

Abstract
Autoreactive lymphocytes that escape central immune tolerance may be silenced via an endogenous peripheral tolerance mechanism known as anergy. Antigen-specific therapies capable of inducing anergy may restore patients with autoimmune diseases to a healthy phenotype while avoiding deleterious side effects associated with global immunosuppression. Inducing anergy in B cells may be a particularly potent intervention, as B cells can contribute to autoimmune diseases through multiple mechanisms and offer the potential for direct antigen-specific targeting through the B cell receptor (BCR). Our previous results suggested autoreactive B cells may be silenced by multivalent ‘soluble antigen arrays’ (SAgAs), which are polymer conjugates displaying multiple copies of autoantigen with or without a secondary peptide that blocks intracellular cell-adhesion molecule-1 (ICAM-1). Here, key therapeutic molecular properties of SAgAs were identified and linked to the immunological mechanism through comprehensive cellular and in vivo analyses. We determined non-hydrolyzable ‘cSAgAs’ displaying multivalent ‘click’-conjugated antigen more potently suppressed experimental autoimmune encephalomyelitis (EAE) compared to hydrolyzable SAgAs capable of releasing conjugated antigen. cSAgAs restored a healthy phenotype in disease-specific antigen presenting cells (APCs) by inducing an anergic response in B cells and a subset of B cells called autoimmune-associated B cells (ABCs) that act as potent APCs in autoimmune disease. Accompanied by a cytokine response skewed towards a Th2/regulatory phenotype, this generated an environment of autoantigenic tolerance. By identifying key therapeutic molecular properties and an immunological mechanism that drives SAgA efficacy, this work guides the design of antigen-specific immunotherapies capable of inducing anergy.
Keywords: Tolerance, Autoimmunity, Autoimmune-associated B cells, Antigen-specific B cells, EAE
1. INTRODUCTION
Many current therapies for autoimmune diseases such as multiple sclerosis (MS) act through nonspecific suppression of the immune response, resulting in global immunosuppression and adverse side effects. Creation of antigen-specific immunotherapies (ASIT) that target and suppress only the offending autoreactive immune cells would address a pressing need for safer and more effective autoimmune therapies. Endogenous mechanisms for maintaining immunological tolerance, such as deletion and regulation of autoreactive B and T lymphocytes, would suggest restoration of immune tolerance is plausible. Therapeutic interventions capable of inducing tolerance, however, remain elusive.
Central immune tolerance is coordinated through silencing by deletion and receptor editing in the bone marrow and thymus, while peripheral tolerance is achieved through a state of antigen unresponsiveness called anergy.1 Anergy occurs when B or T cells mount an initial response to primary antigenic signal but do not receive sufficient secondary signal to sustain activation.1 For T cells, successful antigen-specific activation requires receipt of both a primary antigenic signal (via MHCII) and secondary costimulatory signal (from receptors such as CD80 and CD86) presented by an antigen presenting cell (APC).2–5 B cells, however, are unique in that they can both be activated in this manner and also play the role of APC.6–7 In fact, B cells may act as the main APC during induction of many autoimmune diseases because they more efficiently uptake low concentrations of soluble antigen via high affinity B cell receptors (BCRs) compared to pinocytosis mechanisms of dendritic cells (DCs) and macrophages.7–10 B cells also contribute to the induction and pathogenesis of autoimmune diseases such as MS and type 1 diabetes (T1D) through autoantibody production, cytokine secretion, and dysregulated B cell signaling leading to a loss of tolerance. 6 11–13
As both an effector cell and APC with potential for direct antigen-specific targeting, B cells have emerged as an especially promising target for ASIT.1, 14 B cells can be modulated in an antigen-specific manner either indirectly by targeting other APCs or CD4+ T helper cells, or directly by targeting the BCR, a surface receptor with inherent antigen specificity that can bind soluble antigen.1 Since B cell anergy is the primary endogenous mechanism for silencing autoreactive B cells to maintain immune tolerance, it also presents an attractive strategy for ASIT.15–16 Anergy can be induced through chronic antigen exposure and/or continuous BCR binding, occupation, and clustering in the absence of secondary costimulatory signal, resulting in reduced calcium flux, downregulation of costimulatory markers CD80 and CD86, and thus impaired capacity for T cell stimulation.17–21 Importantly, B cell fate can be directed by the form of self-antigen that is encountered, depending on antigen avidity and the degree of antigen receptor crosslinking.22 For example, oligomeric soluble antigen is reported to induce anergy whereas highly multivalent membrane-bound antigen induces deletion.16, 23 Thus, design and development of an effective B cell-targeted ASIT should take into account appropriate physicochemical properties of delivered antigen in light of known molecular mechanisms for inducing peripheral tolerance.
In previous studies, we investigated multivalent soluble antigen arrays (SAgAPLP:LABL) designed to induce tolerance to a specific multiple sclerosis (MS) autoantigen. SAgAPLP:LABL consists of a hyaluronic acid (HA) polymer conjugated with multiple copies of autoantigen (PLP139.151) and cell adhesion inhibitor (LABL, specific for intracellular adhesion molecule-1, ICAM-1) peptides. Employing a degradable (hydrolyzable) linker to codeliver PLP and LABL, SAgAPLP:LABL was therapeutic in vivo in a murine model of MS (experimental autoimmune encephalomyelitis (EAE))24–28 and exhibited antigen-specific binding with B cells, targeted the B cell receptor (BCR), and dampened BCR-mediated signaling in vitro29 Motivated by results that pointed to sustained BCR engagement as the SAgAPLP:LABL cellular mechanism, we developed a new version of the SAgA molecule using non-hydrolyzable conjugation chemistry. “Click SAgAs” (cSAgAPLP:LABL), employing hydrolytically stable covalent conjugation chemistry to link PLP and LABL to HA, achieved higher avidity B cell binding, greater reduction and inhibition of BCR-mediated signaling, and significantly enhanced in vivo efficacy compared to hydrolyzable SAgAPLP:LABL.30 We concluded that non-hydrolyzable conjugation increased the avidity of cSAgAPLP:LABL to drive in vivo efficacy through dampened BCR-mediated signaling via a mechanism of sustained action (BCR binding and clustering) on the cell surface.
Here, we identified SAgA immunological mechanisms using the EAE mouse model to define arising immune tolerance pathways to the PLP antigen used to induce the disease. Splenocyte immune responses were evaluated following in vivo and ex vivo treatment with click-conjugated (non-hydrolyzable) and hydrolyzable soluble antigen arrays, mixtures of the components, and controls. Targeted immune cell subtypes were identified through flow cytometry binding assays and microfluidic imaging of live cells. Reduced costimulatory signaling was evaluated as a marker of anergy and lasting antigenic tolerance in EAE splenocytes following ex vivo and in vivo treatment. Cytokines, PLP-specific autoantibody production, and splenic immune cell composition were also evaluated following in vivo treatment to determine the shape of the effector response (Th1 vs. Th2, Treg vs. Th17). Through these comprehensive cellular and in vivo analyses, we determined that multivalent polymer arrays displaying click-conjugated PLP induced an anergic response in B cells and a subset of B cells that act as potent APCs in autoimmune disease called autoimmune-associated B cells (ABCs), accompanied by a cytokine response skewed towards a Th2/regulatory phenotype. By identifying cornerstone molecular properties and an immunological mechanism that drives SAgA efficacy, this work guides our understanding of the antigen-specific immune response and informs the future design of ASIT.
2. MATERIALS AND METHODS
2.1. Materials
Hyaluronic acid (HA) sodium salt (MW 16 kDa) was purchased from Lifecore Biomedical (Chaska, MN). 11-azido-3,6,9-trioxaundecan-1-amine (NH2-PEG3-N3), N-hydroxysuccinimide, N-(3-dimethylaminopropyl)-N´-ethylcarbodiimide hydrochloride (EDC), 2-(N-morpholino)ethane-sulfonic acid sodium salt (MES), tris(3-hydroxypropyltriazolylmethyl)amine, and sodium ascorbate (NaAsc) were purchased from Sigma-Aldrich (St. Louis, MO) and used as received without further purification. Copper(II) sulfate pentahydrate (CuSO4 • 5H2O) was purchased from Acros Organics (Geel, Belgium). Alkyne-functionalized peptides bearing an N-terminal 4-pentynoic acid (homopropargyl, hp) modification, hpPLP139–151 (hp-HSLGKWLGHPDKF-OH) and hpLABL (hp-ITDGEATDSG-OH), were originally synthesized in our laboratory via solid phase peptide synthesis. Larger quantities of both hpPLP139–151 and hpLABL peptides were obtained from Biomatik USA, LLC (Wilmington, DE). Unmodified PLP (NH2-HSLGKWLGHPDKF-OH) peptide was purchased from PolyPeptide Laboratories (San Diego, CA). Incomplete Freund’s adjuvant (IFA) and killed Mycobacterium tuberculosis strain H37RA were purchased from Difco (Sparks, MD). Pertussis toxin was purchased from List Biological Laboratories (Campbell, CA). R-phycoerythrin (PE)/Cy7-conjugated anti-mouse CD3, AlexaFluor647-conjugated anti-mouse CD19, Pacific Blue-conjugated anti-mouse CD11c, PerCP-conjugated anti-mouse B220, PE-conjugated anti-mouse CD86, FITC-conjugated anti-mouse CD80, and respective isotype control antibodies were purchased from BioLegend (San Diego, CA). All other chemicals and reagents were analytical grade and used as received.
2.2. Synthesis of Click Soluble Antigen arrays (cSAgAs)
Penn Green-Alk, HA-N3, and click soluble antigen arrays (cSAgAs) were prepared as previously reported.30 Briefly, cSAgAs were constructed using a two-step procedure starting from sodium hyaluronate. 3-(ethyliminomethyleneamino)-N,N-dimethylpropan-1-amine (EDC) and N-hydroxysuccinimide (NHS) neat were added to a solution of sodium hyaluronate in MES buffer. After 5 min of stirring, H2N-PEG3-N3 was added and the solution was stirred at room temperature for 24 hrs before being dialyzed and lyophilized to isolate HA-N3. The appropriate ligands (hpPLP139–151, hpLABL, Penn Green-Alk) were added to a solution of HA-N3 in deionized water, followed by a premixed solution of tris(3-hydroxypropyl-triazolylmethyl)amine (THPTA) and copper (II) sulfate (CuSO4-5H2O) in deionized water. After stirring for 1–2 min, sodium ascorbate (NaAsc) was added and the reaction was allowed to proceed at elevated temperature until the desired conjugation levels were achieved. Following completion of the reaction, the reaction solution was dialyzed and lyophilized. cSAgAs were analyzed qualitatively by FTIR and NMR, and quantitatively by RP-HPLC to determine extent of conjugation (Figure S1).
2.3. Synthesis of Soluble Antigen Arrays
Soluble antigen arrays (SAgAPLP:LABL) were synthesized and characterized as previously reported.24–25 Aminooxy peptides AoPLP and/or AoLABL were linked to HA using oxime conjugation chemistry. Peptide conjugation was determined through gradient reverse-phase analytical high-performance liquid chromatography (RP-HPLC) following cleavage of peptides in 0.1N HCl.
2.4. EAE Mouse In Vivo Studies
In vivo studies were carried out with 4–6 week old SJL/J (H-2) female mice purchased from Envigo Laboratories (Indianapolis, IN). Mice were housed under specified, pathogen-free conditions at the University of Kansas and all experiments were approved by the University’s Institutional Animal Care and Use Committee. Complete Freund’s adjuvant (CFA) was made by combining IFA and killed M. tuberculosis strain H37RA at a final concentration of 4 mg/mL. Animals were induced with experimental autoimmune encephalomyelitis (EAE), the PLP-specific mouse model of relapsing-remitting MS, on day 0 of the study. Immunization was accomplished using a 0.2 mL emulsion containing 200 μg PLP139–151 peptide, plus equal volumes of PBS and CFA. The emulsion was administered subcutaneously (s.c.) as a total of four 50 μL injections, located above each shoulder and each hind flank. Pertussis toxin (100 ng in 100 μL) was injected intraperitoneally on day 0 and day 2 post-immunization.
Treatments were administered on days 4, 7, and 10 as 100 μL subcutaneous injections at the nape of the neck (n=5 mice per treatment group). This three-day dosing schedule was found to be efficacious in previous studies.24–26 Samples were administered at a dose equivalent to 50 nmol PPP per 100 μL (0.5 mM PPP) based on previously reported dosing studies.30 Disease progression was evaluated by a single observer using the following clinical score system: 0, no clinical disease symptoms; 1, weakness or limpness of the tail; 2, weakness or partial paralysis of one or two hind limbs (paraparesis); 3, full paralysis of both hind limbs (paraplegia); 4, paraplegia plus weakness or paralysis of forelimbs; 5, moribund (at which point mice were euthanized). In addition to animal scoring, body weight measurements were performed daily for the 26-day duration of the EAE study.
2.5. Mouse Splenocyte Isolation
Spleens were harvested from healthy control mice and EAE mice on day 12 at peak of disease following disease induction for ex vivo treatment studies, or from EAE in vivo-treated mice on day 25 for ex vivo rechallenge studies. Spleens were collected in 5 mL of RPMI 1640 media supplemented with P-glutamine and 1% Penicillin-Streptomycin on ice, then disrupted by passing through a sterile wire mesh using the rubber end of a 1 mL syringe plunger. Crude cellular extract was collected in 5 mL cRPMI (RPMI 1640 media supplemented with P-glutamine, 1% Penicillin-Streptomycin, and 10% fetal bovine serum (FBS)) and centrifuged at 1100xg for 5 minutes. The cell pellet was resuspended in 5 mL Gey’s lysis solution for 5 minutes on ice to lyse red blood cells, quenched with 10 mL cRPMI, and spun down again at 1100xg for 5 minutes. Cells were resuspended in cRPMI, counted in 0.04% trypan blue, and cultured at 37°C and 5% CO2 in the presence or absence of ex vivo treatment or PLP rechallenge for each respective assay, as follows.
2.6. Day 12 Splenocyte Assays: Evaluating Cell Targeting and Cellular Response to Ex Vivo Treatment at Peak of Disease
2.6.1. Flow Cytometry Binding
Splenocytes isolated from EAE mice on day 12 at peak of disease were seeded at 3×106 cells/mL (9×106 cells/well) in a 12-well plate and cultured in the presence of 25 μM PLP antigen challenge for 72 hours. Healthy splenocytes were cultured in the absence of PLP. Cells were then stained with PE/Cy7 anti-mouse CD3, AlexaFluor647 anti-mouse CD19, and Pacific Blue anti-mouse CD11c per manufacturer guidelines and resuspended in serum-free clear RPMI. Association binding studies were performed in live cells by flow cytometry (BD FACSFusion, BD Biosciences, San Jose, CA), as previously reported.29–30 Cell samples were warmed to 37°C for 2 minutes prior to each flow cytometry run. To observe maximum steady state binding, cells were first run on the cytometer for 30s to establish a cell baseline before adding PennGreen-labeled treatment to achieve a final concentration of 1×106 cells/mL. Data acquisition was continued for an additional 3 minutes after maximum steady state (max. SS) equilibrium was reached (Figure S2B). Treatments were added at an equimolar PLP dose to mimic the dosing scheme from in vivo studies (141.3 μΜ PLP for fcHAPLP and fcSAgAPLP:LABL; 141.3 μΜ LABL for fcHALABL; 18.11 μΜ HA, or the HA molar equivalent to a 141.3 μΜ PLP dose of fcHAPLP). Concentration was determined from preliminary saturation studies which established a Bmax of 141.3 μM PLP for fcSAgAPLP:LABL (Figure S3A).
Flow cytometry binding data was first gated to remove doublets, dead cells, and debris using Kaluza Flow Analysis software (Beckman Coulter, Inc., Brea, CA) (Figure S2A). Max. SS binding for each subpopulation was defined as PennGreen mean fluorescence intensity (MFI) in the max. SS binding region gated on CD19+ B cells, CD3+ (CD19−) T cells, CD11c+ DCs, or CD19+B220+CD11c+ ABCs, minus the cell baseline (Figure S2B).
2.6.2. Fluorescence Microscopy of Binding in Live Cells
Splenocytes were isolated, cultured, and stained as described above. Live cell imaging of splenocytes binding PennGreen-labeled treatments was observed under fluorescence microscopy (Olympus IX81 Inverted Epifluorescence Microscope) using the same concentrations from flow cytometry association binding experiments. CellASIC ONIX M04S Microfluidics Switching Plates and Microfluidics Platform (EMD Millipore, Billerica, MA) were utilized for controlled perfusion of fluorescent samples and media with cells during real-time imaging. Stained cells were first loaded into the imaging chamber, then PennGreen-labeled treatments were perfused into the chamber for 10 minutes (3 psi for 5 minutes, 0.25 psi for 5 minutes) to allow binding with cells, followed by gentle media perfusion (0.25 psi for 5 minutes) to rinse unbound treatment and immediate image capture. Images were processed using Slidebook 5.5 (Intelligent Imaging Innovations, Inc., Denver, CO).
2.6.3. Costimulatory Signaling and Cell Composition following Ex Vivo Treatment
Splenocytes isolated from healthy controls and EAE mice on day 12 at peak of disease were seeded at 3×106 cells/mL (3×106 cells/well) in a 12-well plate and cultured in the presence or absence of 25 μM PLP antigen challenge plus treatment (dosed at 25 uM PLP) for 72 hours. Cells were then stained with PE/Cy7 anti-mouse CD3, AlexaFluor647 anti-mouse CD19, Pacific Blue anti-mouse CD11c, PerCP anti-mouse B220, PE anti-mouse CD86, and FITC anti-mouse CD80 per the manufacturer guidelines and resuspended in serum-free clear RPMI. Marker expression was evaluated by flow cytometry (BD FACSFusion).
2.7. Day 25 Splenocyte Assays: Evaluating Cellular Response to Ex Vivo Antigen Rechallenge following In Vivo Treatment
2.7.1. Costimulatory Signaling and Cell Composition following In Vivo Treatment
Splenocytes isolated from healthy controls and EAE mice on day 25 following in vivo treatment were seeded at 3×106 cells/mL (3×106 cells/well) in a 12-well plate and cultured in the presence or absence of 25 μM PLP antigen rechallenge for 72 hours. Cells were then stained with PE/Cy7 anti-mouse CD3, AlexaFluor647 anti-mouse CD19, Pacific Blue anti-mouse CD11c, PerCP anti-mouse B220, PE anti-mouse CD86, and FITC anti-mouse CD80 per the manufacturer guidelines and resuspended in serum-free clear RPMI. Marker expression was evaluated by flow cytometry (BD FACSFusion).
2.7.2. Cytokine Response
Splenocytes isolated from healthy controls and EAE mice on day 25 following in vivo treatment were seeded at 6×106 cells/mL (6×106 cells/well) in a 24-well plate and cultured in the presence or absence of 25 μM PLP antigen rechallenge for 120 hours. Plates were then spun down at 1100xg for 10 minutes to collect supernatants for cytokine analysis. Secreted cytokines (IL-4, IL-10, IL-6, GM-CSF, IL-2, IFN-γ, IL-17, IL-15, IL-23, and TNF-α) were detected using a U-Plex Mouse Biomarker Group 1 Multiplex Assay ELISA kit (Meso Scale Discovery (MSD), Rockville, MD) according to manufacturer guidelines, as previously described.31
2.7.3. ELISPOT Detection of PLP-specific Autoantibody Response
Immulon 2HB 96-well ELISPOT plates (Thermo Fisher Scientific, Waltham, MA) were coated with PLP antigen at a concentration of 50 μg/ml in 100 μL PBS, incubated overnight at 4°C, then blocked with cRPMI for 1 hour at 37°C. Splenocytes isolated from healthy controls and EAE mice on day 25 following in vivo treatment were seeded at 1×107 cells/mL (1×106 cells/well) in the coated and blocked plates and incubated (37°C, 5% CO2) for 48 hours. Plates were then washed 4X with PBS-Tween (containing 0.05% Tween 20) and 2X with PBS for 2 minutes each. Horseradish peroxidase (HRP)-conjugated anti-mouse IgG secondary antibody in PBS containing 0.5% FBS was added to each well (100 μL) at a concentration of 1 μg/mL and incubated at 37°C for 1 hour. After a second wash with PBS-Tween and PBS, detection buffer containing a 1:1 ratio of TrueBlue Peroxidase Substrate (Kirkegaard & Perry Laboratories, Inc) and agarose heated to 56°C was added to each well (100 μL) and incubated at 4°C overnight. Plates were read on a CTL Ultimate S6 ImmunoSpot Analyzer (Cellular Technology Limited) and analyzed using CTL ImmunoSpot software (Cellular Technology Limited).
2.8. Statistical Analysis
GraphPad Prism was used to perform statistical analyses including ordinary oneway or two-way analysis of variance (ANOVA) and unpaired t-test, followed by Tukey’s, Dunnett’s, or Sidak’s post-hoc test where appropriate. The threshold for statistical significance was set to p<0.05.
3. RESULTS
3.1. Click-conjugated soluble antigen arrays achieved significant therapeutic efficacy in vivo
We first sought to identify key therapeutic molecular properties by testing various combinations of HA, LABL, and PLP antigen in multivalent polymer array or mixture form in vivo against EAE. Polymer alone (cHA), click-conjugated homopolymer arrays with PLP alone (cHAPLP) or LABL alone (cHALABL), click-conjugated arrays with both PLP and LABL (cSAgAPLP:LABL), hydrolyzable arrays with both PLP and LABL (SAgAPLP:LABL), a 1:1 mixture of click-conjugated homopolymer arrays (cHAPLP+cHALABL), and a stoichiometrically equivalent noncovalent mixture of the individual components (cHA+PLP+LABL) were administered at an equimolar 50 nmol PLP dose and compared to mice that received PBS (vehicle) as a negative control (Table 1). From Table 1, groups (3) cHA and (4) cHALABL were included as controls that contain polymer and LABL but lack PLP; (5) cHA+PLP+LABL and (6) SAgAPLP:LABL were included as groups that deliver all components including PLP but not in click-conjugated polymer array form; (7) cHAPLP, (8) cSAgAPLP:LABL, and (9) cHAPLP+cHALABL were included as groups that deliver PLP in click-conjugated form with or without LABL and on one or two HA backbones to probe the importance of these characteristics.
Table 1.
Peptide molar conjugation and approximate molecular weight of treatment groups (numbered 3-9), as determined by RP-HPLC.a
| Treatment Group | Approx. MW (kDa)b |
Average Molar Ratio per Polymerc |
|||
|---|---|---|---|---|---|
| PLP:HA | LABL:HA | ||||
| without PLP | |||||
| 3 | cHAd | 24.7 | 0 | 0 | |
| 4 | cHALABL | 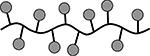 |
39.2 | 0 | 14 |
| with PLP (mixture or hydrolyzable) | |||||
| 5 | cHA+PLP+LABL | 24.7 (cHA), 1.5 (PLP), 1.0 (LABL) |
10 | 10 | |
| 6 | SAgAPLP:LABL | 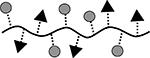 |
31.6 | 5 | 8 |
| with PLP (click-conjugated) | |||||
| 7 | cHAPLP | 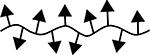 |
50.1 | 15 | 0 |
| 8 | cSAgAPLP:LABL | 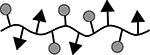 |
50.3 | 11 | 8 |
| 9 | cHAPLP + cHALABL |  |
50.1 (cHAPLP), 39.2 (cHALABL) |
15 | 14 |
Results are an average of triplicate injections from a single batch preparation. In the molecule schematics, dotted lines represent hydrolyzable oxime linker chemistry while solid lines represent non-hydrolyzable ‘click’ linker chemistry.
Molecular weights were calculated from RP-HPLC. MW, molecular weight.
HA, hyaluronic acid; PLP, proteolipid protein peptide; LABL, adhesion inhibitor peptide derived from leukocyte function associated antigen-1. All experiments were dosed based on constant PLP molar concentration.
cHA, azide functionalized HA (16 kDa)
Therapeutic efficacy was measured by daily clinical disease score (Figure 1A and C), daily percent weight change (Figure 1B and D), clinical disease score area under the curve (AUC) (Figure 1E), incidence of disease (Figure S4A and C), and mortality rate (Figure S4B and D). AUC representation of clinical data was included as an informative secondary measure for overall extent of disease in EAE, as it provides a cumulative measure that is not weighted by scaling or time course.32
Figure 1.
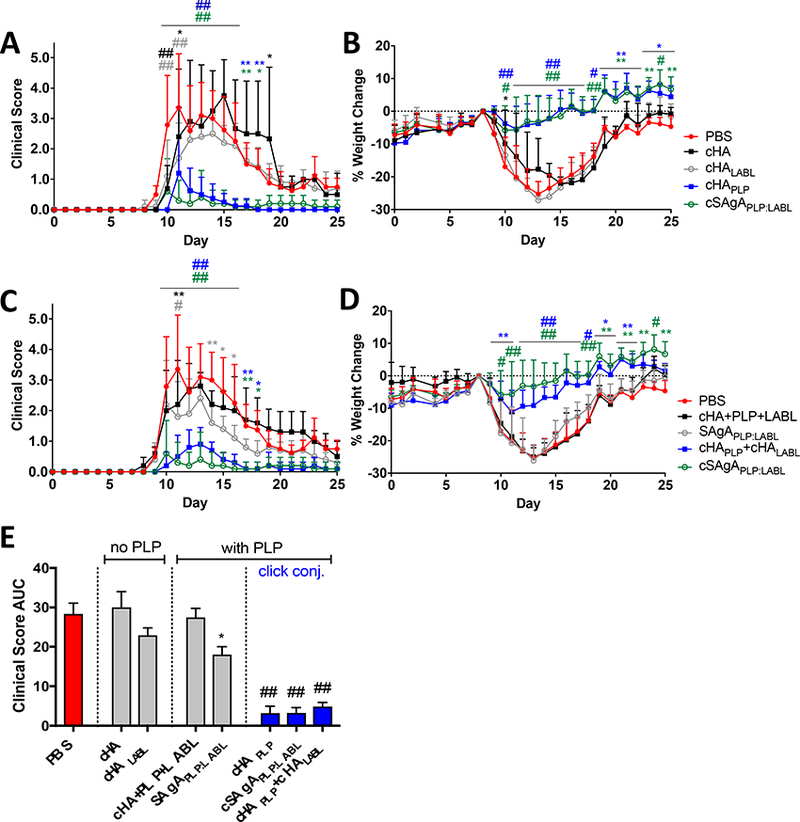
EAE in vivo response to click conjugates (cHA, cHALabl, cHAPLP, and cSAgAPLP:LABL) as measured by A) clinical disease score and B) percent weight loss. EAE in vivo response to groups containing both PLP and LABL (cHA+PLP+LABL, SAgAPLP:LABL, cHAPLP+cHALABL, and cSAgAPLP:LABL) as measured by C) clinical disease score and D) percent weight loss. Data represent mean ± SD (n=5); statistical significance compared to PBS negative control was determined by two-way ANOVA. E) Cumulative EAE in vivo response as measured by clinical disease score area under the curve (AUC) derived from subfigures A and C. Data represent mean ± SEM (n=5); statistical significance compared to PBS negative control was determined by ordinary one-way ANOVA followed by Dunnett’s post hoc test. (*p<0.05, **p<0.01, #p<0.001, ##p<0.0001, color coded according to group)
The in vivo results established that click-conjugated polymer arrays containing PLP antigen were potent therapeutics for EAE. cSAgAPLP:LABL, cHAPLP, and homopolymer mixture cHAPLP+cHALABL significantly alleviated clinical disease scores compared to the PBS negative control on days 10–18, particularly during peak of disease over days 10–16 (p<0.0001) (Figure 1A and C). Similarly, these groups inhibited severe weight loss (indicative of worsening disease) from day 10 onward, particularly during peak of disease (p<0.0001) (Figure 1B and D). The hydrolyzable array (SAgAPLP:LABL) was minimally therapeutic at the same dose, alleviating clinical disease score on days 11 (p<0.001), 14 (p<0.01), and 15–16 (p<0.05), but not affecting weight loss. Clinical disease score AUC also showed significant efficacy for cSAgAPLP:LABL, cHAPLP, and cHAPLP+cHALABL (p<0.0001) and lesser efficacy for SAgAPLP:LABL (p<0.05) compared to PBS (Figure 1E). By completion of the study at day 25, incidence of disease was reduced from 100% (PBS) to 0% (cHAPLP) or 20% (cSAgAPLP:LABL, cHAPLP+cHALABL) in these treated groups (Figure S4A and C). In contrast, the noncovalent component mixture (cHA+PLP+LABL) and polymer arrays lacking PLP (cHA, cHALABL) were not therapeutic compared to the PBS negative control by any of these metrics. It is also important to note that PLP alone (at a dose of 200 nmol) did not significantly alleviate disease compared to PBS (Figure S4E and F), achieving an equivalent effect as the component mixture (HA+PLP+LABL) at the same dose (Figure S4G). From this, we can conclude that molecular properties of (1) multivalent presentation of antigen in a polymer array and (2) non-hydrolyzable conjugation resulted in significant therapeutic efficacy in EAE.
3.2. Targeting disease-specific antigen presenting cells
cSAgA binding was then evaluated ex vivo in splenocytes isolated from healthy or EAE mice at peak of disease (day 12) to determine which immune cell subtypes were being targeted and whether cells associated with EAE were targeted over healthy cells. Splenocytes were antibody-labeled to identify B cells (CD19+), T cells (CD3+), DCs (CD11c+), and autoimmune-associated B cells (ABCs), which express a unique surface phenotype (CD19+B220+CD11c+ or CD19+CD11c+) with CD11c as the singularly characteristic identification marker (Figure S2, Figure S3F). ABCs are a subclass of B cells that have recently been identified as potent antigen presenting cells found in the spleen during autoimmune disease, expressing high levels of CD80/CD86 and MHCII, and have been implicated in human autoimmune diseases as well as murine models of autoimmunity.33–37 fcHAPLP and fcSAgAPLP:LABL exhibited disease-specific targeting in APCs, as binding was significantly higher in EAE compared to healthy subsets for B cells (p<0.0001), ABCs (p<0.01 and p<0.001, respectively), and DCs (p<0.01 and p<0.0001, respectively) (Figure 2B-D). Of these, CD19+ B cells exhibited the greatest difference in binding between EAE and healthy cells. In contrast, there was no difference in binding between T cells from EAE and healthy mice (Figure 2A). Comparing lymphocyte populations, the extent of binding in both EAE- and healthy-associated T cells was equivalent to that in B cells from healthy mice (Figure S3E). Among APC populations, binding was highest in ABCs (Figure 2C and F), followed by DCs and B cells. Overall, fcSAgAPLP:LABL exhibited the highest amount of binding followed by fcHAPLP, reflecting binding trends observed previously in a model B cell line.29 Importantly, both fcSAgAPLP:LABL and fcHAPLP exhibited significantly higher binding than polymer arrays lacking PLP (fcHA and fcHALABL) in B cells (p<0.0001), ABCs (p<0.0001 and p<0.001, respectively), and DCs (p<0.0001).
Figure 2.
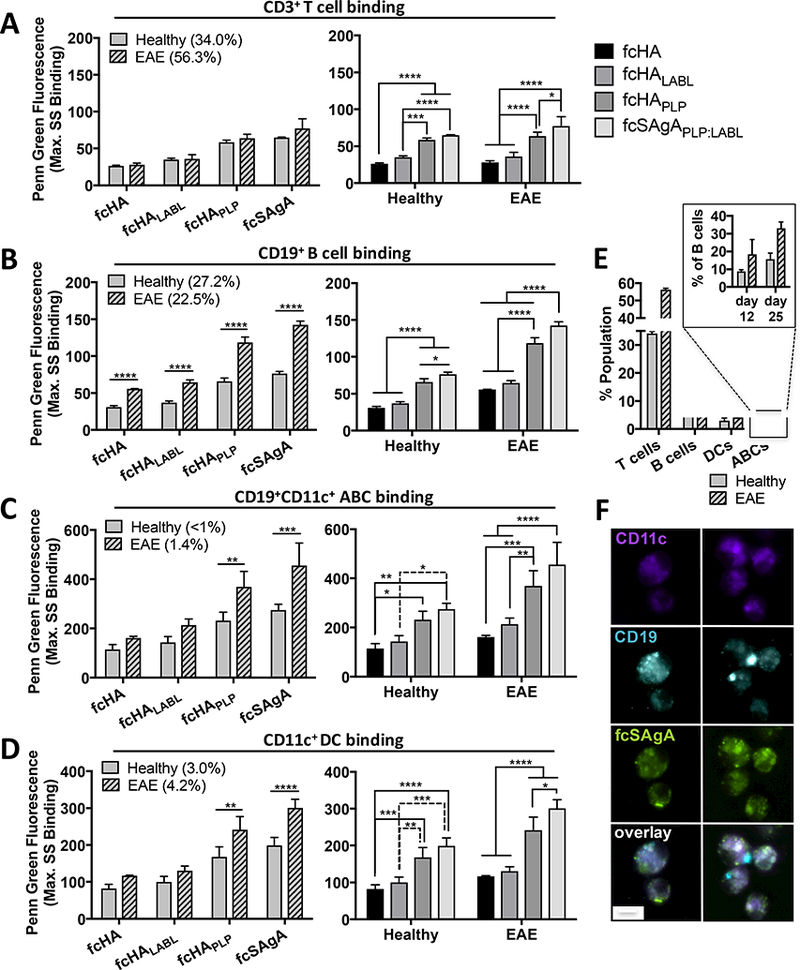
Click conjugate binding (max. SS fluorescence) in EAE versus healthy splenocyte subpopulations isolated at peak of disease (day 12), determined by flow cytometry: A) T cells (CD3+CD19−), B) B cells (CD19+), C) autoimmune-associated B cells (ABCs) (CD19+CD11c+), and D) dendritic cells (DCs) (CD11c+). E) Relative subpopulation composition (% total isolated splenocytes) in healthy versus EAE splenocytes at peak of disease; subset shows ABCs as % of gated B cells on day 12 and 25. Data represent mean ± SD (n=3); statistical significance was determined by two-way ANOVA followed by Sidak’s or Tukey’s post hoc test (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001). F) Fluorescence microscopy shows high fcSAgAPLP:LABL binding (green) on EAE splenocytes that co-express CD11c (violet) and CD19 (blue), identified as ABCs. Real time binding images of live cells were captured using the M04S plate and CellASIC Onyx Microfluidics platform on an Olympus IX81 inverted Epifluorescence microscope. Magnification: 60X air. Scale bar equals 10 μm.
While the amount of binding in DCs exceeded binding in B cells, DCs composed <5% of splenocytes at peak of disease compared to B cells which composed 20–30% (Figure 2E). Given that B cells make up the majority APC population in the spleen and act as the major APC during induction of many autoimmune diseases, it is likely that B cells and ABCs are the predominant APC populations targeted.7–10 It is also significant to note that while the ABC population made up <2% of all splenocytes total, it comprised ~20% of B cells (CD19+B220+) at peak of disease (day 12) and 30–40% of B cells at day 25 (Figure 2E) in EAE mice, consistent with reported literature values. 34, 36
A Resazurin assay was performed to evaluate metabolism in EAE versus healthy splenocytes following 72h ex vivo treatment in the presence or absence of PLP rechallenge. cHAPLP, cSAgAPLP:LABL, and cHAPLP+cHALABL reduced cell metabolism in both PLP-challenged and unchallenged EAE splenocytes but had no effect in healthy splenocytes, providing additional evidence that EAE cells were targeted over healthy cells (Figure S5). Together, these results support our hypothesis that PLP-conjugated soluble antigen arrays target APCs and specifically B cells (including ABCs) over other cell types, and do so in a disease-specific manner.
3.3. Click-conjugated soluble antigen arrays reduced antigen-specific costimulatory signaling in antigen presenting cells
We previously reported that cSAgAPLP:LABL caused significant reduction and inhibition of BCR-mediated calcium flux signaling in a model B cell line, accompanied by sustained binding and BCR clustering on the cell surface and enhanced in vivo efficacy.30 These observations were consistent with B cell anergy which is marked by reduced calcium flux and downregulation of costimulatory markers CD80 and CD86.16–19 Motivated by our previous observations, we therefore investigated CD80/CD86 expression in splenocytes under two conditions: (1) harvested from EAE mice at peak of disease and treated ex vivo during simultaneous PLP rechallenge to evaluate the effect on costimulatory signaling, and (2) harvested from mice treated in vivo and rechallenged with PLP ex vivo to evaluate lasting tolerance.
First, a significant increase in CD86 expression was observed upon ex vivo rechallenge with PLP in B cells (p<0.01), ABCs (p<0.05), and DCs (p<0.05) isolated from spleens of EAE mice at peak of disease (Figure 3). In contrast, CD86 expression was not elevated upon PLP challenge in cells from healthy mice. Increased costimulatory signaling in APCs from EAE mice reflected a memory response to PLP antigen, while cells from healthy mice had never been exposed to antigen and thus did not possess antigenic memory. However, APCs from EAE mice treated ex vivo with cSAgAPLP:LABL, cHAPLP, and cHAPLP+cHALABL did not exhibit increased costimulatory signaling upon PLP rechallenge; CD86 expression was significantly downregulated in B cells, ABCs, and DCs in these treatment groups compared to the untreated control (p<0.001 or p<0.0001). SAgAPLP:LABL treatment also caused CD86 downregulation to a lesser extent in B cells and ABCs (p<0.05), and cHALABL caused CD86 downregulation in B cells (p<0.001), ABCs (p<0.01), and DCs (p<0.01). Conversely, cHA and cHA+PLP+LABL had no effect on CD86 expression in PLP-challenged cells, while cHA+PLP+LABL stimulated unchallenged cells. Thus, ex vivo treatment with cHAPLP, cSAgAPLP:LABL, cHAPLP+cHALABL and to a lesser extent SAgAPLP:LABL and cHALABL caused autoimmune APCs to behave more like healthy cells irresponsive to antigen, despite being taken from EAE mice at peak of disease.
Figure 3.
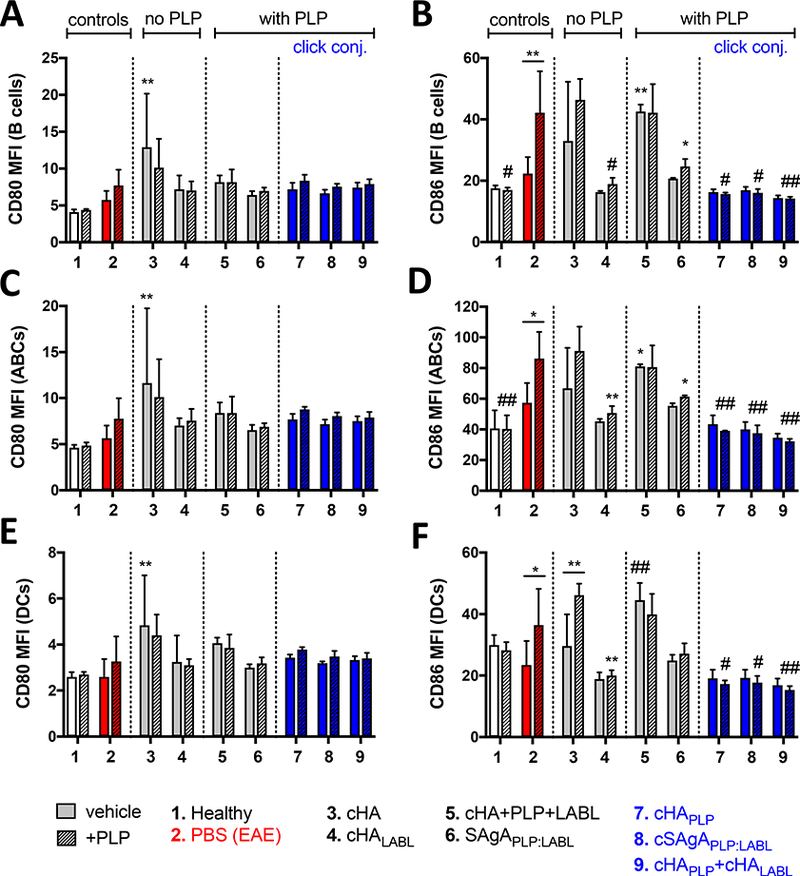
CD80/CD86 costimulatory marker expression in splenocytes isolated from EAE mice at peak of disease (day 12) followed by 72h ex vivo treatment with groups 3–9 in the presence or absence of PLP (25 μM) antigen rechallenge, determined by flow cytometry: A/B) B cells (CD19+), C/D) ABCs (CD19+B220+CD11c+), and E/F) DCs (CD11c+). Data represent mean ± SD (n=3). Statistical significance compared to PBS negative control was determined by two-way ANOVA followed by Dunnett’s post hoc test; statistical significance between +/− PLP (underlined) was determined by two-way ANOVA followed by Sidak’s post hoc test (*p<0.05, **p<0.01, #p<0.001, ##p<0.0001). MFI = mean fluorescent intensity.
Costimulatory marker expression was then evaluated in mice treated in vivo and rechallenged with PLP ex vivo to evaluate lasting tolerance to antigen. Here, a significant increase in CD80 and CD86 expression was observed (p<0.0001) upon ex vivo rechallenge with PLP in B cells, ABCs, and DCs isolated from spleens of EAE mice on day 25, while neither CD80 nor CD86 expression was elevated upon PLP challenge in cells from healthy mice (Figure 4). Again, APCs from EAE mice treated in vivo with cSAgAPLP:LABL, cHAPLP, and cHAPLP+cHALAbl did not exhibit increased costimulatory signaling upon PLP rechallenge, suggesting lasting tolerance to antigen 18 days after treatment. CD80 expression was significantly downregulated in PLP-challenged B cells, ABCs, and DCs in response to these treatments compared to the PBS negative control (p<0.0001). Similarly, CD86 expression was significantly downregulated in PLP-challenged B cells, ABCs, and DCs in response to cHAPLP (p<0.0001), cSAgAPLP:LABL (B cells and ABCs p<0.0001, DCs p<0.001), and cHAPLP+cHALABL (B cells p<0.001, ABCs p<0.0001, DCs p<0.01). Additionally, the number of CD80-expressing B cells (% CD80+) was significantly reduced following treatment with cHAPLP (p<0.01), cSAgAPLP:LABL (p<0.05), and cHAPLP+cHALABL (p<0.05), while CD80 and CD86 expression in other APC subsets was constitutive (Figure S6). Interestingly, CD80 and CD86 expression were also dampened in B cells, ABCs, and DCs following treatment with cHA (B cells and ABCs p<0.05, DCs ^<0.001), cHALABL (p<0.0001), cHA+plp+LABL (p<0.001), and SAgAPLP:LABL (B cells and ABCs p<0.0001, DCs p<0.001). CD86 expression in APCs was dampened to a similar extent following treatment with these groups, with the exception of SAgALp:LABL-treated DCs. However, of all these groups, only the polymer arrays cHALABL, SAgAPLP:LABL, cHAPLP, cSAgAPLP:LABL, and cHAPLP+cHALABL dampened costimulatory signaling in both cases of ex vivo treatment at peak of disease and in vivo treatment.
Figure 4.
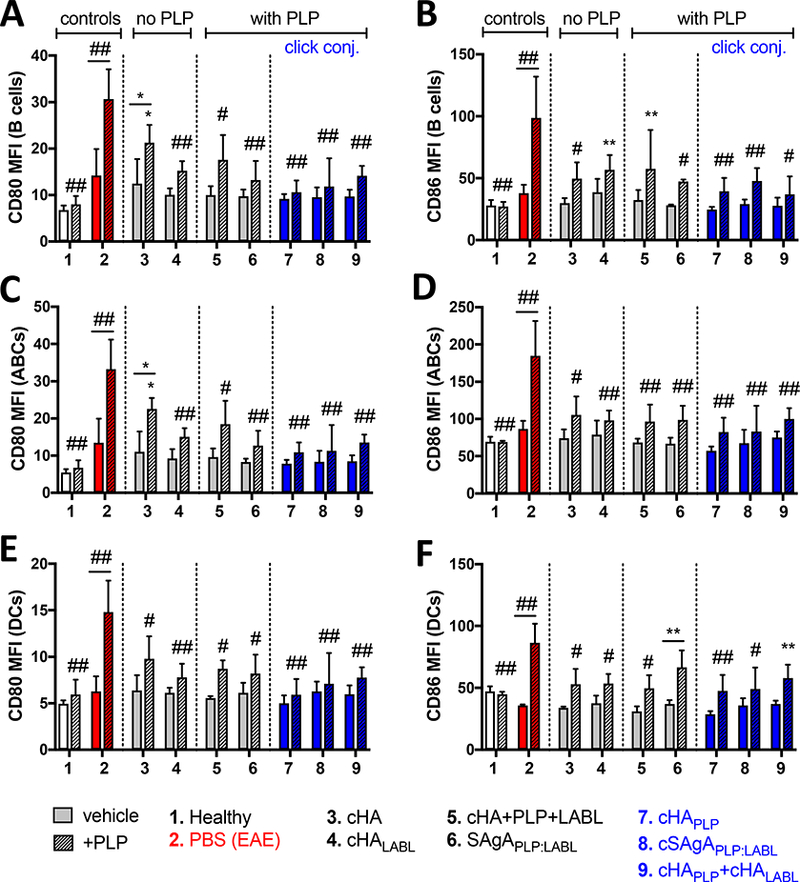
CD80/CD86 costimulatory marker expression in splenocytes isolated from EAE mice on day 25 following in vivo treatment with groups 3–9 and 72h ex vivo antigen rechallenge +/− PLP (25 μM), determined by flow cytometry: A/B) B cells (CD19+), C/D) ABCs (CD 19+B220+CD 11c+), and E/F) DCs (CD11c+). Data represent mean ± SD (n=3). Statistical significance compared to PBS negative control was determined by two-way ANOVA followed by Dunnett’s post hoc test; statistical significance between +/− PLP (underlined) was determined by two-way ANOVA followed by Sidak’s post hoc test (*p<0.05, **p<0.01, #p<0.001, ##p<0.0001). MFI = mean fluorescent intensity.
Flow cytometry forward scatter (FSC), which is a measure of cell volume and can be indicative of activation as lymphocytes mature into larger lymphoblasts, was also evaluated as a measure of cell stimulation after treatment. Indeed, FSC was significantly increased upon plp rechallenge in untreated T cells (p<0.01) and ABCs (p<0.05) isolated from EAE mice at peak of disease (Figure S7), and in T cells (p<0.05), B cells (p<0.01), ABCs (p<0.05), and DCs (p<0.0001) isolated from untreated EAE mice on day 25 (Figure S8). ABCs in particular are characterized by high FSC and side scatter (SSC) profiles.34 A high FSC ABC population was observed exclusively in EAE untreated cells and nontherapeutic groups (3–6) following ex vivo treatment at peak of disease (Figure S7E and F). In contrast, cells treated ex vivo with cHAPLP, cSAgAPLP:ABL, and cHAPLP+cHALABL lacked a high FSC ABC population, exhibiting scatter profiles that closely resembled that of healthy cells. Similarly, in vivo treatment with cHAPLP, cSAgAPLP:LABL, and cHAPLP+cHALABL caused a significant reduction in ABC FSC compared to the PBS negative control, producing ABC scatter profiles that resembled healthy cells (Figure S8E and F). Furthermore, T cell FSC was significantly reduced compared to the untreated control following ex vivo treatment with cHALABL, SAgAPLP:LABL, cHAPLP, cSAgAPLP:LABL, and cHAPLP+cHALABL (p<0.0001) (Figure S7A), directly mirroring CD86 expression in APCs (Figure 3). T cell and B cell FSC were also significantly reduced following in vivo treatment with SAgAPLP:LABL, cHAPLP, cSAgAPLP:LABL, and cHAPLP+cHALABL (Figure S8A and B). These FSC trends align with our observations of reduced costimulatory signaling and suggest inhibited T cell and B cell activation in these treatment groups.
3.4. Click-conjugated soluble antigen arrays caused a shift toward a tolerogenic cytokine response
Key Th1 and Th2 cytokines were measured in EAE splenocytes upon PLP rechallenge on day 25 following in vivo treatment to evaluate the T cell effector response. Regulatory cytokines IL-4 and IL-10 were significantly increased following in vivo treatment with cHA+PLP+LABL, SAgAPLP:LABL, cHAPLP, cSAgAPLP:LABL, and cHAPLP+cHALABL (Figure 5A and B), while IL-6 and GM-CSF were significantly increased following treatment with cHA+PLP+LABL, SAgAPLP:LABL, cSAgAPLP:LABL, and cHAPLP+cHALABL (Figure 5C and D). Trends were similar across these four cytokines, with the highest levels observed after treatment with cSAgAPLP:LABL and cHAPLP+cHALABL. Compared to the PBS negative control, IL-10 was significantly increased following treatment with cHAPLP (p<0.05), cSAgAPLP:LABL (p<0.05), and cHAPLP+cHALABL (p<0.0001); IL-4 was significantly increased following treatment with cHAPLP+cHALABL (p<0.05); GM-CSF was significantly increased following treatment with cSAgAPLP:LABL (p<0.05) and cHAPLP+cHALABL (p<0.01). Inflammatory cytokines IL-2, IFNγ, and IL-17 were unchanged compared to the PBS negative control and ΓΡΝγ was increased upon PLP rechallenge following treatment with all groups (Figure 5E, F, and G). However, treatment with cHAPLP, cSAgAPLP:LABL, and cHAPLP+cHALABL led to the smallest increase in IL-17 upon PLP rechallenge compared to other groups (Figure 5G and S8A) and the overall cytokine balance was weighted toward a regulatory response as indicated by the ratio of IL-10/IL-17 (Figure 5H), IL-17/IL-4 (Figure S9B), and IL-10/IL-2 (Figure S9C).
Figure 5.
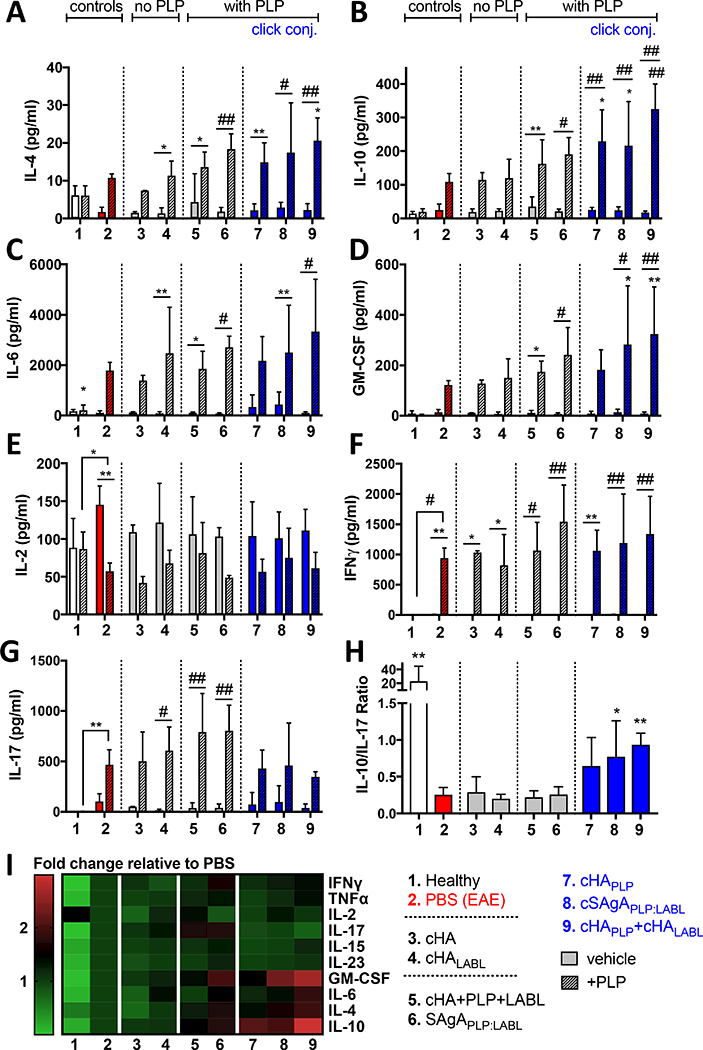
Cytokine response in EAE splenocytes isolated on day 25 following in vivo treatment of EAE mice with groups 3–9 and rechallenged ex vivo +/− PLP (25 μM) for 120h: A) IL-4, B) IL-6, C) IL-10, D) GM-CSF, E) IL-2, F) IFNy, G) IL-17, H) ratio of IL-10 to IL-17 in presence of PLP (Treg:Th17 balance), I) heat map of all cytokine responses in presence of PLP (fold change relative to PBS control). Data represent mean ± SD (n=5). Statistical significance compared to PBS negative control was determined by two-way ANOVA (A-G) or ordinary one-way ANOVA (H) followed by Dunnett’s post hoc test; statistical significance between +/− PLP (underlined) was determined by twoway ANOVA followed by Sidak’s post hoc test (*p<0.05, **p<0.01, #p<0.001).
3.5. Cytokine shift was accompanied by altered B cell compartment and increased humoral response
Alongside cytokines, composition and antibody response were evaluated in EAE splenocytes upon PLP rechallenge following in vivo treatment. The frequency of PLP-specific IgG antibody-secreting cells was significantly increased following in vivo treatment with SAgAPLP:LABL, cHAPLP, cSAgAPLP:LABL, and cHAPLP+cHALABL compared to the PBS negative control (p<0.05) (Figure 6A). Treatment with these groups also led to an increase in overall splenic B cells (% CD19+ splenocytes) (SAgAPLP:LABL, p<0.05) (Figure 6B) accompanied by a decrease in ABCs within the B cell compartment (% CD19+B220+CD11c+ B cells) upon PLP rechallenge (cHAPLP and cSAgAPLP:LABL, p<0.05) (Figure 6C) compared to the negative control. Of note, the composition within the B cell compartment following treatment with SAgAPLP:LABL, cHAPLP, cSAgAPLP:LABL, and cHAPLP+cHALABL resembled that of healthy splenocytes, where % B cells was significantly increased and % ABCs significantly decreased compared to the EAE PBS negative control (p<0.01). While T cell composition remained unchanged following in vivo treatment with these groups (Figure S10F), % T cells (CD3+ splenocytes) was significantly reduced following ex vivo treatment with cHAPLP (p<0.05) and cHAPLP+cHALABL (p<0.01) at peak of disease (Figure S10A). Overall, the concurrent increase in antibody production, expansion of B cells, and reduction of Th1-associated ABCs aligned with observations of increased Th2-associated cytokines in cHAPLP, cSAgAPLP:LABL, and cHAPLP+cHALABL treatment groups.
Figure 6.
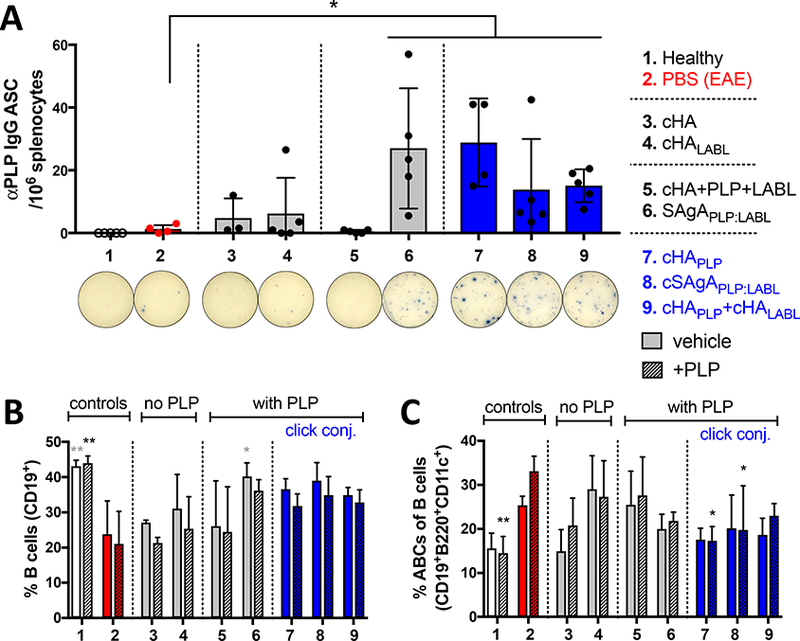
B cell composition and humoral response in EAE splenocytes isolated on day 25 following in vivo treatment of EAE mice with groups 3–9, compared to healthy and PBS negative controls: A) ELISPOT for PLP IgG antibody secreting cells (ASC) per 106 splenocytes, showing representative wells. Data represent mean ± SD (n=5). B/C) Splenocyte composition following in vivo treatment and 72h ex vivo antigen rechallenge +/− PLP (25 μM), determined by flow cytometry: B) % B cells (CD19+) overall, and C) % ABCs (CD19+B220+CD11c+) of gated B cells. Data represent mean ± SD (n=3). Statistical significance compared to PBS negative control was determined by paired t-test (A) or two-way ANOVA followed by Dunnett’s post hoc test (B,C) (*p<0.05, **p<0.01).
4. DISCUSSION
In our previous work, we evaluated the cellular mechanism of SAgAs using an immortalized B cell line. Our results suggested SAgAs induced B cell anergy, as indicated by BCR targeting, sustained SAgA binding on the cell surface, and dampened BCR-mediated signaling.29–30 Here, we utilized the EAE mouse model to elucidate therapeutic and immunological mechanisms of SAgAs by comparing HA, PLP, and LABL controls to various multimeric SAgA constructs (Table 1).
Our in vivo results established that click-conjugated soluble antigen arrays containing PLP (cSAgAPLP:LABL, cHAPLP+cHALABL, and cHAPLP) were significantly therapeutic against EAE (Figure 1). The hydrolyzable array (SAgAPLP:LABL) was less potent at the same dose, while the physical mixture (HA+PLP+LABL) and polymer arrays lacking PLP (cHA, cHALABL) were not therapeutic. From this, we concluded the molecular properties of (1) multivalent presentation of antigen and (2) non-hydrolyzable conjugation resulted in significant therapeutic efficacy in EAE. Our previous results would suggest this therapeutic effect was driven by high avidity, antigen-specific binding to the BCR, which inhibited BCR-mediated signaling.29–30 We previously showed that non-hydrolyzable click-conjugated cSAgAPLP:LABL could achieve greater efficacy than hydrolyzable SAgAPLP:LABL at the same and even lower doses,30 which was substantiated by the results shown here (Figure 1E, Figure S4G). Of note, cHAPLP (conjugation of ~15 PLP per backbone) exhibited significant therapeutic efficacy in vivo, to a similar extent as cSAgAPLP:LABL (conjugation of ~10 PLP and ~10 LABL per backbone). We previously proposed that cSAgAPLP:LABL’s enhanced binding avidity might be attributed to the combined affinities of PLP and LABL (binding to BCR and ICAM-1, respectively).30 These new results suggest that non-hydrolyzable conjugation of PLP antigen at a slightly higher valency may enhance binding avidity to a similar extent as the two signals combined. Thus, it will be important to further investigate valency effects in the future.
Antigen arrays fcSAgAPLP:LABL and fcHAPLP preferentially targeted APCs including B cells, ABCs, and DCs, exhibiting the highest amount of binding to ABCs (Figure 2). B cells were the largest population targeted given their high frequency in the spleen, and likely the predominant APC population targeted given emerging evidence that supports their critical role as APC during induction and propagation of autoimmune diseases such as MS.7–10, 38 Importantly, all APC subtypes were targeted in a disease-specific manner, as binding was significantly higher in EAE APCs than healthy APCs.
Furthermore, both in vivo and ex vivo treatment with cHAPLP, cSAgAPLP:LABL, and cHAPLP+cHALABL reduced costimulatory signaling in APCs, causing them to behave more like healthy cells that were non-responsive to antigen rather than activated APCs that exhibited antigenic memory. In B cells and ABCs, this response was indicative of anergy, which is induced when B cells encounter antigen in the absence of a secondary costimulatory signal (i.e., from TLRs or CD40L on T cells) in concert with continuous BCR binding and clustering, leading to impaired BCR signaling and downregulation of costimulatory markers CD80 and CD86.17–21 This immunological mechanism is consistent with our previously published work where cSAgAs exhibited BCR binding and clustering on the cell surface, resulting in B cells with reduced calcium flux.29–30 Importantly, as CD80 and CD86 expression must be upregulated upon antigen stimulation for B cells to function as effective APCs, anergic B cells are no longer able to activate antigen-specific T cells.22 Based on the two-signal requisite of the immunological synapse, if a T cell receives primary antigenic signal (via MCHII) in the absence of secondary costimulatory signal (i.e. CD80/86), it presumes self-antigen ‘recognition’ and becomes anergic, failing to mount a response to the antigen in future encounters.2–5 Induction of B cell anergy can therefore have a two-fold therapeutic effect, by inducing both an effector B cell population that is not responsive to autoantigen and B cell APCs with reduced antigen presentation capacity leading to insufficient T cell activation.
It is particularly significant to note that costimulatory marker expression was downregulated to a healthy level even in ABCs, which typically express very high levels of CD80/86 upon activation - much higher than normal B cells, DCs, or follicular B cells.33–35 Localized at the T:B cell border in the spleen, ABCs are potent APCs that exhibit enhanced ability to present antigen and stimulate antigen-specific T cell activation and proliferation compared to follicular B cells. Beyond their in vivo localization, in vitro studies have shown that ABCs are able to efficiently present antigen not because they uptake and present antigen at a higher rate, but rather because they express higher levels of MHCII and CD80/86.33 Thus, downregulation of costimulatory signals on ABCs should have a profound inhibitory effect on antigen presentation and T cell activation. To our knowledge, this is the first example of an application that targets and modulates the effector phenotype of this newly recognized ABC population. Combined, these results indicated that treatment with cSAgAPLP:LABL, cHAPLP, and cHAPLP+cHALABL induced PLP-specific anergy in B cells and ABCs, resulting in reduced APC capacity and tolerance to autoantigen.
DCs also showed reduced costimulatory signaling following treatment with click-conjugated PLP arrays, phenomena that could arise from several pathways. One possibility is through DC phagocytosis of apoptotic anergic B cells or T cells, resulting in presentation of molecular signatures on the DC surface that signal to phagocytic APCs to process and present cell contents in a tolerogenic context.4 Alternatively (or in conjunction), dampened costimulatory marker expression may be a downstream effect of elevated regulatory cytokines such as IL-10 and GM-CSF, which have been associated with reduced expression of CD80 and CD86 on DCs.39–41 Modulation of DC costimulatory marker expression could also contribute to the dampened B cell response through altered DC antigen presentation to B cells.
In addition to signals 1 and 2, cytokines are recognized as a third ‘chemical signal’ that is essential for providing context to antigen presentation for T cell activation, as cytokine milieus shape the resulting T cell effector response. In MS, a cell-mediated Th1 and inflammatory Th17 response is associated with worsening disease while a humoral Th2 and tolerogenic Treg response is associated with improving disease.42–43 IL-2, TNFα, ΓΡΝγ, and IL-17 are key cytokines associated with a Th1/Th17 response while IL-10, IL-4, and IL-6 are associated with anergy and a Th2/Treg response.44–46 Lower levels of IL-10 in particular are observed in untreated MS patients compared to healthy individuals, implying that upregulation has a therapeutic effect against the disease.47 Administration of recombinant IL-10 has even been shown to prevent the development of EAE in rats.48 GM-CSF has also been shown to promote expansion of regulatory lymphocytes, induce high levels of Th2-associated cytokines such as IL-10, and favor the development of ‘tolerogenic’ DCs with reduced CD80 and CD86 expression.39–41 Notably, in vivo treatment with cSAgAPLP:LABL, cHAPLP, and cHAPLP+cHALABL induced high levels of these cytokines associated with anergy and a regulatory Th2 response (IL-10, IL-4, IL-6, and GM-CSF) while reducing relative levels of IL-17.
A Th2 response is also associated with B cell expansion and increased antibody production, largely regulated by IL-10 and IL-4 producing B cells.45, 49–51 We observed changes in the B cell compartment consistent with elevated IL-10 and IL-4 where the frequency of B cells increased and yet ABCs decreased in therapeutic treatment groups (cHAPLP, cSAgAPLP:LABL, and cHAPLP+cHALABL). Notably, this shift resembled the composition of healthy splenocytes and contrasted that of EAE splenocytes. The accompanying increase in PLP-specific IgG production may reflect an expansion of antibody-secreting plasma cells while antigen-presenting B cells (such as ABCs) were reduced. A correlation was also observed in IL-6 and GM-CSF, which support B cell survival and expansion and have been identified to promote regulatory B cell differentiation and IL-10 production.40–41, 46
In contrast to Th2-associated B cells, ABCs are stimulated by Th1 inflammatory cytokines and the resulting ABC-mediated antigen presentation tends to skew CD4 T cells toward Th17 polarization.36–37 In aged mice, ABCs have been shown to produce high levels of the inflammatory cytokine TNFα, which can be inhibited by IL-10. Increased numbers of ABCs within the B cell compartment may even contribute to increasing inflammation with age.52 While we did not observe a reduction in TNFa (Figure S9D), we did observe a relative decrease in IL-17 levels alongside increased IL-10 in therapeutic groups, which may have driven alteration of the B cell compartment by reducing Th1-associated ABCs.
Regulatory cytokine trends were mirrored by an increase in autoantibody production following in vivo treatment with cSAgAPLP:LABL, cHAPLP, and cHAPLP+cHALABL. Along with the altered B cell compartment and decrease in APC costimulatory signaling, this increase in PLP autoantibody production further supported a shift away from a Th1 cell-mediated response toward a Th2 humoral-mediated response. Unlike humoral-mediated autoimmune diseases such as SLE and rheumatoid arthritis, increased autoantibody production in Th1-mediated autoimmune diseases such as MS and T1D has been associated with a shift toward a therapeutic Th2 response rather than propagation of disease.7,31,53–55 We have observed a dose-dependent correlation between autoantibody response and reduced EAE clinical score (Figure S11). Others have made similar observations; for example, IL-10 treatment in rats both prevented the development of EAE and caused a coinciding elevation of myelin-specific autoantibodies.48 Likewise, Wraith et al reported various effective allergen-specific immunotherapies that caused increased IL-10 and IgG secretion, even elevated levels of IFNγ and TNFα, but still induced allergen-specific tolerance.56 Interestingly, B cells from mice treated with glatiramer acetate (an FDA-approved drug with clinical success against MS) exhibited reduced cell surface expression of CD80/86, higher levels of IL-10, and increased IgG1 autoantibody production, similar to our observations with cHAPLP, cSAgAPLP:LABL, and cHAPLP+cHALABL.38
Thus, the groups that achieved significant therapeutic efficacy shared cornerstone molecular properties (click-conjugated, non-hydrolyzable, multivalent PLP in a soluble polymer array) and exhibited a unique immune response ‘fingerprint’: (1) high avidity binding and disease-specific targeting of APCs, (2) downregulation of costimulatory signals in APCs, (3) a shift toward regulatory cytokines, and (4) an increased humoral response. Building upon our previous work, we can thus propose a probable therapeutic immunological mechanism (Figure 7). Click-conjugated PLP arrays first target antigen-specific APCs, in particular B cells and ABCs, through high avidity binding driven by the antigen (PLP) for the BCR. The arrays act through sustained BCR engagement, exhibiting prolonged residence on the cell surface (observed previously).29 Continuous antigen occupation and clustering of the BCR in the absence of costimulatory signal leads to B cell anergy, resulting in dampened calcium flux and impaired BCR signaling (observed previously)30 followed by downregulation of costimulatory markers CD80 and CD86. Downregulation of CD80/CD86 (critical ‘signal 2’ for antigenic T cell activation) generates APCs with reduced capacity to activate T cells. This is accompanied by an increase in regulatory cytokines IL-10 and IL-4 (context ‘signal 3’ for antigenic T cell activation) and increased humoral response, shifting the cytokine balance from Th1/Th17 towards a Th2/regulatory phenotype. Therefore, click-conjugated soluble antigen arrays target disease-specific B cells using signal 1 (antigen) to downregulate signal 2 (costimulation), shifting the balance in signal 3 (cytokines) and the resulting effector response to induce an environment of autoantigenic tolerance.
Figure 7.
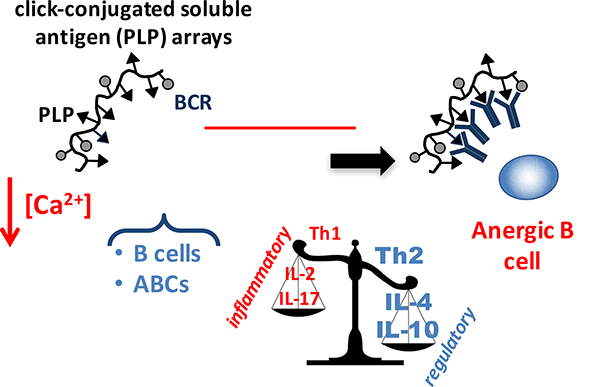
Schematic showing proposed therapeutic mechanism: Click-conjugated soluble antigen arrays containing PLP (cSAgAPLP:LABL, cHAPLP, and cHAPLP+cHALABL) target EAE-specific B cells and autoimmune-associated B cells (ABCs) through high avidity binding that is driven by the antigen (PLP) for the B cell receptor (BCR). The molecule is retained on the cell surface and through continuous engagement and clustering of BCR, induces B cell anergy, resulting in reduced ability to mobilize calcium and impaired BCR signaling. This leads to downregulated expression of costimulatory markers CD80 and CD86 (critical ‘signal 2’ for antigenic T cell activation) and is accompanied by a shift from a Th1/Th17 to a Th2/Treg weighted cytokine response (context ‘signal 3’ for antigenic T cell activation). Thus, the induced anergic B cell phenotype produces APCs without the capacity to activate T cells against autoantigen and results in an environment of autoantigenic tolerance.
5. CONCLUSIONS
In summary, we have demonstrated that constructing soluble antigen arrays utilizing non-hydrolyzable linkers imbues key molecular properties that lend to therapeutic efficacy in vivo and antigenic tolerance in a murine model of multiple sclerosis. Cellular response assays indicated the effect was driven by multivalent SAgAs binding to APCs, in particular a subset of autoimmune B cells, which reduced expression of costimulatory signals on APCs and skewed cytokines toward a Th2/regulatory phenotype. This ‘fingerprint’ indicative of autoantigenic tolerance was observed when EAE mice were treated in vivo and also when splenocytes from EAE mice were treated ex vivo. To expand upon the core findings in this paper, future studies could test the durability of tolerance during relapse as well as extrapolation of SAgA-induced anergy as a mechanism of therapeutic tolerance in other models of autoimmunity. Here, design features of soluble antigen arrays have been linked to immunological mechanisms to inform and guide the design of future ASITs.
Supplementary Material
HIGHLIGHTS.
Professional antigen presenting cells and B cells in particular, contribute to autoimmunity by recognizing and presenting autoantigen, directing effector cell response, and contributing to the cytokine milieu.
Delivering autoantigen epitope as soluble antigen arrays (SAgAs) skewed disease-specific B cells toward anergy to restore a healthy phenotype in mice with EAE.
SAgAs engaged a potent subset of B cells called autoimmune-associated B cells with high avidity specificity, and subsequently downregulated costimulatory signaling and shifted cytokines toward a Th2/regulatory immune response.
SAgA induction of anergy was driven by linking multiple copies of autoantigen epitope to hyaluronan via a non-degradable linker, which should provide insight into the design of novel antigen-specific immunotherapies.
ACKNOWLEDGEMENTS
We gratefully acknowledge support from the National Institutes of Health Graduate Training Program in Dynamic Aspects of Chemical Biology Grant (T32 GM008545) from the National Institutes of General Medical Sciences (C.J.P. and M.L.), the Madison and Lila Self Graduate Fellowship at the University of Kansas (B.L.H. and J.D.G.), and the Howard Rytting pre-doctoral fellowship from the Department of Pharmaceutical Chemistry at the University of Kansas (C.J.P.). We would like to thank Dr. Blake Peterson for graciously donating the Pennsylvania Green parent compound. Additionally, we thank the Macromolecule and Vaccine Stabilization Center, KU NMR Lab, Microscopy and Analytical Imaging Core Lab, and the Kansas Vaccine Institute at the University of Kansas for their collaboration and instrument use.
Footnotes
CONFLICT OF INTEREST
C.J.B. is a co-founder and Chairman of the Board of Orion BioScience. Orion has licensed the rights to patents claiming soluble antigen arrays.
SUPPORTING INFORMATION AVAILABLE
The synthesis, purification, and analytical characterization of Penn Green-Alk, HA-N3, and cSAgAs is detailed in the Supplementary Methods, along with protocols for the EAE in vivo dosing study and ex vivo Resazurin assay. Figure S1 presents a schematic of the cSAgAPLP:LABL synthesis and a sample chromatogram to illustrate quantification of peptide and fluorophore conjugation. Figure S2 details the flow cytometry gating scheme used to identify antibody-labeled splenocytes and maximum SS binding. Figure S3 elaborates upon the methods used to evaluate binding. Figure S4 presents incidence of disease and mortality rate from the EAE in vivo studies in Figure 1, along with in vivo results from a separate study evaluating treatment with soluble PLP compared to SAgAPLP:LABL and cSAgAPLP:LABL. Figure S5 shows results of a Resazurin assay to evaluate EAE and healthy murine splenocyte metabolism following ex vivo treatment. Figure S6 shows CD80 expression in B cells following ex vivo and in vivo treatment. Figures S7 and S8 detail the FSC of splenocyte subpopulations determined by flow cytometry following ex vivo and in vivo treatment, respectively. Figure S9 includes additional cytokine responses (TNFα, IL-15, IL-23) following in vivo treatment. Figure S10 shows the relative composition of splenocyte subpopulations following ex vivo and in vivo treatment. Figure S11 presents a correlation between ELISPOT and EAE clinical disease score AUC results from a cSAgAPLP:LABL in vivo dosing study used to inform the 50 nmol PLP dose used in this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Kontos S; Grimm AJ; Hubbell JA, Engineering antigen-specific immunological tolerance. Curr. Opin. Immunol 2015, 35, 80–88. [DOI] [PubMed] [Google Scholar]
- 2.Mueller DL; Jenkins MK; Schwartz RH, Clonal expansion versus functional clonal inactivation: a costimulatory signalling pathway determines the outcome of T cell antigen receptor occupancy. Annu. Rev. Immunol 1989, 7 (1), 445–480. [DOI] [PubMed] [Google Scholar]
- 3.Feldmann M; Steinman L, Design of effective immunotherapy for human autoimmunity. Nature 2005, 435 (7042), 612. [DOI] [PubMed] [Google Scholar]
- 4.Miller SD; Turley DM; Podojil JR, Antigen-specific tolerance strategies for the prevention and treatment of autoimmune disease. Nat. Rev. Immunol 2007, 7 (9), 665. [DOI] [PubMed] [Google Scholar]
- 5.Mueller DL, Mechanisms maintaining peripheral tolerance. Nat. Immunol 2010,11 (1), 21–27. [DOI] [PubMed] [Google Scholar]
- 6.Hampe CS, B cells in autoimmune diseases. Scientifica 2012, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wong FS; Wen L; Tang M; Ramanathan M; Visintin I; Daugherty J; Hannum LG; Janeway CA; Shlomchik MJ, Investigation of the role of B-cells in type 1 diabetes in the NOD mouse. Diabetes 2004, 53 (10), 2581–2587. [DOI] [PubMed] [Google Scholar]
- 8.Rivera A; Chen C-C; Ron N; Dougherty JP; Ron Y, Role of B cells as antigen-presenting cells in vivo revisited: antigen-specific B cells are essential for T cell expansion in lymph nodes and for systemic T cell responses to low antigen concentrations. International immunology 2001,13 (12), 1583–1593. [DOI] [PubMed] [Google Scholar]
- 9.O’Neill SK; Shlomchik MJ; Glant TT; Cao Y; Doodes PD; Finnegan A, Antigen-specific B cells are required as APCs and autoantibody-producing cells for induction of severe autoimmune arthritis. J. Immunol. 2005, 174 (6), 3781–3788. [DOI] [PubMed] [Google Scholar]
- 10.Serreze DV; Fleming SA; Chapman HD; Richard SD; Leiter EH; Tisch RM, B lymphocytes are critical antigen-presenting cells for the initiation of T cell-mediated autoimmune diabetes in nonobese diabetic mice. J. Immunol. 1998,161 (8), 3912–3918. [PubMed] [Google Scholar]
- 11.Jackson SW; Kolhatkar NS; Rawlings DJ, B cells take the front seat: dysregulated B cell signals orchestrate loss of tolerance and autoantibody production. Curr. Opin. Immunol. 2015, 33, 70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Youinou P; Hillion S; Jamin C; Pers J-O; Saraux A; Renaudineau Y, B lymphocytes on the front line of autoimmunity. Autoimmunity reviews 2006, 5 (3), 215–221. [DOI] [PubMed] [Google Scholar]
- 13.Claes N; Fraussen J; Stinissen P; Hupperts R; Somers V, B cells are multifunctional players in multiple sclerosis pathogenesis: insights from therapeutic interventions. Frontiers in immunology 2015, 6, 642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Franks SE; Getahun A; Hogarth PM; Cambier JC, Targeting B cells in treatment of autoimmunity. Curr. Opin. Immunol 2016, 43, 39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goodnow CC; Crosbie J; Adelstein S; Lavoie TB; Smith-Gill SJ; Brink RA; Pritchard-Briscoe H; Wotherspoon JS; Loblay RH; Raphael K, Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature 1988, 334 (6184), 676. [DOI] [PubMed] [Google Scholar]
- 16.Getahun A; O’Neill SK; Cambier JC, Establishing anergy as a bona fide in vivo mechanism of B cell tolerance. J. Immunol. 2009, 183 (9), 5439–5441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gauld SB; Benschop RJ; Merrell KT; Cambier JC, Maintenance of B cell anergy requires constant antigen receptor occupancy and signaling. Nat. Immunol. 2005, 6 (11), 1160–1167. [DOI] [PubMed] [Google Scholar]
- 18.Cambier JC; Gauld SB; Merrell KT; Vilen BJ, B-cell anergy: from transgenic models to naturally occurring anergic B cells? Nat. Rev. Immunol. 2007, 7 (8), 633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Merrell KT; Benschop RJ; Gauld SB; Aviszus K; Decote-Ricardo D; Wysocki LJ; Cambier JC, Identification of anergic B cells within a wild-type repertoire. Immunity 2006, 25 (6), 953–962. [DOI] [PubMed] [Google Scholar]
- 20.Healy JI; Dolmetsch RE; Timmerman LA; Cyster JG; Thomas ML; Crabtree GR; Lewis RS; Goodnow CC, Different nuclear signals are activated by the B cell receptor during positive versus negative signaling. Immunity 1997, 6 (4), 419–428. [DOI] [PubMed] [Google Scholar]
- 21.Yarkoni Y; Getahun A; Cambier JC, Molecular underpinning of B-cell anergy. Immunol. Rev. 2010, 237 (1), 249–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gauld SB; Merrell KT; Cambier JC, Silencing of autoreactive B cells by anergy: a fresh perspective. Curr. Opin. Immunol. 2006, 18 (3), 292–297. [DOI] [PubMed] [Google Scholar]
- 23.Hartley SB; Crosbie J, Elimination from peripheral lymphoid tissues of selfreactive B lymphocytes recognizing membrane-bound antigens. Nature 1991, 353 (6346), 765. [DOI] [PubMed] [Google Scholar]
- 24.Sestak J; Mullins M; Northrup L; Thati S; Forrest ML; Siahaan TJ; Berkland C, Single-step grafting of aminooxy-peptides to hyaluronan: a simple approach to multifunctional therapeutics for experimental autoimmune encephalomyelitis. J. Controlled Release 2013, 168 (3), 334–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sestak JO; Sullivan BP; Thati S; Northrup L; Hartwell B; Antunez L; Forrest ML; Vines CM; Siahaan TJ; Berkland C, Codelivery of antigen and an immune cell adhesion inhibitor is necessary for efficacy of soluble antigen arrays in experimental autoimmune encephalomyelitis. Mol. Ther.--Methods Clin. Dev. 2014, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sestak JO; Fakhari A; Badawi AH; Siahaan TJ; Berkland C, Structure, size, and solubility of antigen arrays determines efficacy in experimental autoimmune encephalomyelitis. AAPS J 2014, 16 (6), 1185–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thati S; Kuehl C; Hartwell B; Sestak J; Siahaan T; Forrest ML; Berkland C, Routes of Administration and Dose Optimization of Soluble Antigen Arrays in Mice with Experimental Autoimmune Encephalomyelitis. J. Pharm. Sci. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hartwell BL; Smalter Hall A; Swafford D; Sullivan BP; Garza A; Sestak JO; Northrup L; Berkland C, Molecular Dynamics of Multivalent Soluble Antigen Arrays Support a Two-Signal Co-delivery Mechanism in the Treatment of Experimental Autoimmune Encephalomyelitis. Mol. Pharmaceutics 2016, 13 (2), 330–343. [DOI] [PubMed] [Google Scholar]
- 29.Hartwell BL; Martinez-Becerra FJ; Chen J; Shinogle H; Sarnowski M; Moore DS; Berkland C, Antigen-Specific Binding of Multivalent Soluble Antigen Arrays Induces Receptor Clustering and Impedes B Cell Receptor Mediated Signaling. Biomacromolecules 2016, 17 (3), 710–722. [DOI] [PubMed] [Google Scholar]
- 30.Hartwell BL; Pickens CJ; Leon M; Berkland C, Multivalent Soluble Antigen Arrays Exhibit High Avidity Binding and Modulation of B Cell Receptor-Mediated Signaling to Drive Efficacy against Experimental Autoimmune Encephalomyelitis. Biomacromolecules 2017, 18 (6), 1893–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Northrup L; Griffin JD; Christopher MA; Antunez LR; Hartwell BL; Pickens CJ; Berkland C, co-delivery of autoantigen and dexamethasone in incomplete Freund’s adjuvant ameliorates experimental autoimmune encephalomyelitis. J. Controlled Release 2017, 266, 156–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fleming KK; Bovaird JA; Mosier MC; Emerson MR; LeVine SM; Marquis JG, Statistical analysis of data from studies on experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2005, 170 (1), 71–84. [DOI] [PubMed] [Google Scholar]
- 33.Rubtsov AV; Rubtsova K; Kappler JW; Jacobelli J; Friedman RS; Marrack P, CD11c-expressing B cells are located at the T cell/B cell border in spleen and are potent APCs. J. Immunol. 2015, 195 (1), 71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rubtsov AV; Rubtsova K; Fischer A; Meehan RT; Gillis JZ; Kappler JW; Marrack P, Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c+ B-cell population is important for the development of autoimmunity. Blood 2011,118 (5), 1305–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rubtsov AV; Rubtsova K; Kappler JW; Marrack P, TLR7 drives accumulation of ABCs and autoantibody production in autoimmune-prone mice. Immunol. Res 2013, 55 (1–3), 210–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Naradikian MS; Hao Y; Cancro MP, Age-associated B cells: key mediators of both protective and autoreactive humoral responses. Immunol. Rev. 2016, 269 (1), 118–129. [DOI] [PubMed] [Google Scholar]
- 37.Hao Y; O’Neill P; Naradikian MS; Scholz JL; Cancro MP, A B-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood 2011,118 (5), 1294–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kala M; Rhodes SN; Piao W-H; Shi F-D; Campagnolo DI; Vollmer TL, B cells from glatiramer acetate-treated mice suppress experimental autoimmune encephalomyelitis. Exp. Neurol 2010, 221 (1), 136–145. [DOI] [PubMed] [Google Scholar]
- 39.Parmiani G; Castelli C; Pilla L; Santinami M; Colombo M; Rivoltini L, Opposite immune functions of GM-CSF administered as vaccine adjuvant in cancer patients. Annals of Oncology 2006, 18 (2), 226–232. [DOI] [PubMed] [Google Scholar]
- 40.Pulendran B; Banchereau J; Burkeholder S; Kraus E; Guinet E; Chalouni C; Caron D; Maliszewski C; Davoust J; Fay J, Flt3-ligand and granulocyte colony-stimulating factor mobilize distinct human dendritic cell subsets in vivo. J. Immunol. 2000, 165 (1), 566–572. [DOI] [PubMed] [Google Scholar]
- 41.Sheng JR; Quan S; Soliven B, CD1dhiCD5+ B Cells Expanded by GM-CSF In Vivo Suppress Experimental Autoimmune Myasthenia Gravis. J. Immunol. 2014, 193 (6), 2669–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fletcher JM; Lalor S; Sweeney C; Tubridy N; Mills K, T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin. Exp. Immunol. 2010, 162 (1), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Legroux L; Arbour N, Multiple sclerosis and T lymphocytes: an entangled story. J. Neuroimmune Pharmacol. 2015, 10 (4), 528–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mauri C; Bosma A, Immune regulatory function of B cells. Annu. Rev. Immunol 2012, 30, 221–241. [DOI] [PubMed] [Google Scholar]
- 45.Carter NA; Rosser EC; Mauri C, Interleukin-10 produced by B cells is crucial for the suppression of Th17/Th1 responses, induction of T regulatory type 1 cells and reduction of collagen-induced arthritis. Arthritis research & therapy 2012, 14 (1), R32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rosser EC; Oleinika K; Tonon S; Doyle R; Bosma A; Carter NA; Harris KA; Jones SA; Klein N; Mauri C, Regulatory B cells are induced by gut microbiota-driven interleukin-ΐβ and interleukin-6 production. Nat. Med. 2014, 20 (11), 1334. [DOI] [PubMed] [Google Scholar]
- 47.Ozenci V; Kouwenhoven M; Huang Y; Xiao B; Kivisakk P; Fredrikson S; Link H, Multiple Sclerosis: Levels of Interleukin-10-Secreting Blood Mononuclear Cells are Low in Untreated Patients but Augmented During Interferon-b-1b Treatment. Scandinavian journal of immunology 1999, 49 (5), 554. [DOI] [PubMed] [Google Scholar]
- 48.Rott O; Fleischer B; Cash E, Interleukin-10 prevents experimental allergic encephalomyelitis in rats. Eur. J. Immunol. 1994, 24 (6), 1434–1440. [DOI] [PubMed] [Google Scholar]
- 49.Lund FE, Cytokine-producing B lymphocytes—key regulators of immunity. Curr. Opin. Immunol. 2008, 20 (3), 332–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hurdayal R; Ndlovu HH; Revaz-Breton M; Parihar SP; Nono JK; Govender M; Brombacher F, IL-4-producing B cells regulate T helper cell dichotomy in type 1-and type 2-controlled diseases. Proc. Natl. Acad. Sci. U. S. A. 2017, 201708125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Luu VP; Vazquez MI; Zlotnik A, B cells participate in tolerance and autoimmunity through cytokine production. Autoimmunity 2013, 47 (1), 1–12. [DOI] [PubMed] [Google Scholar]
- 52.Ratliff M; Alter S; Frasca D; Blomberg BB; Riley RL, In senescence, age-associated B cells secrete TNFa and inhibit survival of B-cell precursors. Aging cell 2013, 12 (2), 303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Y; Krieg AM, Induction of autoantibody production but not autoimmune disease in HEL transgenic mice vaccinated with HEL in combination with CpG or control oligodeoxynucleotides. Vaccine 2004, 22 (20), 2641–2650. [DOI] [PubMed] [Google Scholar]
- 54.Lin MS; Hubert MT; Delmastro MM; Bertera S; Wong CT; Lakomy R; He J; Sklavos MM; Coudriet GM; Pietropaolo M, A multivalent vaccine for type 1 diabetes skews T cell subsets to Th2 phenotype in NOD mice. Immunol. Res 2011, 50 (2–3), 213–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Orban T; Farkas K; Jalahej H; Kis J; Treszl A; Falk B; Reijonen H; Wolfsdorf J; Ricker A; Matthews JB, Autoantigen-specific regulatory T cells induced in patients with type 1 diabetes mellitus by insulin B-chain immunotherapy. Journal of autoimmunity 2010, 34 (4), 408–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sabatos-Peyton CA; Verhagen J; Wraith DC, Antigen-specific immunotherapy of autoimmune and allergic diseases. Curr. Opin. Immunol. 2010, 22 (5), 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.