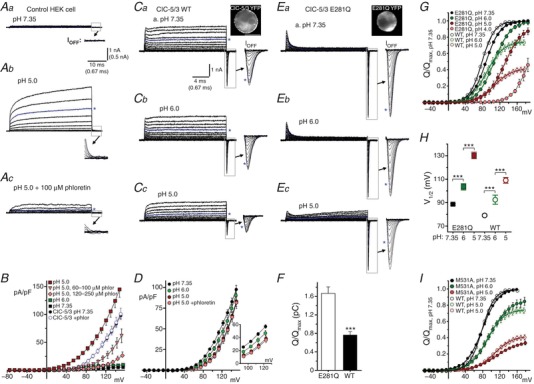
A and B, endogenous acid‐activated anion currents are functionally distinct from ClC‐3‐mediated transport currents. A, leak‐subtracted currents (50 ms test pulses from −40 to +160 mV, in 20 mV increments) recorded from an untransfected HEK cell. Asterisks indicate traces recorded at +120 mV applied potential. Aa, current at pH 7.35 is insignificant. At pH 5.0 (Ab), a slowly‐activating I Cl,acid appears; this current is partially blocked by 100 μM phloretin (Ac). No transient current components are detectable (insets, showing boxed regions at magnified scale) in control or acid‐activated currents. B, I SS–V relationships for endogenous pH‐dependent current (pH 7.35, 6.0, 5.0) in untransfected cells. I Cl,acid is strongly activated at pH 5.0. Increasing concentrations of phloretin (>120 μM) block 80–90% of current at +115 mV, with progressive relief of block evident with greater depolarization. By comparison, currents in ClC‐5/3‐expressing cells (circles, pH 7.35) are insensitive to phloretin. C, ClC‐5/3 wild‐type I SS and transient gating currents in pH 7.35 (a), 6.0 (b) and 5.0 (c) external saline (−60 to +160 mV test pulse range). Image (Ca, top right) shows YFP‐fusion protein expression. Transport is progressively inhibited by increasing external proton concentrations (Cb and c). In parallel, gating charge magnitude is decreased (insets show transients at magnified scale). Asterisks indicate traces recorded at +120 mV. D, I SS–V relationship for ClC‐5/3 wild‐type currents, showing pH‐dependent inhibition in the range of +60 to +160 mV (inset). Phloretin (150–250 μM) effectively suppressed endogenous I Cl,acid at voltages below +140 mV. The M531A mutant displayed similar pH‐dependent inhibition (not shown; see text). E, the E281Q mutation essentially abolished transport, leaving large gating charge movements; image (a, top right) shows protein localization. With increased proton concentration (Eb and c), the voltage dependence of gating is shifted, but maximal Q undergoes little decrement. A small residual I Cl,acid is evident in this recording at pH 5.0 (Ec). F, overall Q max (pC) for ClC‐5/3 wild‐type (WT) and E281Q‐expressing cells. In the absence of transport, E281Q Q max is increased by over 2‐fold (*** P < 0.0001 vs. wild‐type; Mann–Whitney test; n ≥ 25). G, pH dependence of wild‐type (open symbols) and E281Q (filled symbols) Q–V relationships (external pH 4.0–7.35). Values are normalized to Q max at pH 7.35; continuous lines show Boltzmann functions fitted to the data. H, pH dependence of V 1/2 of Q–V for wild‐type (open symbols) and E281Q (filled symbols; *** P < 0.0005; one‐way ANOVA). I, the M531A mutant (filled symbols, n = 8) displays a pH‐dependent inhibition of charge movement highly similar to that of wild‐type (open symbols). [Color figure can be viewed at http://wileyonlinelibrary.com]
External protons neutralize Gluext and inhibit ClC‐5/3 transport
Gluext (ClC‐3 E224) is proposed to act as an external gate for both the Cl− and H+ pathways (Miller & Nguitragool, 2009; Basilio et al. 2014; Khantwal et al. 2016). In a hyperpolarized voltage field, unprotonated and negatively charged Gluext occupies the external anion binding site, blocking the ion pathway (“closed” conformation). A depolarizing field drives inward rotation of Gluext (associated with Q On charge movement) into close proximity with the central anion tyrosine gate (ClC‐3 Y572) (Feng et al. 2010; Grieschat & Alekov, 2012; Basilio et al. 2014). Upon receiving an internally supplied H+, Gluext rotates outwardly, concurrently opening the pore to external Cl− while transferring a proton to the external milieu (Feng et al. 2012; Basilio et al. 2014; Khantwal et al. 2016). A requirement of this model is that Gluext is accessible to external protons. External proton binding was proposed (Picollo et al. 2010) to account for ClC‐5 transport inhibition observed at lowered external pH (Picollo & Pusch, 2005; Picollo et al. 2010; De Stefano et al. 2013). However, the two‐electrode oocyte recordings used in these studies were inadequate to resolve gating currents or a predicted pH‐dependent regulation of charge movement.
To test the prediction that external protons bind Gluext to inhibit charge movement in a pH‐ and voltage‐dependent manner, we assessed ClC‐5/3 I SS and Q Off at external pH of 7.35, 6.0 and 5.0. We asked whether external protons in this pH range inhibit overall transport, and/or are able to “hold open” Gluext in an external position and facilitate anion transport. In parallel, we recorded from the E281Q mutant lacking the internal “proton glutamate”, to assess Gluext gating behaviour in the absence of internally supplied protons (Fig. 5). Phloretin (150–250 μM) was included to minimize I Cl,acid activation. Wild‐type ClC‐5/3 I SS and gating currents were progressively decreased with increasing external proton concentration (Fig. 4 C). At pH 5.0, I SS was inhibited by 45% (26 ± 3 pA/pF, +115 mV; P < 0.0005 vs. control; Fig. 4 Cc and Table 1), while Q max decreased by ∼25% at pH 6.0 and by ∼60% at pH 5.0 (Fig. 4 G and Table 1). In parallel, the V 1/2 of Q–V shifted by +30 mV (Fig. 4 H and Table 1). The M531A mutant, which exhibits higher‐efficiency transport (Fig. 3), likewise demonstrated a similar pH‐dependent inhibition of steady‐state transport and charge mobilization (Fig. 4 I).
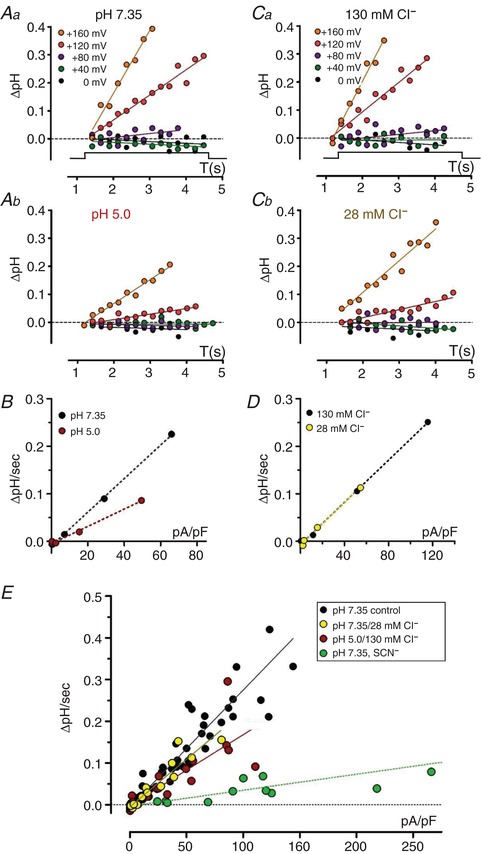
Figure 5. Increased external proton concentration reduces H+ exchange coupling.
A, pH changes recorded during depolarizing pulses (0, +40, +80, +120, +160 mV) in BCECF‐loaded ClC‐5/3 wild‐type‐expressing cell in normal 130 mM Cl− saline. Aa, pH 7.35. Ab, pH 5.0. Lines are fitted to the data by linear regression. Alkalization rates (ΔpH/s) are decreased in pH 5.0. B, plots of ΔpH/s vs. I SS (pA/pF) for the cell recording shown in A; dotted lines were fitted by linear regression. The slope in pH 5.0 is significantly decreased (*** P < 0.001 vs. pH 7.35), indicating an increased Cl−/H+ exchange ratio. C, pH changes recorded in a ClC‐5/3 wild‐type‐expressing cell in external pH 7.35 saline containing 130 mM Cl− (Ca) and 28 mM Cl− (Cb). Alkalization rates (ΔpH/s) are reduced in 28 mM Cl−. D, rates (ΔpH/s) plotted vs. I SS (pA/pF) for the cell recording shown in C. Fitted slopes (dotted lines) for the two conditions are identical, indicating an unchanged coupling ratio. E, ΔpH/s plotted vs. I SS density (pA/pF) for altered external pH and Cl− conditions. Data are fitted by linear regression. In 28 mM Cl−, slope is insignificantly altered compared to normal saline (P > 0.13 vs. 130 mM Cl−; see also Table 2). In contrast, in pH 5.0 saline, slope is reduced to 60% of pH 7.35 control (*** P < 0.0001 vs. pH 7.35). Overall slope in external pH 5.0 corresponds to a Cl−/H+ coupling ratio of 3.3:1 (see also Table 2). [Color figure can be viewed at http://wileyonlinelibrary.com]
The E281Q mutant showed insignificant I SS at pH 7.35 (Fig. 4 E and Table 1), but large Q On and Q Off movements. E281Q Q max was over 2‐fold greater than for wild‐type (Fig. 4 F and Table 1), presumably reflecting the full population of gating charge mobilized in the absence of internally supplied protons. In contrast to wild‐type, E281Q Q max was relatively insensitive to pH, decreasing only to 87% of control at pH 5.0 (Fig. 4 G). E281Q V 1/2 of Q–V was significantly more positive at pH 7.35 (88 ± 1 mV; P < 0.0001 vs. wild‐type), and V 1/2 was strongly pH dependent, shifting by over +40 mV at pH 5.0 (131 ± 3 mV; Fig. 4 H and Table 1). An analogous ClC‐5 (E268A) Gluin mutant was previously found to demonstrate a pH‐insensitive Q max, and a pH‐dependent positive shift in V 1/2 of Q–V (Zifarelli et al. 2012). These results support a direct interaction of external protons with E224 (Picollo et al. 2010) that is able to neutralize and reduce gating Q movements. Moreover, they show that external protons and voltage modulate gating Q differently in cycling (wild‐type) vs. non‐transporting (E281Q) conditions. For E281Q, external protons significantly depress Q at submaximal potentials (e.g. +115 mV; Fig. 4 E and G), consistent with proton binding to E224. However, with increasing depolarization, ∼90% of the maximal charge available at pH 7.35 can be recruited at pH 5.0, indicating that the E224–proton interaction is voltage dependent. The relative value of wild‐type Q max indicates that ∼50% of Gluext is charged (unprotonated and incompletely cycled) during steady‐state transport, and thus recorded upon repolarization as Q Off. External protons diminish the proportion of Q that can be mobilized by voltage, potentially by slowing E224 deprotonation (Fig. 4 G).
We were unable to detect any gating or ion transport behaviour consistent with a reversed polarity of transport (Cl− outward, H+ inward) under physiological recording conditions. Hyperpolarizing steps from V H (−40 mV or 0 mV) to voltages as negative as −160 mV elicited no detectable Q On or Q Off movements for either ClC‐5/3 wild‐type or E281Q mutant, and no net inward wild‐type I SS. The rectifying transport demonstrated for ClC‐5 and ClC‐3 contrasts with recent molecular models of ClC cycling that assume reversibility of Cl−/H+ transport (Feng et al. 2012; Basilio et al. 2014; Khantwal et al. 2016). Although physiological roles have been proposed that require a reversed polarity of exchange, or charge movement at negative voltages (Guzman et al. 2013, 2014), it remains unclear how such a process could be activated by voltage.
Increased external proton concentration reduces the efficiency of Cl−/H+ coupling
The above results raise additional questions about the nature of pH‐dependent inhibition of transport current (I SS). The reduced I SS at pH 5.0 may reflect a slowed rate of 2:1 Cl−/H+ coupled exchange (“turnover”). Alternatively, external protonation of E224 may increase the likelihood of E224 adopting an outward, “open” configuration, permitting Cl− “slippage” current and increasing exchange ratio. Since wild‐type transport behaviour at external pH 5.0 bears little resemblance to that of the uncoupled “gateless” E224A mutant at normal pH (Fig. 2 E), we recorded cellular pH changes to assess the impact of external protons on coupling. To minimize any contribution by endogenous I Cl,acid, phloretin (250–350 μM) was included in the external saline, and I SS was quantified 5 ms into each 20 ms test pulse. As observed above, pH 5.0 transport was inhibited by over 50% (22 ± 3 pA/pF; n = 7), and corresponding alkalization rates (ΔpH/s) at pH 5.0 were strongly reduced (Fig. 5 A and Table 2). Moreover, the normalized slope (ΔpH/s vs. I SS) at pH 5.0 was decreased compared to pH 7.35 in same‐cell recordings (n = 4; Fig. 5 B). Overall, the slope at pH 5.0 was decreased by 40% (P < 0.0001 vs. pH 7.35; Fig. 5 E), corresponding to a Cl−/H+ exchange ratio of ∼3.3 (Table 2). These results suggest that external protons impair H+ coupling in addition to slowing turnover rate, presumably through interaction with E224.
The question arises as to whether measurements of pH‐dependent fluorescence changes (ΔpH/s) are sufficiently sensitive to discriminate between conditions of partial uncoupling vs. slowed rates of normally coupled exchange. To address this question, we assessed transport and coupling at pH 7.35 in 28 mM external Cl−, which produces a comparable predicted shift in transporter equilibrium potential (Fig. 5 C and D). In 28 mM Cl−, I SS amplitude was significantly reduced (P < 0.0002 vs. 130 mM Cl− at +115 mV; Fig. 6 E and Table 1), and Q/I ratio increased by over 2‐fold (P < 0.0001 vs. control; Fig. 6 F and Table 1). Alkalization rates in 28 mM Cl− were accordingly slowed compared to 130 mM Cl− in same‐cell recordings (Fig. 5 C), but normalized slopes (ΔpH/s vs. I SS; Fig. 5 D) were unchanged (104 ± 2% of control, n = 3; P > 0.13 vs. control) (Fig. 5 E and Table 2), indicating that coupling stoichiometry was maintained.
Mutations removing the internal tyrosine “anion gate” increase Cl− transport
The ClC‐5/3 Y572 tyrosine represents one of two hydroxyl‐bearing side chains (Y572, S181, homologous to ClC‐ec1 Y445, S107) proposed to act as the internal anion selectivity filter, with the internal tyrosine considered to be the “Cl− gate” (Miller & Nguitragool, 2009). Located between Gluin and Gluext, Y445/572 has been proposed to interact with the central Cl− binding site and coordinate H+/Cl− exchange (Accardi et al. 2006; Walden et al. 2007; Miller & Nguitragool, 2009). Mutations to ClC‐ec1 Y445 reduce H+ coupling in bilayer and liposome studies (Accardi et al. 2006; Walden et al. 2007).
We assessed transport and gating in two tyrosine mutants (Y572S and Y572V) that eliminate the large phenol group. Y572S and Y572V I SS amplitudes in 130 mM external Cl− were greatly increased over wild‐type I SS level, with an over 6‐fold increase in average current amplitude at +115 mV (Fig. 6 A and Table 1). In contrast to wild‐type, I SS amplitudes in external SCN− decreased significantly compared to Cl− (Fig. 6 A and B, and Table 1). Both mutations resulted in greatly reduced magnitude of gating transients in external Cl− (Fig. 6 A and F, and Table 1). Y572S Q max increased significantly in external SCN−, potentially because a decrease in I SS amplitude improved Q resolution (Fig. 6 A and Table 1). To better resolve transient current gating components, we slowed Y572S transport rate by reducing external Cl−, replacing Cl− with the non‐transported anion methanesulfonate (Smith & Lippiat, 2010b ). Y572S I SS in 13 mM Cl− was reduced by ∼75%, to a level comparable to wild‐type I SS in control saline (Fig. 6 B and E, and Table 1). In parallel, Q max and the Q/I ratio were increased by severalfold (P ≤ 0.03 vs. 130 mM Cl−; Fig. 6 F and Table 1), consistent with a reduced probability of Gluext protonation. However, Y572S Q movements in 13 mM Cl− remained significantly smaller and briefer than wild‐type (Fig. 6 B, C and F). These results show that Y572 regulates anion transport, as well as the relative selectivity between Cl− and the “slippery” SCN− anion.
We next investigated the potential interaction between the internal and external gates using an E224A/Y572S double mutant lacking both gates. In Cl−‐containing saline, E224A/Y572S resulted in a greatly magnified E224A‐like non‐rectifying conductance (>250 pA/pF at +115 mV; Fig. 6 G and Table 1). Outward I SS exceeded that of the Y572S single mutant for voltages up to +150 mV (Fig. 6 H). In SCN−‐containing saline, E224A/Y572S outward I SS was further increased by severalfold (Fig. 6 Gb and Table 1). I SS in SCN− was not significantly larger than for the E224A single mutant (P > 0.19; Fig. 6 H). These results indicate that E224 and Y572 individually represent significant barriers for anion movement. E224 is required for outwardly rectifying transport, while Y572 is a major barrier for anion influx in balanced Cl−, and regulates central anion “slippage”. In the absence of Gluext (E224A), SCN− is highly conductive and its transport rate is essentially independent of Y572 status.
The Y572 hydroxide regulates transport and exchange coupling
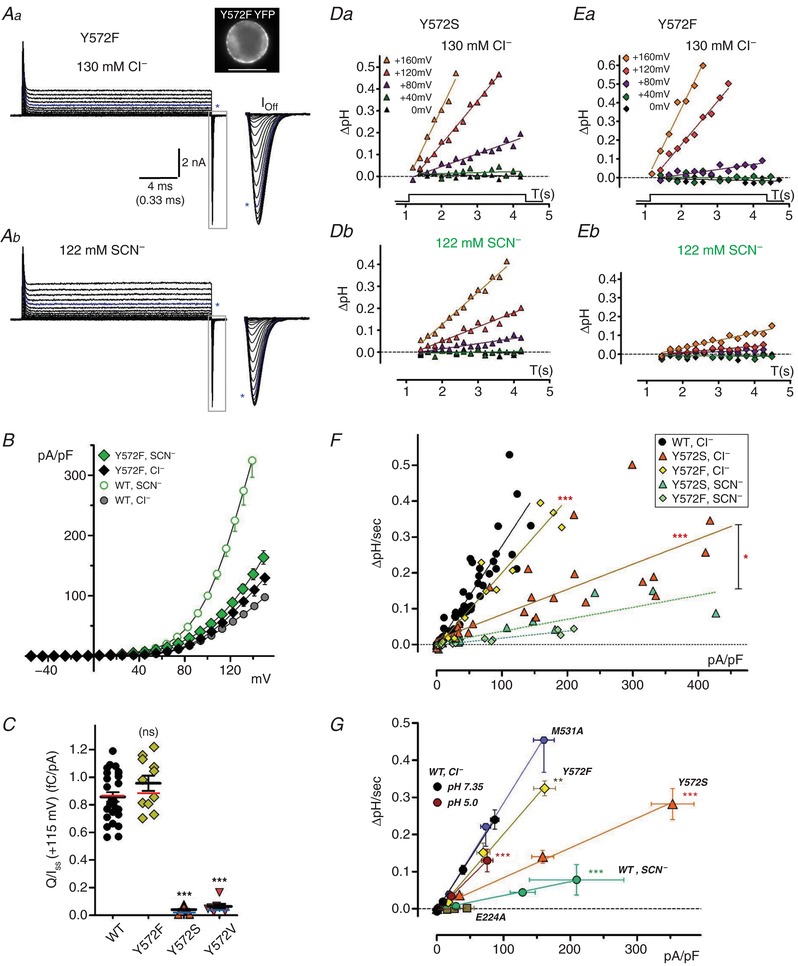
A potential hydrogen bonding interaction between the Gluext carboxylate and the central tyrosine hydroxide has been proposed to create a structurally closed “common” gate of the ClC‐1 Cl− channel (Bennetts & Parker, 2013). In bacterial ClC‐ec1, a Y445F mutation that removes only the terminal –OH group reduced Cl− and H+ transport in liposomes, but Cl−/H+ exchange remained strongly coupled (Walden et al. 2007). We investigated transport, gating and coupling for the homologous ClC‐5/3 Y572F mutant. Y572F‐expressing cells exhibited slightly larger I SS (n = 11; Fig. 7 A and B) and larger Q max (Table 1) than measured for wild‐type ClC‐5/3, but these increases are consistent with higher overall membrane expression in the selected cells (inset, Fig. 7 A). Y572F Q/I ratio was not significantly altered (Fig. 7 C; P > 0.05 vs. wild‐type), consistent with largely normal functional transport. However, Y572F altered behaviour in two notable respects. First, gating transients displayed larger amplitudes but briefer durations than observed for wild‐type (Fig. 7 A), with a V 1/2 of Q–V that was shifted by +12 mV (P < 0.0005 vs. wild‐type; Table 1). Secondly, in external SCN− Y572F I SS was increased by only ∼25% (Fig. 7 A and B, Table 1), in contrast with a 3‐fold increase displayed by wild‐type. Removal of only the tyrosine terminal –OH therefore impacts voltage‐dependent gating charge movement, as well as relative Cl− and SCN− transport rates. The kinetics of gating charge movement is examined in further detail below.
Figure 7. Y572F mutation alters anion transport and H+ coupling.
A, Y572F transport and gating currents in Cl−‐ (a) and SCN−‐containing external saline (b). Image at top right shows Y572F‐YFP expression in the recorded cell. Steady‐state (I SS) currents are displayed to a voltage of +160 mV; Off‐gating transients (boxed regions, expanded at right) are displayed to a voltage of +180 mV. Asterisks indicate traces at +115 mV. Y572F results in large, rapid gating currents. B, quantified Y572F I SS–V relationships (n = 10). Y572F I SS is slightly larger than wild‐type (circles) in Cl−, but Y572 current undergoes little increase in SCN− compared to wild‐type. C, Q/I SS ratios (fC/pA, +115 mV) compared for wild‐type and Y572 mutants (Y572F, Y572S, Y572V). Coloured lines overlaid on plots indicate mean ratios after correction of Q and I values to a corrected voltage of +115 mV. Q/I SS is unaltered in Y572F, but greatly decreased in Y572S and Y572V (*** P < 0.0001). D and E, pH changes recorded during depolarizing pulses in BCECF‐loaded Y572S‐ (D) and Y572F‐expressing cells (E). Y572S exhibits robust H+ transport rates (ΔpH/s) in external Cl− (Da) and slowed rates in external SCN− (Db; same‐cell recording). Y572F (E) likewise exhibits robust H+ transport in Cl− (Ea), and markedly slower transport in external SCN− (Eb, same‐cell recording). Continuous lines are fitted to the data by linear regression. F, rates of pH change (ΔpH/s) plotted vs. corresponding current densities (pA/pF) for Y572S and Y572F, compared to wild‐type (filled circles). Data for each genotype are fitted by linear regression (lines). Y572S slope in Cl− is reduced to ∼25% of wild‐type (*** P < 0.0001, n = 8; see also Table 2), and further reduced in external SCN− (** P = 0.011 vs. Cl−, n = 3). Y572F slope in Cl− is significantly reduced compared to WT (*** P < 0.0001; n = 4), indicating an increased exchange ratio of ∼2.7:1, while H+ transport is nearly eliminated in SCN−. G, relationships between rate of pH change (DpH/sec) and mean current density (pA/pF) are summarized for ClC‐5/3 wild type (WT; pH 7.35 and pH 5.0 in external Cl−, and pH 7.35 in external SCN− ) and mutant genotypes (external Cl−, pH 7.35). Symbols show mean values (± S.E.M.) recorded at each applied potential level (0, +40, +80, +120 and +160 mV). Solid lines are fit to each data group by linear regression. Asterisks denote significantly reduced slope (** P<.001; *** P<0.0001) compared to WT in control saline (pH 7.35, external Cl−). [Color figure can be viewed at http://wileyonlinelibrary.com]
A role for Y572 in regulating Cl−/H+ exchange coupling was confirmed by cellular pH recordings. In external Cl−, Y572S transport drove intracellular alkalization at rates (ΔpH/s) comparable to, or exceeding, rates recorded from wild‐type at corresponding voltages (Fig. 7 D–G and Table 2). However, the normalized slope of the ΔpH/s vs. I SS relationship decreased to ∼25% of wild‐type (P < 0.0001; Fig. 7 F–G), indicating a Cl−/H+ exchange ratio of approximately 8:1 (Table 2). In SCN−, Y572S ΔpH/s rates (Fig. 7 Db) and corresponding slope (Fig. 7 F and G) decreased by an additional ∼50% (P < 0.02 vs. Cl−). The degree of uncoupling observed for Y572S in SCN− does not differ significantly from wild‐type (Table 2). Y572F likewise demonstrated robust proton transport in external Cl−, consistent with the transport ability exhibited by ClC‐ec1 Y445F (Walden et al. 2007), with alkalization rates (ΔpH/s) equal to, or exceeding, wild‐type at corresponding voltages (Fig. 7 G, Table 2). Nevertheless, Y572F normalized slope (ΔpH/s vs. I SS) was significantly decreased by ∼25% compared to wild‐type (P < 0.0001; Fig. 7 F and G), corresponding to a Cl−/H+ exchange ratio of ∼2.7 (Table 2). In external SCN−, pH increases were nearly undetectable, indicating an ∼90% loss of coupling (Fig. 7 Eb and F). These results suggest that the terminal tyrosine hydroxide additionally regulates anion “slippage”. Figure 7 G summarizes ion transport and coupling behaviour for wild‐type ClC‐5/3 under the ionic and pH conditions analysed above, and for the mutant genotypes E224A, M531A, Y572S and Y572F.
The Y572 hydroxide regulates gating charge movements
We hypothesized that a hydrogen‐bonding interaction between the Y572 –OH and the E224 carboxylate may establish a tight, “inward‐closed” inner gate conformation necessary to prevent inward Cl− “slippage”. Such a charge interaction might additionally impact gating charge movement in the voltage field. To test these possibilities, we isolated wild‐type and Y572 mutant Q movements in the E281Q mutant background to eliminate any disruption of this interaction by protons. The kinetics of Q Off movement following depolarizing test pulses were first assessed by measuring transient duration at 50% of peak amplitude (half‐width). As described above, the E281Q mutant (wild‐type Y572) displays prominent On‐ and Off‐gating transients (Fig. 8 Aa). At strongly depolarized voltages, E281Q I SS was increased slightly above that of non‐transfected cells (P < 0.03, +160 mV; Fig. 8 B). Off‐transient durations were 0.2–0.3 ms, lengthening slightly with increasing depolarization (Fig. 8 C). As described earlier, the absence of E224 in the E281Q condition (E224A/E281Q) permits substantial bidirectional current (Fig. 8 B). Thus, while Y572 acts as an anion barrier, its presence alone is insufficient for inner gate closure.
Y572S/E281Q unexpectedly displayed a sharply outwardly rectifying I SS at strongly depolarized voltages (Fig. 8 Ab) (11.4 pA/pF at +115 mV; P < 0.0003 vs. E281Q; Fig. 8 Ab and B). This result demonstrates that Y572 is required to effectively block uncoupled Cl− influx in the inward/closed gate conformation. Y572S/E281Q Off‐transient duration was shortened to < 50% that of E281Q (Fig. 8 C), confirming our preliminary observations above. In contrast, Y572F/E281Q I SS was not significantly increased over that of E281Q (Fig. 8 Ac and B, and Table 1), indicating that the tyrosine –OH is not essential for effective gate closure. However, the Y572F Off‐transient duration was similarly shortened by ∼50% (Fig. 8 C). Additionally, Y572S and Y572F mutations resulted in visibly briefer On‐transient charge movements (Fig. 8 A). Y572S Q max was decreased by ∼40% compared to E281Q wild‐type (P < 0.01, Table 1), indicating that the tyrosine impacts the overall magnitude of charge movement. Relative to E281Q, Y572S V 1/2 of Q–V exhibited a pronounced positive shift (>+25 mV), while Y572F V 1/2 exhibited a milder +15 mV shift (Table 1). Together, these results suggest that side‐chain bulk and charge‐dependent interactions contribute differently to inner gate closure and charge movement kinetics. To test this idea, we replaced tyrosine with histidine (Y572H), which contributes similar bulk and a potential charge‐based interaction between the imidazole –NH and E224 carboxyl. Y572H/E281Q displayed a prolonged transient outward current at depolarized voltages (Fig. 8 Ad) that appeared to be a combination of gating and ionic currents. This current declined within 10 ms to a level indistinguishable from that of E281Q (Fig. 8 B and Table 1), consistent with a delayed inner gate closure. Y572H Off‐gating transients had intermediate durations that lengthened with increasing depolarization, approaching wild‐type values at +160 to +180 mV (Fig. 8 C).
To examine the voltage dependence of charge movement, we first applied a +180 mV maximally activating depolarization, followed by repolarizing tail pulses to increasingly negative voltages (+20 mV to −180 mV; Fig. 8 Da). E281Q transient duration was prolonged (350–400 μs) at 0 mV, and exhibited pronounced voltage‐dependent shortening with increasingly negative tail pulse voltage (Fig. 8 Da and E). In contrast, the Y572F (Fig. 8 Dc) and Y572S mutations (Fig. 8 Db) resulted in progressively briefer transient durations (Fig. 8 E). Voltage dependence was reduced in Y572F, and virtually absent in Y572S (Fig. 8 E). Y572H mutant transient duration was comparable to wild‐type at 0 mV and strongly voltage dependent, shortening to resemble Y572F with increasing tail pulse repolarization (Fig. 8 Dd and E). For all genotypes, total Q was nearly independent of negative tail pulse voltage (ranging from 0 mV to −160 mV; Fig. 8 F), indicating that the inwardly mobilized charge is completely recovered upon repolarization at a voltage‐dependent rate.
Within each genotype, transients exhibited a 2‐ to 3‐fold range in peak amplitudes between individual recordings, but we found no significant correlation between transient duration and amplitude at a given voltage level (data not shown). Since transient peak amplitudes at strongly hyperpolarized tail pulses exceeded 10–15 nA in some cases, we carried out additional experiments to rule out the possibility that differences in charge movement kinetics are influenced by I Off size or associated voltage errors. Recordings were made from cells expressing lower levels of E281Q or Y572/E281Q protein, and a +115 mV activation pulse was applied, thereby reducing maximal I Off to several nanoamps. R S compensation was additionally increased (75–80% correction), resulting in faster gating transient kinetics in both groups. Under these conditions, E281Q Off‐transient duration (Fig. 8 H) and decay time (from 90 to 10% peak amplitude; Fig. 8 I) were prolonged at 0 mV, and exhibited marked voltage‐dependent shortening. Y572S/E281Q transient duration and decay time were markedly briefer and completely insensitive to voltage, as observed previously. These results indicate that the central tyrosine side chain interacts with Gluext to regulate central gate closure, as well as the rate of Gluext movement in the voltage field.
Discussion
ClC‐5/3 displays transport and gating features highly consistent with those of ClC‐3a13–19A mutants (Zhao et al. 2007; Stauber & Jentsch, 2010; Guzman et al. 2013). The large gating charge (Q) facilitates analysis of the relationships between gating, I SS and Cl−/H+ exchange coupling. Several conclusions can be drawn from these data: (1) ClC‐3 transport is functionally unidirectional and activated only at depolarized potentials under physiological recording conditions; (2) with key limitations, the Q/I ratio reflects the efficiency of cytoplasmic proton transfer Gluext; (3) SCN− uncouples ClC‐3 H+/Cl− exchange and slows proton transport; (4) Gluext can be protonated from the exterior, which reduces transport rate and impairs Cl−/H+ coupling; (5) the inner tyrosine gate (Y572) regulates anion movement and coupling, but is not essential for proton transport; (6) Y572 interacts with E224 to form an effectively “closed” inner gate. We incorporate these findings into a functional model of transport (Fig. 9).
Figure 9. Proposed model for ClC‐3 physiological transport.
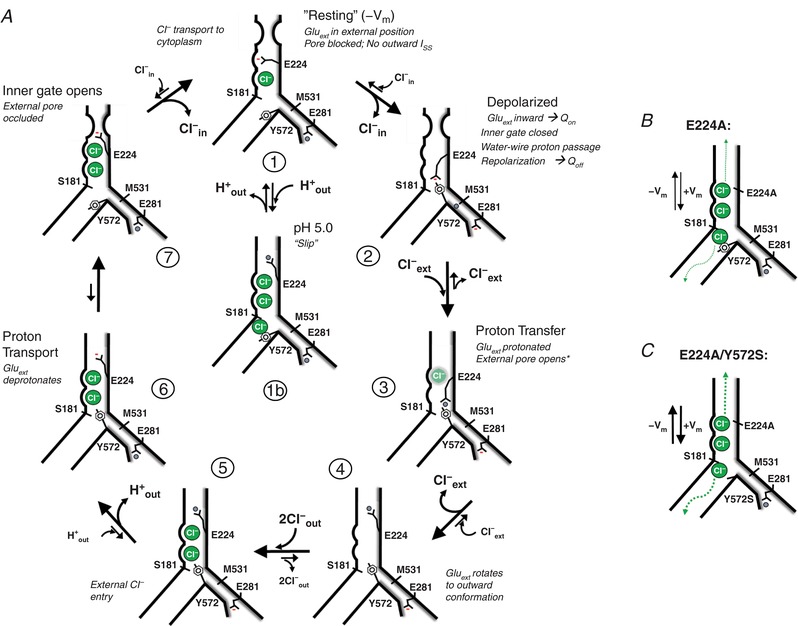
A, the depicted cycle incorporates concepts from recently published models (Bennetts & Parker, 2013; Basilio et al. 2014; Khantwal et al. 2016). Our results demonstrate that voltage‐dependent transport is essentially unidirectional (Cl− influx, H+ efflux) on a millisecond–second time scale. In a negative voltage field (State 1; −V m), E224 (Gluext) is charged and externally rotated, and occupies the Cl− ext site). State 1 is equivalent to the “outer‐facing closed” state described in Khantwal et al. (2016), in which the pore is effectively blocked by E224. From a negative “resting” V m, hyperpolarization does not elicit detectable gating charge movement or ion current. Depolarization drives inward E224 rotation (Q On) into a central position (State 2). This charge movement is prominent in ClC‐3. A large Q Off/I SS ratio is consistent with a relatively low efficiency of proton transport along the internal water‐wire to Gluext (Han et al. 2014). The data support an interaction between E224 and Y572 to create a tightly closed inner gate conformation (Bennetts & Parker, 2013). In the absence of internal proton supply (e.g. E281Q), this gate is tightly closed to Cl− flux, but is leaky in the absence of Y572 (Y572S). In State 3, an internal proton is transferred to E224, disrupting the E224–Y572 association. Protonated E224 rotates outwardly to its preferred outward conformation (State 4), allowing entry of two extracellular Cl− anions into the pore (State 5). E224 deprotonation (State 6; corresponding to State 1 of Khantwal et al. (2016) may trigger a conformational occlusion of the outer pore, and the opening of the internal anion gate (Bennetts & Parker, 2013; Basilio et al. 2014; Khantwal et al. 2016) (State 7), allowing sequential influx of two Cl− anions (States 1–2). With increasing concentration of external protons, the protonation of E224 in State 1 is proposed to cause E224 to readopt its outward conformation (State 1b), allowing uncoupled Cl− entry in the presence of the open internal gate. Transition to State 1b slows the exchange cycle, resulting in reduced transport and less tightly coupled exchange. Uncoupled Cl− transport in this state may be limited by innately poor anion conductance, similar to E224A. In the absence of Y572 (Y572S), anion “slippage” may occur in E224 inward‐ (States 2–3) and outward‐facing states (States 4–7) where the entry of extracellular Cl− is permitted. B, the absence of Gluext (E224A) abolishes gating charge movement and proton transport, and allows bidirectional anion transport, irrespective of the presence/absence of E281. The limited transport rate of E224A indicates that Y572, or a conformational barrier (Khantwal et al. 2016), limits Cl− conductance. C, the “gateless” condition (E224A/Y572S) creates a large, channel‐like anion conductance. [Color figure can be viewed at http://wileyonlinelibrary.com]
Implications of ClC‐3 transport rectification
ClC‐3, ‐4 and ‐5 mammalian transporters demonstrate pronounced outward rectification (Li et al. 2000, 2002; Picollo & Pusch, 2005; Zdebik et al. 2008; Alekov & Fahlke, 2009; Picollo et al. 2010; Smith & Lippiat, 2010a ; Grieschat & Alekov, 2012; De Stefano et al. 2013; Guzman et al. 2013, 2015; Okada et al. 2014; Alekov, 2015). Following subtraction of linear “leak” and capacitance, gating charge movements and ion transport are essentially completely rectifying, and activated only at depolarized potentials (Smith & Lippiat, 2010a ; Grieschat & Alekov, 2012; De Stefano et al. 2013; Guzman et al. 2013, 2015; Alekov, 2015). We likewise observe ClC‐5/3 gating charge movements (outward On‐transient currents) and I SS only upon depolarization to positive voltages. Hyperpolarization to voltages as negative as −160 mV elicits no detectable inward transients or net inward I SS. Inward gating currents occur only upon repolarization (Q Off) from a positive voltage. These data indicate that in a negative V m field, charged E224 side chains are overwhelmingly positioned in an external conformation, likely associating with the external anion binding site (Clext) (State 1, Fig. 9 A). In this “resting” state, E224 effectively blocks the channel pore even though the inner Y572 gate is open (Basilio et al. 2014; Khantwal et al. 2016). Accordingly, removal of Y572 (Y572S) when E224 is present does not allow anion efflux, while removal of both gates (E224A/Y572S; Fig. 9 C) dramatically increases the non‐rectifying anion transport exhibited by the E224A mutant. Structural studies of the bacterial ClC exchanger indicate that Gluext preferentially adopts an outward‐facing conformation when mutated to Gln to mimic its protonated (neutral) state (Dutzler et al. 2003; Feng et al. 2010). This conformational preference accounts for the lack of physiological evidence for cytoplasmically directed proton transport, regardless of voltage or pH. Physiological transporter rectification can thus be attributed predominantly to E224.
Our model emphasizes the role of a depolarizing voltage field in activating transport by driving inward Gluext movement (Q On; State 1 to State 2; Fig. 9 A), and facilitating ion exchange with the physiologically favoured polarity (clockwise arrows, Fig. 9 A). Internal protonation of Gluin (State 1) is insensitive to Cl− binding (Picollo et al. 2012), while proton transfer to Gluext (States 2–3) is facilitated by binding of Cl− to the central site (Cl− cent) (Wang & Voth, 2009; Lee et al. 2016). ClC‐3 voltage‐dependent transport behaviour is thus consistent with E224 binding and export of a cytoplasmic proton (States 3–4), followed by extracellular (or vesicular) Cl− entry into the transporter (State 5), deprotonation of outwardly facing E224 (State 6), and internal gate opening allowing Cl− passage into the cytoplasm (States 7–1). External E224 protonation in its Clext conformation is predicted to cause it to readopt the outward‐facing open position (State 1 to State 1b). This may account for the strong positive shift in V 1/2 of Q–V at lowered pH, and significantly impact coupling in the endosomal environment. Recently, a ClC‐5 mutant (D76H) with greatly slowed gating was identified that transiently permits (several milliseconds) a reversed polarity of voltage‐dependent Cl−/H+ exchange upon repolarization from an activating depolarizing step (De Stefano et al. 2013; Alekov, 2015). The response of Gluext to voltage and protonation under native physiological conditions may therefore fundamentally underlie the rectifying nature of ClC‐3 and ClC‐5 transport (Smith & Lippiat, 2010b ; De Stefano et al. 2013; Guzman et al. 2013, and this study).
Relative Q/I SS reflects ClC transport efficiency
ClC‐3 and ClC‐5 exhibit greatly differing Q/I SS ratios. The intrinsically large ClC‐3 gating Q reflects a lower cycling efficiency, despite maintaining a 2:1 ratio of Cl−/H+ exchange. Q max of wild‐type ClC‐5/3 is ∼50% that of the non‐transporting E281Q mutant, indicating that ∼50% of the E224 population at steady‐state resides in a charged, inwardly rotated position (State 2) awaiting proton delivery. This incompletely cycled, charged population is evident upon repolarization as Q Off (State 2 to State 1, Fig. 9 A). In ClC‐ec1, addition of a large methionine side chain to a key water‐wire component (A404M) inhibited H+ transport (Han et al. 2014). We predicted that improving water‐wire‐mediated proton transfer should increase ClC‐3 transport efficiency. Accordingly, substitution of alanine for methionine at the analogous ClC‐3 residue (M531A) substantially reduced the Q/I SS ratio, and increased the H+ transport rate. This residue cannot account for the inherent transport difference between ClC‐3 and ClC‐5, which both have methionine at this position. However, this finding supports the possibility that other structural differences exist that confer a lower probability of water‐wire formation in ClC‐3.
SCN− reduces coupling and slows the rate of ClC‐3 proton transport
Extracellular SCN− and NO3 − anions are well established to increase ClC‐ec1, ClC‐4 and ClC‐5 transporter currents (Nguitragool & Miller, 2006; Zdebik et al. 2008; Alekov & Fahlke, 2009; Orhan et al. 2011; Grieschat & Alekov, 2012). These anions create a “slippage” mode of exchange that is associated with 15–20 anions transported per proton (Alekov & Fahlke, 2009; Grieschat & Alekov, 2012). We find that SCN− increases ClC‐5/3 I SS by over 3‐fold while markedly slowing cellular alkalization rate (ΔpH/Δt) compared to Cl−, with similar behaviour exhibited by M531A. These results are consistent with the reported behaviour of ClC‐4, which exhibited larger whole‐cell current but decreased ΔpH/Δt rate in external NO3 − and SCN− (Alekov & Fahlke, 2009). In contrast, a previous ClC‐5 whole‐cell study found that SCN− increased ClC‐5 currents by 10‐ to 15‐fold without changing ΔpH/Δt (Grieschat & Alekov, 2012), while an earlier oocyte recording study found ClC‐5 proton transport rate to be decreased by SCN− (Zdebik et al. 2008). Our results do not resolve these earlier findings. Compared to ClC‐5/3, we find that SCN− causes a larger (>5‐fold) increase in ClC‐5 I SS, while moderately decreasing H+ transport rate. However, interpretation of these data is limited by the voltage corrections necessitated by the large ClC‐5 I SS amplitudes, particularly in SCN−. Our results confirm that SCN− strongly uncouples ClC‐3 exchange, as previously documented for both ClC‐5 and ‐4. These related ClC exchangers nevertheless appear to exhibit differences in their behaviour in “slippage” mode.
ClC‐3 Q max was not appreciably changed in SCN−, suggesting a maintained efficacy of proton transfer to E224 (State 2 to State 3; Fig. 9 A). This suggests the slowing of a subsequent cycle step(s) where E224 is outwardly rotated (e.g. States 4–7), permitting anion “slippage”. In these states, Y572 represents a particularly poor barrier to SCN− movement; this is demonstrated by the enormous (∼20‐fold) increase in SCN− current in the E224A mutant (Table 1). The effect of SCN− on Y572S and Y572V mutant transport is more complicated. In external Cl−, Y572S Cl−/H+ coupling ratio is increased to ∼8:1, but I SS is increased sufficiently to maintain a wild‐type rate of H+ transport (Table 2). SCN− further uncouples mutant exchange (∼17:1) while slowing H+ transport by > 60% (Table 2). SCN− thus acts similarly to uncouple wild‐type and Y572S Cl−/H+ exchange, but has opposite effects on I SS amplitude. These data highlight the importance of accounting for both coupling and proton transport rate when assessing changes in I SS.
Effect of external protons on gating charge and H+ transport coupling
The impact of external protons on ClC exchange has been extensively examined. The current results rule out the previous conclusion that ClC‐3 accounts for I Cl,acid (Matsuda et al. 2008, 2010). Our findings are consistent, however, with the ability of external protons to access Gluext in its “closed” Cl− ext conformation. This is demonstrated by a pH‐dependent reduction in wild‐type gating Q, and a strong positive shift in Q voltage dependence for wild‐type as well as the non‐transporting (E281Q) mutant. I SS is reduced by lowering external pH (pH 5.0), consistent with the inhibition previously reported for ClC‐5 currents (Picollo & Pusch, 2005; Picollo et al. 2010; De Stefano et al. 2013). An unanticipated finding is that external protons additionally introduce Cl− “slippage”, increasing the Cl−/H+ exchange ratio to over 3:1. In contrast, when external Cl− concentration is reduced at pH 7.35, 2:1 coupling is maintained despite a similarly slowed cellular alkalization rate. The pH‐dependent Cl− slippage supported by our results may result from external protonation of E224 in its external conformation (State 1), or reprotonation while in an outward “open” state (States 4–7, Fig. 9 A), favouring or prolonging the “open” configuration. Because Cl− slippage is effectively prevented while the inner Y572 gate remains closed (States 5–6), we suggest that slippage is likely to occur as a result of E224 protonation in State 1, favouring transition to State 1b (Fig. 9 A, centre). In this state, both gates are open, allowing uncoupled anion influx. At pH 5.0, coupled proton exchange clearly continues, as evidenced by slowed voltage‐dependent intracellular pH increases. However, transitions into State 1b would both slow coupled transport, as well as introduce uncoupled Cl− flux. The pH dependence of the proposed State 1b is consistent with recent evidence for a structural occlusion of the outer ClC‐ec1 pore that is opened by external protonation of Gluext, allowing anion passage (Khantwal et al. 2016). External protons therefore may regulate E224 position, allowing anion influx under conditions where an open inner gate permits uncoupled anion movement.
This proposed mechanism of pH‐dependent uncoupling of wild‐type transport differs substantially from the uncoupling effect of NO3 − and SCN− anions, which greatly increase anion transport. We interpret transport at pH 5.0 to reflect both normal 2:1 cycling, in addition to a Cl− “slippage” component that contributes up to about one‐third of the overall recorded I SS (∼26 pA/pF at +115 mV), in order to account for an increase in coupling ratio to > 3:1 (Table 2). The pH‐dependent reduction in Q suggests that the protonated population of E224 is increased by ∼60% at pH 5.0. Increasingly acidic conditions that constitutively protonate and open E224 could conceivably lead to a “slippage” rate comparable to the modest Cl− transport rate observed in the E224A mutant (20–25 pA/pF per 100 mV), where Gluext is completely absent (Fig. 9 B). Implications of this possibility are discussed further below.
The Y572 inner gate regulates ClC‐3 anion selectivity and H+ coupling
We have considered Y572 function in the context of proton transport and cycling. A hydrogen bond between the tyrosine hydroxide and Gluext carboxylate has been proposed to be critical for ClC channel gating (Bennetts & Parker, 2013). The transfer of an internal proton to E224 in this “inward‐closed” conformation could therefore trigger both inner gate opening and H+ transport (Bennetts & Parker, 2013). To determine how these two gates interact to obstruct anion flow, it is necessary first to prevent the interruption of the gate‐closed state by E224 protonation (State 3, Fig. 9 A). We therefore created Y572 double mutants either lacking E224 (E224A/Y572S), or with E224 intact in an E281Q background (E281Q/Y572S and E281Q/Y572F). In the E281Q single mutant with both outer and inner gates intact, anion movement at depolarized voltages is effectively obstructed, consistent with the “inward‐closed” conformation (State 1, Fig. 9 A). Removal of either gate individually allows significant inward anion movement, revealing this state to be important for regulating “slippage”. E224 removal (E224A or E224A/E281Q; Fig. 9 B) allows bidirectional anion currents regardless of the presence of E281. Y572 removal (E281Q/Y572S) permits only a degree of anion influx, because E224 is sufficient to prevent efflux. Therefore, neither E224 nor Y572 individually is sufficient to obstruct depolarization‐driven Cl− influx. Significantly, however, removal of both gates (E224A/Y572S; Fig. 9 C) allows far larger (5‐ to 10‐fold) currents than the summed individual mutant currents. This suggests an interaction between the two gates is required to fully close the pathway. H‐bonding does not appear to be essential for this role, since the pathway remains effectively closed to current when Y572 phenolic –OH is removed (E281Q/Y572F), or when tyrosine is replaced with a side chain of similar bulk (E281Q/Y572H).
The importance of the –OH group in a functional cycling context is nevertheless demonstrated by weakened coupling and substantially altered Cl−/SCN− selectivity in the Y572F mutant. Removal of the entire side chain (Y572S) dramatically increases currents and profoundly impairs coupling, consistent with the properties of the homologous Y445 gate in ClC‐ec1 (Accardi et al. 2006; Walden et al. 2007). However, in contrast to the uncoupling effect of SCN−, Y572S supports normal, if not greater, rates of alkalization than the wild‐type exchanger, indicating that proton transport is essentially preserved. The diminished Q max and Q/I ratio for Y572S and Y572V mutants therefore do not indicate an altered proton transfer “efficiency” per se, further supporting the conclusion that meaningful evaluations of transport “efficiency” must include coupling assays, as well as comparison of relative Q and I SS.
Interaction between Y572 and E224 regulates the rate of gating charge movements
Analysis of gating transient kinetics in the E281Q mutant background confirmed that gating transient durations are shortened in Y572S and Y572F mutants. If E224 and Y572 closely interact to form a closed inner gate (Bennetts & Parker, 2013) (State 2, Fig. 9 A), the Q Off transition (State 2 to State 1; Fig. 9 A) requires the “dissociation” of this conformation. With both gates intact, the rate of charge movement (transient duration) upon repolarization is strongly voltage dependent (Fig. 8 E, H and I), suggesting that an E224–Y572 charge interaction affects the rate of E224 charge recovery to its external position. Consistent with this prediction, removal of the Y572 –OH (Y572F) reduced the voltage dependence and markedly shortened transient duration, indicating a more rapid movement of E224 in the voltage field. Removing Y572 entirely (Y572S) had minimal additional effect, suggesting that the –OH regulates this voltage‐dependent process. These findings support a physical interaction between Gluext and the inner tyrosine gate, potentially via H+‐bonding (Bennetts & Parker, 2013). We propose that this conformation is necessary to form the “inward‐closed” inner gate that characterizes E281Q behaviour. Loss of Y572 results in a “partially open” gate when E224 is inward‐facing in a depolarizing field; thus the interaction between both gates appears to be necessary for preventing uncoupled anion entry.
Our model predicts that the inward‐closed gate conformation (Bennetts & Parker, 2013) is necessarily disrupted by Gluext protonation (State 2 to State 3, Fig. 9 A), triggering Gluext outward rotation and proton transport (States 4–6). However, the protonation step does not necessarily trigger opening of the inner gate (Khantwal et al. 2016). Maintaining a closed inner gate while Gluext is “outward facing” (States 4–7) may be important for preventing additional Cl− slippage, as is observed for Y572S. E224 deprotonation (Basilio et al. 2014; Khantwal et al. 2016), or another event, may be required for the conformational reopening of the inner gate (State 7). This requirement may explain the limited uncoupling induced by external protons. However, the (re)protonation of E224 in its external position when the inner gate has reopened would promote the transition to State 1b, permitting anion slippage as discussed above.
Implications for endosome physiology
ClC‐3 plays a role in the acidification of endosomes, lysosomes and secretory vesicles (Li et al. 2002; Hara‐Chikuma et al. 2005b; Maritzen et al. 2008; Smith & Lippiat, 2010a ; Guzman et al. 2014). It remains unresolved, however, whether ClC‐3‐mediated exchange directly contributes to acidification, as proposed for ClC‐5 (Smith & Lippiat, 2010a ). ClC‐3 2Cl−/H+ exchange and gating “capacitance” have both been proposed to provide charge neutralization for V‐ATPase‐mediated proton transport (Guzman et al. 2013, 2014). However, in the endosomal environment where the ClC‐3 exterior faces the vesicle lumen, either proposal would require a fundamentally different voltage dependence, and ion exchange and/or charge movement with a polarity opposite to that observed in physiological recordings.
Assuming that the basic features of ClC‐3 plasma membrane transport are maintained in endosomes, our results suggest that ClC‐3 might contribute to endosome acidification in two successive phases. In phase 1, coupled Cl−/H+ exchange in newly formed endosomes would contribute to lowering vesicular pH and Cl− concentration. Exchange would be highly favourable, assuming a high initial [Cl−] creating a lumen‐to‐cytoplasm gradient, coupled with a highly negative lumen potential (V lumen) created by cell‐surface negative charge (Donnan effects; reviewed by Lamb et al. 2009). Modelling suggests that ClC‐3‐mediated 2Cl−/H+ exchange could reduce vesicle pH by up to 1 unit (Lamb et al. 2009; Smith & Lippiat, 2010a ), as well as rapidly reduce vesicular Cl− to the 15–20 mM range detected in early endosomes (Hara‐Chikuma et al. 2005b). Currently, it remains unclear whether Cl− is excluded during endosome formation, or is rapidly removed by transport (Hara‐Chikuma et al. 2005b).
In phase 2, an acidic lumen created by V‐ATPase‐mediated proton transport would increasingly favour Gluext protonation and an outward/open conformation, while generating a positive V lumen. These conditions may generate an “E224A‐like” non‐rectifying Cl− transport behaviour, allowing lumen‐directed Cl− flux appropriate for charge neutralization. This proposed function is consistent with an observed secondary (15–45 min), ClC‐3‐ and V‐ATPase‐dependent acidification of endosome pH, as well as a secondary increase in endosomal [Cl−] from ∼15 mM to 40–50 mM (Hara‐Chikuma et al. 2005b). The plausibility of this role is supported by the finding that the ClC‐5 E211A mutant, which behaves like a Cl− channel (analogous to ClC‐3 E224A), is able to rescue V‐ATPase‐mediated acidification of endosomes from ClC‐5 null mice (Novarino et al. 2010). ClC‐5 E211A nevertheless failed to rescue other endocytosis defects, indicating that Cl−/H+ exchange is required during endosomal maturation (Novarino et al. 2010; Satoh et al. 2016). An initial reduction in vesicular [Cl−] mediated by ClC‐3 coupled exchange might therefore be critical for establishing a favourable Cl− gradient to support vesicular Cl− uptake in phase 2. Further work is clearly needed to assess the potential significance of uncoupled ClC‐3‐mediated Cl− current under the voltage and ionic conditions present in endosomes.
Additional information
Competing interests
None declared.
Author contributions
This work was conducted in the laboratory of F.S.L., in the Department of Pediatrics, Vanderbilt University School of Medicine. J.R. contributed to the conception and design of the work, data acquisition, analysis and interpretation of data, and drafting and revision of the manuscript and figures. H.‐N.N. contributed to data acquisition and analysis, and drafting and revision of the manuscript and figures. F.S.L. contributed to the conception and design of the work, analysis and interpretation of data, and drafting and revision of the manuscript and figures. All the authors approved the final version of the manuscript, and agree to be accountable for all questions related to the accuracy and integrity of the work. All the qualified authors and contributors to this work are listed.
Funding
This work was funded by National Heart, Lung, and Blood Institute (NHLBI, USA) support to F. S. Lamb (R01 HL128386).
Acknowledgements
We thank Dr Alessio Accardi for encouraging the assessment of Cl−/H+ coupling in the presence of increased external protons. Confocal imaging was performed in part at the Vanderbilt Cell Imaging Shared Resource (supported by NIH grants CA68485, DK20593, DK58404, DK59637 and EY08126).
Biography
Jeffrey Rohrbough is a Research Assistant Professor in the Department of Pediatrics at Vanderbilt University Medical Center. Dr Rohrbough received his training in Neuroscience at the University of California, Los Angeles. His work has focused largely on synaptic development, including the functional development of voltage‐gated ion channels and multiple classes of synaptic transmitter receptors (GluR, GABAR, AChR) in Xenopus spinal neurons, and the development and regulation of glutamatergic synaptic function in Drosophila, using electrophysiological recordings, confocal microscopy and optical recording techniques in live cells. Since 2012, he has studied ion transport properties of the ClC‐3 Cl−/H+ exchanger and the LRRC8 (VRAC) channel.

Edited by: Peying Fong & Yoshihiro Kubo
Linked articles This article is highlighted by a Perspective by Guzman & Fahlke. To read this Perspective, visit https://doi.org/10.1113/JP276670.
References
- Accardi A, Lobet S, Williams C, Miller C & Dutzler R (2006). Synergism between halide binding and proton transport in a CLC‐type exchanger. J Mol Biol 362, 691–699. [DOI] [PubMed] [Google Scholar]
- Accardi A & Miller C (2004). Secondary active transport mediated by a prokaryotic homologue of ClC Cl− channels. Nature 427, 803–807. [DOI] [PubMed] [Google Scholar]
- Alekov AK (2015). Mutations associated with Dent's disease affect gating and voltage dependence of the human anion/proton exchanger ClC‐5. Front Physiol 6, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekov AK & Fahlke C (2009). Channel‐like slippage modes in the human anion/proton exchanger ClC‐4. J Gen Physiol 133, 485–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barg S, Huang P, Eliasson L, Nelson DJ, Obermuller S, Rorsman P, Thevenod F & Renstrom E (2001). Priming of insulin granules for exocytosis by granular Cl− uptake and acidification. J Cell Sci 114, 2145–2154. [DOI] [PubMed] [Google Scholar]
- Basilio D, Noack K, Picollo A & Accardi A (2014). Conformational changes required for H+/Cl− exchange mediated by a CLC transporter. Nat Struct Mol Biol 21, 456–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennetts B & Parker MW (2013). Molecular determinants of common gating of a ClC chloride channel. Nat Commun 4, 2507. [DOI] [PubMed] [Google Scholar]
- Deriy LV, Gomez EA, Jacobson DA, Wang X, Hopson JA, Liu XY, Zhang G, Bindokas VP, Philipson LH & Nelson DJ (2009). The granular chloride channel ClC‐3 is permissive for insulin secretion. Cell Metab 10, 316–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefano S, Pusch M & Zifarelli G (2013). A single point mutation reveals gating of the human ClC‐5 Cl−/H+ antiporter. J Physiol 591, 5879–5893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan D, Winter C, Cowley S, Hume JR & Horowitz B (1997). Molecular identification of a volume‐regulated chloride channel. Nature 390, 417–421. [DOI] [PubMed] [Google Scholar]
- Duan D, Zhong J, Hermoso M, Satterwhite CM, Rossow CF, Hatton WJ, Yamboliev I, Horowitz B & Hume JR (2001). Functional inhibition of native volume‐sensitive outwardly rectifying anion channels in muscle cells and Xenopus oocytes by anti‐ClC‐3 antibody. J Physiol 531, 437–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutzler R, Campbell EB & MacKinnon R (2003). Gating the selectivity filter in ClC chloride channels. Science 300, 108–112. [DOI] [PubMed] [Google Scholar]
- Feng L, Campbell EB, Hsiung Y & MacKinnon R (2010). Structure of a eukaryotic CLC transporter defines an intermediate state in the transport cycle. Science 330, 635–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Campbell EB & MacKinnon R (2012). Molecular mechanism of proton transport in CLC Cl−/H+ exchange transporters. Proc Natl Acad Sci U S A 109, 11699–11704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentzsch M, Cui L, Mengos A, Chang XB, Chen JH & Riordan JR (2003). The PDZ‐binding chloride channel ClC‐3B localizes to the Golgi and associates with cystic fibrosis transmembrane conductance regulator‐interacting PDZ proteins. J Biol Chem 278, 6440–6449. [DOI] [PubMed] [Google Scholar]
- Grieschat M & Alekov AK (2012). Glutamate 268 regulates transport probability of the anion/proton exchanger ClC‐5. J Biol Chem 287, 8101–8109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieschat M & Alekov AK (2014). Multiple discrete transitions underlie voltage‐dependent activation in CLC Cl−/H+ antiporters. Biophys J 107, L13–L15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunther W, Piwon N & Jentsch TJ (2003). The ClC‐5 chloride channel knock‐out mouse – an animal model for Dent's disease. Pflugers Arch 445, 456–462. [DOI] [PubMed] [Google Scholar]
- Guzman RE, Alekov AK, Filippov M, Hegermann J & Fahlke C (2014). Involvement of ClC‐3 chloride/proton exchangers in controlling glutamatergic synaptic strength in cultured hippocampal neurons. Front Cell Neurosci 8, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman RE, Grieschat M, Fahlke C & Alekov AK (2013). ClC‐3 is an intracellular chloride/proton exchanger with large voltage‐dependent nonlinear capacitance. ACS Chem Neurosci 4, 994–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman RE, Miranda‐Laferte E, Franzen A & Fahlke C (2015). Neuronal ClC‐3 splice variants differ in subcellular localizations, but mediate identical transport functions. J Biol Chem 290, 25851–25862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W, Cheng RC, Maduke MC & Tajkhorshid E (2014). Water access points and hydration pathways in CLC H+/Cl− transporters. Proc Natl Acad Sci U S A 111, 1819–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara‐Chikuma M, Wang Y, Guggino SE, Guggino WB & Verkman AS (2005a). Impaired acidification in early endosomes of ClC‐5 deficient proximal tubule. Biochem Biophys Res Commun 329, 941–946. [DOI] [PubMed] [Google Scholar]
- Hara‐Chikuma M, Yang B, Sonawane ND, Sasaki S, Uchida S & Verkman AS (2005b). ClC‐3 chloride channels facilitate endosomal acidification and chloride accumulation. J Biol Chem 280, 1241–1247. [DOI] [PubMed] [Google Scholar]
- Jentsch TJ (2007). Chloride and the endosomal‐lysosomal pathway: emerging roles of CLC chloride transporters. J Physiol 578, 633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch TJ, Lutter D, Planells‐Cases R, Ullrich F & Voss FK (2016). VRAC: molecular identification as LRRC8 heteromers with differential functions. Pflugers Arch 468, 385–393. [DOI] [PubMed] [Google Scholar]
- Khantwal CM, Abraham SJ, Han W, Jiang T, Chavan TS, Cheng RC, Elvington SM, Liu CW, Mathews II, Stein RA, McHaourab HS, Tajkhorshid E & Maduke M (2016). Revealing an outward‐facing open conformational state in a CLC Cl−/H+ exchange transporter. Elife 5, e11189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb FS, Moreland JG & Miller FJ Jr (2009). Electrophysiology of reactive oxygen production in signaling endosomes. Antioxid Redox Signal 11, 1335–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Swanson JM & Voth GA (2016). Multiscale simulations reveal key aspects of the proton transport mechanism in the ClC‐ec1 antiporter. Biophys J 110, 1334–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Shimada K, Showalter LA & Weinman SA (2000). Biophysical properties of ClC‐3 differentiate it from swelling‐activated chloride channels in Chinese hamster ovary‐K1 cells. J Biol Chem 275, 35994–35998. [DOI] [PubMed] [Google Scholar]
- Li X, Wang T, Zhao Z & Weinman SA (2002). The ClC‐3 chloride channel promotes acidification of lysosomes in CHO‐K1 and Huh‐7 cells. Am J Physiol Cell Physiol 282, C1483–C1491. [DOI] [PubMed] [Google Scholar]
- Maritzen T, Keating DJ, Neagoe I, Zdebik AA & Jentsch TJ (2008). Role of the vesicular chloride transporter ClC‐3 in neuroendocrine tissue. J Neurosci 28, 10587–10598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda JJ, Filali MS, Collins MM, Volk KA & Lamb FS (2010). The ClC‐3 Cl−/H+ antiporter becomes uncoupled at low extracellular pH. J Biol Chem 285, 2569–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda JJ, Filali MS, Volk KA, Collins MM, Moreland JG & Lamb FS (2008). Overexpression of CLC‐3 in HEK293T cells yields novel currents that are pH dependent. Am J Physiol Cell Physiol 294, C251–C262. [DOI] [PubMed] [Google Scholar]
- Miller C & Nguitragool W (2009). A provisional transport mechanism for a chloride channel‐type Cl−/H+ exchanger. Philos Trans R Soc Lond B Biol Sci 364, 175–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller FJ Jr, Filali M, Huss GJ, Stanic B, Chamseddine A, Barna TJ & Lamb FS (2007). Cytokine activation of nuclear factor κB in vascular smooth muscle cells requires signaling endosomes containing Nox1 and ClC‐3. Circ Res 101, 663–671. [DOI] [PubMed] [Google Scholar]
- Moreland JG, Davis AP, Bailey G, Nauseef WM & Lamb FS (2006). Anion channels, including ClC‐3, are required for normal neutrophil oxidative function, phagocytosis, and transendothelial migration. J Biol Chem 281, 12277–12288. [DOI] [PubMed] [Google Scholar]
- Moreland JG, Davis AP, Matsuda JJ, Hook JS, Bailey G, Nauseef WM & Lamb FS (2007). Endotoxin priming of neutrophils requires NADPH oxidase‐generated oxidants and is regulated by the anion transporter ClC‐3. J Biol Chem 282, 33958–33967. [DOI] [PubMed] [Google Scholar]
- Nguitragool W & Miller C (2006). Uncoupling of a CLC Cl−/H+ exchange transporter by polyatomic anions. J Mol Biol 362, 682–690. [DOI] [PubMed] [Google Scholar]
- Novarino G, Weinert S, Rickheit G & Jentsch TJ (2010). Endosomal chloride‐proton exchange rather than chloride conductance is crucial for renal endocytosis. Science 328, 1398–1401. [DOI] [PubMed] [Google Scholar]
- Okada T, Akita T, Sato‐Numata K, Islam MR & Okada Y (2014). A newly cloned ClC‐3 isoform, ClC‐3d, as well as ClC‐3a mediates Cd‐sensitive outwardly rectifying anion currents. Cell Physiol Biochem 33, 539–556. [DOI] [PubMed] [Google Scholar]
- Orhan G, Fahlke C & Alekov AK (2011). Anion‐ and proton‐dependent gating of ClC‐4 anion/proton transporter under uncoupling conditions. Biophys J 100, 1233–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picollo A, Malvezzi M & Accardi A (2010). Proton block of the CLC‐5 Cl−/H+ exchanger. J Gen Physiol 135, 653–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picollo A & Pusch M (2005). Chloride/proton antiporter activity of mammalian CLC proteins ClC‐4 and ClC‐5. Nature 436, 420–423. [DOI] [PubMed] [Google Scholar]
- Picollo A, Xu Y, Johner N, Berneche S & Accardi A (2012). Synergistic substrate binding determines the stoichiometry of transport of a prokaryotic H+/Cl− exchanger. Nat Struct Mol Biol 19, 525–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch M & Zifarelli G (2015). ClC‐5: Physiological role and biophysical mechanisms. Cell Calcium 58, 57–66. [DOI] [PubMed] [Google Scholar]
- Riazanski V, Deriy LV, Shevchenko PD, Le B, Gomez EA & Nelson DJ (2011). Presynaptic CLC‐3 determines quantal size of inhibitory transmission in the hippocampus. Nat Neurosci 14, 487–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar G, Love R, Styers ML, Werner E, Peden A, Rodriguez S, Gearing M, Wainer BH & Faundez V (2004). AP‐3‐dependent mechanisms control the targeting of a chloride channel (ClC‐3) in neuronal and non‐neuronal cells. J Biol Chem 279, 25430–25439. [DOI] [PubMed] [Google Scholar]
- Satoh N, Yamada H, Yamazaki O, Suzuki M, Nakamura M, Suzuki A, Ashida A, Yamamoto D, Kaku Y, Sekine T, Seki G & Horita S (2016). A pure chloride channel mutant of CLC‐5 causes Dent's disease via insufficient V‐ATPase activation. Pflugers Arch 468, 1183–1196. [DOI] [PubMed] [Google Scholar]
- Scheel O, Zdebik AA, Lourdel S & Jentsch TJ (2005). Voltage‐dependent electrogenic chloride/proton exchange by endosomal CLC proteins. Nature 436, 424–427. [DOI] [PubMed] [Google Scholar]
- Smith AJ & Lippiat JD (2010a). Direct endosomal acidification by the outwardly rectifying CLC‐5 Cl−/H+ exchanger. J Physiol 588, 2033–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AJ & Lippiat JD (2010b). Voltage‐dependent charge movement associated with activation of the CLC‐5 2Cl−/1H+ exchanger. FASEB J 24, 3696–3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauber T & Jentsch TJ (2010). Sorting motifs of the endosomal/lysosomal CLC chloride transporters. J Biol Chem 285, 34537–34548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauber T, Weinert S & Jentsch TJ (2012). Cell biology and physiology of CLC chloride channels and transporters. Compr Physiol 2, 1701–1744. [DOI] [PubMed] [Google Scholar]
- Steinmeyer K, Schwappach B, Bens M, Vandewalle A & Jentsch TJ (1995). Cloning and functional expression of rat CLC‐5, a chloride channel related to kidney disease. J Biol Chem 270, 31172–31177. [DOI] [PubMed] [Google Scholar]
- Stobrawa SM, Breiderhoff T, Takamori S, Engel D, Schweizer M, Zdebik AA, Bosl MR, Ruether K, Jahn H, Draguhn A, Jahn R & Jentsch TJ (2001). Disruption of ClC‐3, a chloride channel expressed on synaptic vesicles, leads to a loss of the hippocampus. Neuron 29, 185–196. [DOI] [PubMed] [Google Scholar]
- Varela D, Simon F, Riveros A, Jorgensen F & Stutzin A (2004). NAD(P)H oxidase‐derived H2O2 signals chloride channel activation in cell volume regulation and cell proliferation. J Biol Chem 279, 13301–13304. [DOI] [PubMed] [Google Scholar]
- Walden M, Accardi A, Wu F, Xu C, Williams C & Miller C (2007). Uncoupling and turnover in a Cl−/H+ exchange transporter. J Gen Physiol 129, 317–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D & Voth GA (2009). Proton transport pathway in the ClC Cl−/H+ antiporter. Biophys J 97, 121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdebik AA, Zifarelli G, Bergsdorf EY, Soliani P, Scheel O, Jentsch TJ & Pusch M (2008). Determinants of anion‐proton coupling in mammalian endosomal CLC proteins. J Biol Chem 283, 4219–4227. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Li X, Hao J, Winston JH & Weinman SA (2007). The ClC‐3 chloride transport protein traffics through the plasma membrane via interaction of an N‐terminal dileucine cluster with clathrin. J Biol Chem 282, 29022–29031. [DOI] [PubMed] [Google Scholar]
- Zifarelli G, De Stefano S, Zanardi I & Pusch M (2012). On the mechanism of gating charge movement of ClC‐5, a human Cl−/H+ antiporter. Biophys J 102, 2060–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zifarelli G & Pusch M (2009). Conversion of the 2 Cl−/1 H+ antiporter ClC‐5 in a NO3 −/H+ antiporter by a single point mutation. EMBO J 28, 175–182. [DOI] [PMC free article] [PubMed] [Google Scholar]