

Abstract
Introduction:
Chromatin remodeling complexes play important roles in the control of genome regulation in both normal and diseased states, and are therefore critical components for the regulation of epigenetic states in cells. Given the role epigenetics plays in cancer, for example, chromatin remodeling complexes are routinely targeted for therapeutic intervention.
Areas covered:
Protein mass spectrometry and proteomics are powerful technologies used to study and understand chromatin remodeling. While impressive progress has been made in this area, there remain significant challenges in the application of proteomic technologies to the study of chromatin remodeling. As parts of large multisubunit complexes that can be heavily modified with dynamic post-translational modifications, challenges in the study of chromatin remodeling complexes include defining the content, determining the regulation, and studying the dynamics of the complexes under different cellular states.
Expert Commentary:
Important considerations in the study of chromatin remodeling complexes include the complexity of sample preparation, the choice of proteomic methods for the analysis of samples, and data analysis challenges. Continued research in these three areas promise to yield even greater insights into the biology of chromatin remodeling and epigenetics and the dynamics of these systems in human health and cancer.
Keywords: quantitative proteomics, mass spectrometry, protein interaction networks, chromatin remodeling, histone modifications, cancer, epigenetics
1. Introduction
Eukaryotic cells pack their massive genetic material into a few microns by forming a highly organized polymeric complex, known as chromatin. The benefits of forming this highly compact chromatin structure is not only to save space, but more importantly, to allow the maintenance and use of genetic information to be tightly and efficiently regulated. Cells containing the same genome can be phenotypically different from one another, a phenomenon known as epigenetics [1]. In order to have the right machinery access the right place on chromatin at the correct time, eukaryotic cells have developed complicated regulation mechanisms. A key route to understand how chromatin activities are regulated is through investigating chromatin associated and remodeling proteins. Chromatin remodeling proteins are typically parts of large multi-subunit complexes with a large variety of functions concentrated on the regulation of histones and their positioning on DNA (Figure 1). Chromatin remodeling complexes have been defined and studied using many experimental approaches [2]. Importantly, chromatin remodeling proteins are commonly mutated in cancer and are important targets for therapeutic intervention [3]. Therefore, further study of the content of chromatin complexes and proteins, their regulation, and their dynamics are important for a better understanding of their roles in human health and cancer (Figure 2). Protein mass spectrometry and proteomics has played an important role in the study of chromatin remodeling. However, given the diversity of chromatin remodeling complexes and chromatin associated proteins, important challenges continue to exist for their study. Aspects like choice of proteomic method, sample preparation, and data analysis are important considerations. Excellent recent reviews on epigenetics [4] and chromatin immunoprecipitation coupled to proteomics [5] have recently been written. There is some overlap of topics with the current review and these recent works, which is the case for nearly all reviews on chromatin and proteomics. Here we sought to cover additional topics including the coupling of proteomics to structural biology, the study of chromatin dynamics using quantitative proteomics, and the methods used to analyze network datasets. In addition, an important consideration covered here is the current and future impact of proteomics for the study of chromatin remodeling complexes and their role in cancer.
Figure 1. Overview of composition and function of selected chromatin remodeling complexes.
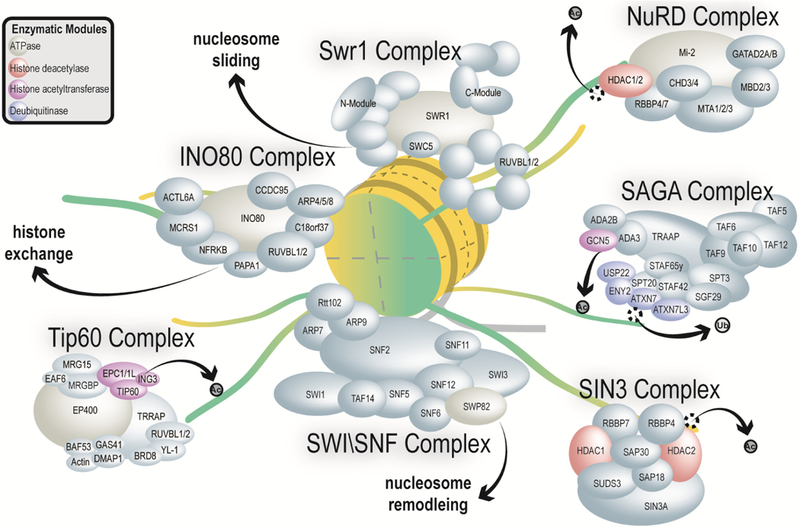
General architectural representations of chromatin remodeling complexes studied using proteomics technologies is shown. These complexes include the Swr1 complex, the NuRD complex, the SAGA complex, the Sin3 complex, the Swi/Snf complex, the Tip60 complex, and the Ino80 complex. A generalized representation of the histone octamer is shown in yellow and green and the general biological function of each complex is shown. HDAC 1 and HDAC2 are highlighted in pink since they are parts of multiple complexes. The drawing is not to scale and only reflects relative sizes of the subunits. A single schematic nucleosome is shown (yellow), an acetyl group is represented by Ac and ubiquitin is represented by Ub.
Figure 2. Overview of chromatin relationship to cancer.
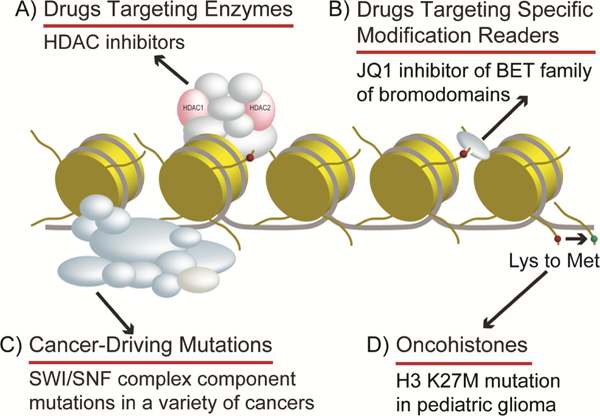
Histones and chromatin associated proteins play a variety of roles in cancer. Histones and many chromatin associated proteins are amongst the most frequently mutated genes in human cancers [83] and this has resulted in great interest in therapeutic targeting of these proteins for the treatment of cancers [3]. A) Drug targeting enzymes like HDAC inhibitor suberoylanilide hydroxamic acid have been approved for the treatment of cutaneous T- cell lymphoma. Many proteomic studies, including AP-MS after drug treatment, have been used to study HDAC inhibitors and their effect on specific HDAC complexes, like Sin3/HDAC complexes [79, 80, 81, 82, 95, 96]. B) Drugs targeting specific modification readers like JQ1 small molecule BET bromodomain inhibitor have emerged as potentially powerful anticancer targeting drugs. Proteomic approaches have recently played important roles in studying cellular response and resistance to these drugs [91]. C) Cancer-driving mutations, like the many found in SWI/SNF complexes, are being identified by proteomic approaches. For example, loss of SWI/SNF complex subunit SNF5 was identified as a cancer-driving mutation through XL-MS perturbation studies [34, 78]. D) Oncohistones, or mutated histones, like histone H3 Lysine-27 to methionine mutation exists in highly aggressive pediatric gliomas. AP-MS studies have been very useful in identifying histone lysine-to-methionine interacting partners [97].
The fundamental unit of chromatin is the nucleosome, which is formed by histone octamers wrapped by ~147bp DNA. Although the basic structure of each nucleosome is very similar, the diversity between each individual nucleosome is a key component of epigenetics. The diversity of nucleosomes largely relies on combinations of modification on histone tails (also referred to as ‘histone codes’), the existence of variants for core histone subunits, and evidence suggests that certain histone mutations can be cancer drivers [6]. Histones with mutations that drive cancer are referred to as oncohistones [7] (Figure 2D). To be able to analyze histone variants and isoforms with different modifications globally, quantitative mass spectrometry (MS) is one of the most powerful tools. The analysis of histones and their modifications by proteomics is a highly active area of research, and we recommend excellent recent reviews for much greater detail on this topic [8, 9].
2. Capturing chromatin proteins for proteomic analysis.
Capturing chromatin protein samples for proteomic analysis can involve multiple challenging steps. Since histones are wrapped by DNA, harsh extraction conditions are sometimes necessary to release histones from insoluble chromatin fractions. However, harsh extraction conditions can also disrupt protein-protein interactions. For this reason, different extraction and purification methods should be applied depending on the protein group of interest. A classical protocol for extracting proteins is the Dignam, or nuclear extraction, method [10]. Briefly, the Dignam method isolates nuclei, and then releases nuclear proteins with relatively harsh extraction conditions and high salt concentrations. Proteins that are not tightly bound to the insoluble chromatin fraction can be obtained in the extract. The advantage of this method is that proteins should stay close to their native state. Further affinity purification and chromatography can be applied to the extract and the purified proteins can be analyzed by mass spectrometry (Figure 3A). For example, Vermeulen et al. (2010) used Dignam extracts incubated with synthesized histone tails to identify proteins bound to various histone states [11]. Likewise, Eberl et al. (2013) used histone peptide purifications from Dignam extracts [12]. Both methods, combined with quantitative MS, are powerful approaches to identify novel chromatin readers. A similar approach is to assemble nucleosomes in vitro containing certain modifications as baits instead of histone tails [13]. This type of proteomics study provides important information about possible ‘readers’ for certain modifications [13]. Studying the features of modification binding proteins can be of great significance since these binding proteins can be potential targets for epigenetic inhibition, which might serve as a therapeutic method for cancer (Figure 2).
Figure 3. Analytical workflows.
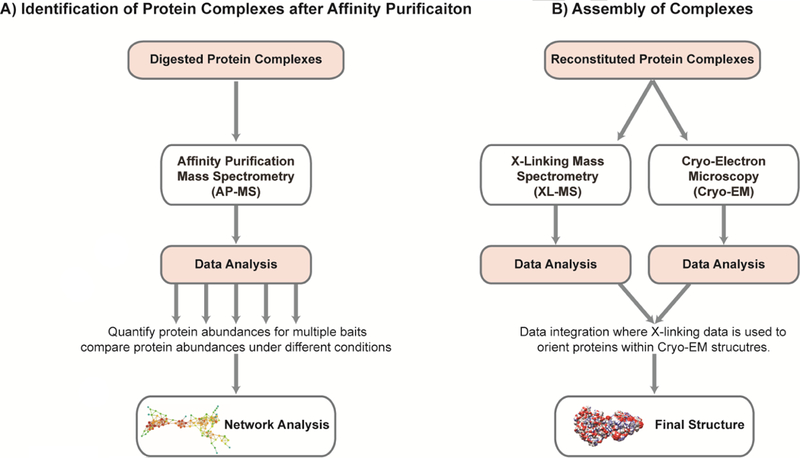
A brief overview of two workflows illustrating how quantitative proteomics is applied to the study of large protein complexes in chromatin biology. Both workflows progress in the same general manner, beginning with sample preparation, followed by mass spectrometry, and culminating in data analysis. However, each workflow utilizes different techniques. The first workflow, A) Identification of Protein Complexes, uses digested proteins and affinity purification mass spectrometry (AP-MS) to quantify and compare the protein abundances through the analysis of protein protein interactions (PPI) networks. The second workflow, B) Assembly of Protein Complexes, uses reconstituted protein complexes with both cryo- Electron Microscopy (cryo-EM) and cross-linking mass spectrometry (XL-MS) to determine the final structure of large protein complexes. Both methods are useful for determine the composition of large protein complexes in chromatin biology.
In order to preserve weak and transient interactions during harsh steps, another approach is crosslinking cells with formaldehyde before protein extraction from the general chromatin immunoprecipitation (ChIP) [14, 15]. Crosslinking allows for the preservation of weak and transient interactions during harsh steps, such as sonication and stringent washing and has been optimized for different applications [16, 17, 18, 19]. From this, numerous methods have been developed that allow for proteomic analysis at specific genomic regions. If the DNA sequence of the genomic region of interest is known, associated proteins can also be enriched by probing for a specific DNA sequence in a method called chromatin affinity purification with mass spectrometry (ChAP-MS) where a LexA DNA affinity handle is inserted upstream of a start codon for analysis by mass spectrometry [20]. A further development of the ChAP-MS approach is termed CRISPR-ChAP-MS where a catalytically inactive protein A tagged version of Cas9 was used with a guide RNA to target a promoter in order specifically enrich this region followed by proteomics analysis [21]. An additional method named Proteomics of Isolated CHromatin segments (PICh) was used to isolate proteins associated with human telomeric chromatin [22]. In PICh a specific oligonucleotide probe containing locked nucleic acid was used to hybridize to chromatin followed by capture on magnetic beads and proteomics analysis [22]. Another interesting approach to investigate proteins that associate with newly synthesized chromatin is named DNA mediated chromatin pull-down (Dm-ChP) [23]. The idea of this method is to label newly synthesized DNA by incorporating 5-ethynyl-2’-deoxyuridine (EdU) [23]. Then the incorporated EdU is linked to biotin with a Click reaction [23]. By affinity purifying biotin, proteins that are crosslinked to the newly synthesized DNA can be enriched for mass spectrometry analysis [23]. A similar approach, called nascent chromatin capture (NCC) directly incorporated biotin-dUTP into replicating DNA and streptavidin was used to capture proteins and DNA from replication forks and nascent chromatin [24]. Both Dm- ChP and NCC are methods useful for capturing specific chromatin states.
3. Coupling structural biology and proteomics
Advances in structural biology, have led to the convergence of cryo-Electron Microscopy (cryo-EM) and cross-linking mass spectrometry (XL-MS) especially for the study of large protein complexes (Figure 3B). In particular, the cross-linking of peptides is used to determine sites of interaction between two proteins, and this is used to facilitate the analysis of protein complex structures from cryo-EM [25, 26]. In XL-MS, crosslinking is used to freeze protein-protein interactions for subsequent identification by mass spectrometry thereby permitting capture of transient interactions and determination of macromolecular structure. Although most cross-linking is chemical- based [25], photo-cross-linking is possible [27]. In chemical XL-MS, crosslinking reagents containing two reactive groups separated by spacer arms. The reactive groups of crosslinkers such as bis(sulfosuccinimidyl) suberate and disuccinimidyl suberate induce nucleophilic attacks on primary amines and couple them to of lysine residues[25]. The length of the spacer between the reactive species of cross-linking reagent provides a spatial constraint on the residues that can be cross-linked hence making XL-MS a useful technique for structural and interaction network information.
Different variations of XL-MS have been developed, from proteome-wide crosslinking strategies for identification of endogenous protein complexes [26] to lysine- specific chemical cross-linking of protein complexes [28] and Cross-Linking-Assisted and SILAC-based Protein Identification (CLASPI) [29]. XL-MS and cryo-EM have been used to study the structure of several chromatin remodeling complexes. This includes the SWR1 complex [30], the INO80 complex [31], the polycomb complex [32], and the chromatin assembly factor 1 complex [33]. Essentially, in these studies XL-MS data is used to orient the location and positioning of individual proteins within the Cryo-EM structure. This serves as a method of constraining all possible predictions of protein positioning in a Cryo-EM structure. XL-MS has also been used without cryo-EM to provide valuable insights into complexes. This includes and analysis of the Swi/Snf complex where the loss of the Snf5 subunit leads to aberrant complexes [34], and an analysis of the NuRD complex where the dynamics, stoichiometry, and inter-complex contacts were determined using XL-MS coupled to other proteomic techniques [35]. It is likely that XL-MS will only grow in use for the study of chromatin remodeling complexes.
4. Revealing the dynamics of the chromatin proteome
Chromatin is highly dynamic, meaning the proteome is constantly changing during cellular activities. Therefore, in chromatin proteomic studies, the comparison of samples from different conditions or time points are frequently involved. In order to compare the proteome between two different conditions, quantitative proteomics approaches are needed. Stable isotope labeling by amino acids in cell culture (SILAC) [36] has become a widely used approach in quantitative proteomics. In the SILAC approach, cells are grown under conditions to incorporate heavy stable isotope amino acids for in vivo integration into the proteome and quantification can be achieved by comparing the ratio of heavy amino acids to light amino acids in labeled sample as compared to unlabeled control [36]. In the past five years, SILAC has been used for the comparison of the chromatin conformation around specific genomic loci and chromatin dynamics during cellular processes. To isolate specific genomic loci, different capture methods have been developed to isolate centrosome-associated chromatin [37], telomeric chromatin [38], dynamics of replication-specific chromatin [24] and cell cycle-specific chromatin [39]. To quantify nascent post-replication chromatin from mature nuclease -resistant chromatin, Alabert et al. have developed nascent chromatin capture in combination with SILAC permitted a quantitative proteomic comparison of post- replication chromatin composition [24]. Similarly, Grolimund et al. have developed quantitative telomeric chromatin isolation protocol and SILAC isolation of telomeric chromatin for comparative quantification of the telomere-specific protein components in cells with different telomere states [38]. Chromatin capture methods for the isolation of specific genomic loci are very helpful for identification and quantification of chromatin signatures around the genome.
SILAC-based approaches have been applied to monitor the turnover of proteins and modifications. Mews et al. monitored the turnover of global histone proteins, as well as newly acetylated and methylated histones using this method [40]. By analyzing the dynamics of histone modifications when cells reenter the cell cycle from quiescence, they found that histone methylation was static during quiescence exit while histone acetylation was more dynamic [40]. In this work, heavy lysine was used to track the overall newly synthesized histones, while heavy glucose and heavy methionine was used to track new acetyl and methyl modifications, respectively [40]. The modification specific tracking was achieved because, as cells metabolize, methyl groups provided by methionine and acetyl groups provided by glucose are used to modify histones. As long as the heavy isotope does not affect cell metabolism, the turnover of histones and specific modifications can be tracked by monitoring the incorporation of heavy labeled residuals with quantitative proteomics [41]. Other examples of Histone PTMs analysis studied using SILAC based approaches include analyzing different functional states in multiple biological samples [42] and studying the degradation of histones in response to DNA damage [43].
In label free quantitative proteomics, stable isotopes are not used, and samples are analyzed independently and quantitatively compared to each other using peak intensities or spectral counts, for example [44]. Label free quantitative proteomics has been routinely used to study chromatin remodeling complexes and protein interaction networks. This includes the analysis of the Tip60, Ino80, and SRCAP complexes [45], histone deacetylase complexes [46], the polycomb complex [47, 48, 49], and the cardiac TBX5 interactome [50], for example. In addition, the stoichiometry of chromatin remodeling proteins has been analyzed using label free quantitative proteomics approaches [51]. Also, new components of DNA damage systems with strong links to chromatin have also been studies using label free quantitative proteomics approaches [52, 53]. Lastly, label free quantitative proteomic methods have been used to study the dynamics of changes in histone modifications during the malaria parasite life cycle [54].
5. Analysis of quantitative protein complex and network datasets
Data analysis is a major challenge for the analysis of chromatin remodeling complexes using proteomics approaches. This is particularly the case with quantitative proteomics datasets and protein protein interaction (PPI) network datasets. Unlike binary datasets, where a value of 1 is used if a protein is present and a value of 0 if a protein is absent, quantitative protein datasets are weighted and correspond to measurements derived from quantitative proteomics. A common way to construct quantitative PPI networks is through differential protein expression by comparing experimental and control replicates to find prey proteins likely enriched in experimental samples. Differential protein expression can be determined through simple, commonly used statistical methods like Student’s t-test, log fold change, p-value thresholds, and ranking systems that rank values based on z-scores of abundance correlations like Spearman correlation coefficients.
With the growing popularity of label-free quantitative proteomics, more advance software packages are using a combination of statistical methods to increase the reliability of differential protein expression. QSpec, and its extension QProt, use Z- score based statistics with log fold change parameters and false discovery rates estimated by Bayes method [55]. The GeneChip statistical tool PLGEM uses the power law global error model combined with the signal-to-noise ratio statistic [56]. D scores use spectra counts obtained from quantitative APMS experiments to assign probabilities to each prey in every bait. These quantitative scores analyze the uniqueness and reproducibility of a bait-prey interaction, normalized to produce a DN score for each prey in every bait [57]. Normalized spectral abundance factors (NSAFs) use spectra counts normalized with prey protein length to assign a probability to each pairwise interaction, quantitatively determining the preference between a prey and bait relative to all other baits [58]. Likewise, the Significance Analysis of INTeractome (SAINT) algorithm uses a mixture modeling approach to determine the likelihood of a protein-protein interaction [59, 60]. These statistical methods have been recently reviewed elsewhere [55, 61, 62], and are important for their analysis of proteomics data in general.
Once individual PPIs are determined, a PPI network can be assembled. Assembling PPI networks for chromatin associated proteins is particularly challenging because, in most cases, chromatin associated proteins appear in large, multisubunit complexes consisting of ‘core’ subunits coupled with different ‘attachment’ modules [63]. In addition to complex composition, subunits interact with several different partners and appear in multiple, distinct complexes. This complexity makes it difficult to identify chromatin associated protein interactions, and thereby requires a myriad of computational solutions to generate and visualize PPI networks (Figure 4).
Figure 4. Network visualization examples.
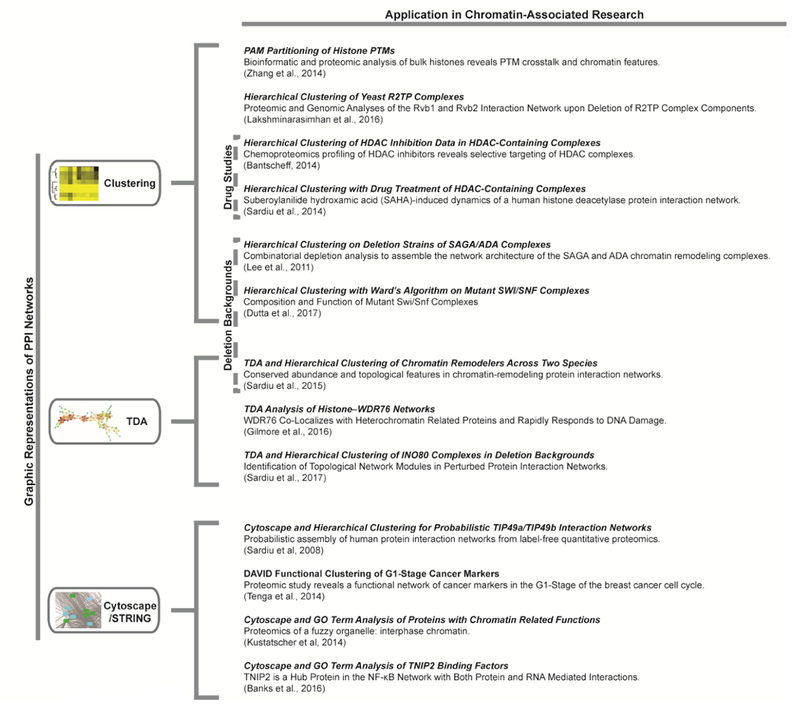
Three data visualization methods for protein protein interaction (PPI) networks are represented: Clustering, Topological Data Analysis (TDA), and Cytoscape/STRING. Each visualization method includes relatively recent examples in chromatin biology from quantitative proteomics. Research examples including AP-MS after drug treatment or protein complexes in genetic deletion backgrounds are also indicated.
When assembling networks, clustering is the process of grouping proteins into clusters such that proteins within a cluster interact with one another, but do not interact with proteins in other clusters. Most networks are represented as a graph where nodes correspond with proteins and edges with pairwise interactions, characterized by a few highly connected nodes, called hubs. Two traditional types of clustering are often used and well accepted in proteomics: hierarchical clustering and partition clustering. Both hierarchical and partition clustering are well studied and utilized in quantitative proteomics. Hierarchical clustering represents each protein as a node with interacting protein linked together to form a tree-like dendrogram. When using hierarchical clustering, the partitioning method determines the proximity between two clusters. Cluster proximity is important and differentiate between various agglomerative hierarchical techniques; for example, group-based proximities such as UPGMA/WPGMA consider the number of points in each cluster making a linkage between groups (UPGMA), or makes linkages within groups by treating all clusters equally (WPGMA). Ward’s method is a prototype-based proximity that measures the proximity between two clusters in terms of the increase in sum of squares when two clusters are merged [64]. In contrast to hierarchical methods, partitioning methods subdivide the data into a predetermined number of subsets, without any implied hierarchical relationship between these clusters. Markov clustering (MCL) algorithm and K-means are well-known partitioning methods. The MCL algorithm can predict protein complexes from protein interaction networks. K-means locally optimizes the average squared distance of points from their nearest cluster center. Both MCL and K- means are available in STRING [65]. Other partition methods include PAM (Partitioning Around Medoids), which applies an absolute error criterion, and FANNY. FANNY is unique in that it allocates membership coefficients for each of the proteins in any given cluster, instead of assigning a protein to only one cluster [64]. The choice of a suitable clustering algorithm depends on the purpose of the experiment and the type of data. For this reason, running more than one algorithm and comparing various clustering approaches is useful. Some clustering algorithms will result in uninformative clusters. On the other hand, one approaches might provide insight into complex assembly, while another is more useful for network organization. For example, a recent quantitative proteomics dataset used a PTM analysis with data with both Ward’s hierarchical method and the PAM clustering algorithms to identify changes in chromatin states and histone PTM patterns [66].
More recent to traditional clustering analysis, other methods are utilized to cluster proteomics data including Topological Data Analysis (TDA) [67]. TDA incorporates geometric approaches to explore the shape of data. Unlike traditional clustering analysis where a node corresponds to a single protein, nodes in topological networks represent clusters of proteins with connections between nodes that contain proteins in common. Using the connectivity between nodes, TDA captures the connection between complexes that share proteins and can reveal interesting features of the data. For this reason, TDA has been especially useful in chromatin biology. For example, Sardiu et al. compared protein abundances between three chromatin remodeling complexes from two species, Saccharomyces cerevisiae and Homo sapiens [68]. Using spectral count values and hierarchical clustering with Ward’s method, Sardiu et al. found that, in general, yeast and human baits that are members of the same complex clustered together [68]. In addition, Topological Data Analysis (TDA) captured the connection between orthologous protein pairs in human and yeast datasets [68]. TDA showed that proteins in the conserved core of the three chromatin complexes are highly correlated, complementing the results obtained from traditional clustering methods, and revealing interesting features of the data [68]. Using both clustering and TDA showed correlation between orthologous proteins; furthermore, these results demonstrated that meaningful biological results can be obtained by comparing quantitative proteomic datasets across species [68]. This is another example of uniting different visualization methods for unique, complementary insights. In this case, TDA was applied to study the conservation of chromatin remodeling complexes [68]. TDA has also been utilized to study chromatin associated proteins [52], and the dynamics of chromatin remodeling complexes [69] (Figure 4).
Biological systems are a collection of highly integrated networks; therefore, to define PPI networks in a biologically relevant fashion, it is important to view PPI networks in a larger biological context. One way to elucidate the biological function of proteins is to integrate data analysis with prior knowledge through functional annotation. Incorporating functional annotation into PPI networks can help determine biological processes and pathways in the data. Annotation tools like the Gene Ontology (GO) project can organize data as a matrix/graph connecting genes to GO molecular function, bioprocess, or cellular component terms [70, 71]. GO can be mined for known molecular functions, biological processes, and cellular components in which genes participate. Oher annotation tools, like the DAVID annotation tool [72] and GeneMANIA [73], annotate proteins with their known biological functions. Cytoscape is an open source software platform used to visualize interaction networks and biological pathways [74]. It is a diverse and open source tool for visualizing networks and a wide variety of plugins have been developed for the platform [75]. It is valuable to integrate PPI networks derived from quantitative proteomics with annotative tools. Functional annotation can highlight whether proteins involved in similar biological pathways are clustered together in a biologically relevant fashion. For example, Tenga, et al. explored the biological mechanisms that control cancer cell cycle progression [76]. Using spectral count data from the MCF-7 breast cancer cell line arrested in G1-phase, Tenga, et al. used the DAVID annotation tool to identify proteins that control chromatin maintenance, including HDAC1/2 [76] (Figure 4).
6. Expert commentary
Many of the large multiprotein complexes involved in chromatin remodeling have been studied using several proteomic techniques (Figure 1). Ino80 [31, 68, 69], Swr1 [30, 68], NuRD [35], SAGA [77], Tip60 [45], SWI/SNF [34, 78], and Sin3 [79, 80, 81, 82] are a few examples. Proteomic analysis of chromatin remodeling has provided key insights in to the content of these complexes and their important roles in regulating the genome. However, chromatin remodeling proteins and complexes also play very important roles in human cancer, and extensive research is focused on devising therapies to disrupt or inhibit chromatin associated proteins [3]. Several chromatin remodeling proteins, histone modifiers, and histones themselves are amongst the most significantly mutated genes across several cancer types [83]. Components of the Swi/Snf complex are part of this list [83], and coupled with other studies have led to great interest in the Swi/Snf complex as a target for therapeutic intervention [84] (Figure 2C). Proteomics based approaches have emerged as key approaches to study chromatin remodeling complexes in this context, and should continue to be a major focus of ongoing and future research. Here, the key is to study the perturbation of chromatin remodeling complexes and the effects these perturbations have on PPIs within these complexes.
Proteomics approaches are commonly used method for measuring the relative abundance of proteins in different biological conditions like during different phases of the cell cycle or after chromatin activation/repression [85, 86, 87]. To understand how complexes are assembled, regulated, and modulated it is useful to study, not only the conservation of networks and complexes, but also perturbed networks and complexes. The transcriptional activation/repression of chromatin results from a collection of dynamic, integrated activities. Therefore, to fully understand the dynamic nature of chromatin remodeling complexes, it is essential to study PPI networks in the context of changing environments. For example, Kustatscher et al. constructed a PPI network by comparing relative protein abundances across may perturbed systems, including cell cycle phases and drug treatments, to determine a protein’s probability to function in chromatin [85].
Another method often utilized to construct perturbed networks is protein complexes in genetic deletion backgrounds. Deletions affect the interaction between bait-prey protein pairs such that deletion data sets are more likely to reflect the direct relationship between bait and prey proteins. In this approach, the protein interactions of complexes are studied after deleting individual genes in a complex from the genome. This is especially useful for protein in large, multisubunit complexes where proteins in the same complex will have very similar quantitative values. A comprehensive deletion network analysis has been successfully completed on the yeast R2TP complex [88], Ino80 complex [69], and the SAGA chromatin remodeler complex [77]. In all these cases, the deletion network analysis identified new complexes that associate the complex, including several interesting proteins that co-purified with the complex only in the absence of another gene. In addition to finding new interactions, deletion network analysis is important in detecting hub proteins, which are known to be essential for protein complex organization. Analysis of mutated Swi/Snf complexes is also emerging providing new insights regarding how these complexes may function in cancers [34, 78]. In addition, point mutations can have significant impacts on PPIs. For example, histone H3 Lysine-27 to methionine mutations exist in highly aggressive pediatric gliomas, and this mutation has a significant impact on the PPIs of histone H3 [89] (Figure 2D). type of mutated histone is considered an oncohistone [7], and affinity purification coupled to quantitative proteomics approaches can play an important role in determining if they have unique protein protein interactions, for example. Another comprehensive proteomic approach in cancer biology is the study of misregulation via protein-protein interactions. For example, Alekseyenko et al. used BRD4-NUT, a fusion oncoprotein that drives NUT midline carcinoma (NMC), and chromatin crosslinking AP-MS to identify BRD4-NUT fusion oncoprotein interactions in NMC patient samples [90]. Alekseyenko et al. found that NMC is dependent on ectopic NUT- mediated interactions between acetyltransferase EP300 and components of BRD4 regulatory complexes, leading to a cascade of misregulation.
Therapeutic targeting of chromatin remodeling complexes is an active area of research with drugs available or under development, and proteomics approaches are contributing to these studies. For example, bromodomain inhibitors, like JQ1, have emerged as potentially powerful anticancer drugs (Figure 2B) and proteomic approaches have recently played important roles in studying cellular response and resistance to these drugs [91, 92]. In addition, a PPI study of a the mixed-lineage leukemia (MLL) protein played an important role in the determination that suppressing the degradation of wild type MLL in order to compete with mutant MLL chimeric proteins could be a powerful approach to treating this form of leukemia [93].
The analysis of HDAC inhibitors is perhaps the most prominent example of the use of proteomics to study chromatin remodeling proteins, their complexes and their targeting in cancer. HDAC inhibitors have been shown to induce tumor cell apoptosis and cell-cycle arrest [94]. HDAC inhibitors like suberoylanilide hydroxamic acid and romidepsin have been approved for the treatment of cutaneous T-cell lymphoma and chemical proteomics approaches have been used to study these inhibitors and their effect on specific HDAC complexes (Figure 2A) [79, 80, 81, 82]. Furthermore, proteomic approaches have been used to further study the effect HDAC inhibitors have on the proteome of a cell [95, 96]. The studies described here regarding the analysis of chromatin remodeling complexes and their relationship to cancer incorporate many, if not all, of the key sample preparation, quantitative proteomic analysis, and data analysis techniques and considerations described earlier in this review.
7. Five-year view
Proteomic analysis of chromatin remodeling proteins and complexes will remain important studies for the near term and for many years to come. They play key roles in the regulation of the genome in human health and disease. Since chromatin remodeling proteins are typically parts of large multiprotein complexes (Figure 1) efforts coupling XL-MS and cryo-EM will play a major role elucidating the structure and function of these complexes. In addition, the dynamics of chromatin remodeling complexes will be the major focus of research. This will include changes in complexes during development, in different cell types, and due to perturbations like the application of a drug. With increasing research into the role these complexes play in cancer, significant additional research is needed regarding the effect inhibitors have on chromatin remodeling complexes and their function.
8. Key issues.
The use of quantitative proteomic approaches to determine protein levels in samples is essential and provides deeper insight into the relationship of individual proteins in a complex. Methods using stable isotopes or label free methods have been successfully applied to the study of chromatin remodeling and epigenetics.
Chromatin remodeling proteins are typically part of large protein complexes. This makes their analysis in a complete complex difficult using structural biology techniques. However, the coupling of cross-linking mass spectrometry and cryo-EM has proven a powerful approach to determine the architecture of chromatin remodeling complexes.
The key for future studies of chromatin remodeling complexes will be to study the dynamics of these complexes in different cell types, under different disease states, the effects of mutations on their organization, and the effects of small molecule inhibitors on their networks.
Histones and chromatin remodeling proteins are frequently mutated in cancer. Understanding the effects of these mutations on protein interactions, for example, will be important for understanding of the molecular mechanisms of cancer and potential therapeutic interventions.
Given the complexity of the data generated in these studies, state of the art computational pipelines need to be used.
Acknowledgments
Funding
This work was supported by the Stowers Institute for Medical Research and by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number RO1GM112639. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Declaration of interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
- 1.Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet. 2016. August;17(8):487–500. doi: 10.1038/nrg.2016.59. PubMed PMID [DOI] [PubMed] [Google Scholar]
- 2.Li B, Carey M, Workman JL. The role of chromatin during transcription.cell. 2007. February 23;128(4):707–19. doi: 10.1016/j.cell.2007.01.015. PubMed PMID:;eng [DOI] [PubMed] [Google Scholar]
- 3.Dawson MA. The cancer epigenome: Concepts, challenges, and therapeuticopportunities. Science 2017. March 17;355(6330):1147–1152. doi: 10.1126/science.aam7304. PubMed PMID: ; eng. [DOI] [PubMed] [Google Scholar]
- 4.Noberini R, Sigismondo G, Bonaldi T. The contribution of mass spectrometry- based proteomics to understanding epigenetics. Epigenomics. 2016. March;8(3):429–45. doi: 10.2217/epi.15.108. PubMed PMID: ; eng. [DOI] [PubMed] [Google Scholar]
- 5.Wierer M, Mann M. Proteomics to study DNA-bound and chromatin-associated gene regulatory complexes. Human molecular genetics. 2016. October 01; 25(R2):R106–R114. doi: 10.1093/hmg/ddw208. PubMed PMID: ; PubMed Central PMCID: PMC5036873. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lewis PW, Muller MM, Koletsky MS, et al. Inhibition of PRC2 activity by a gain- of-function H3 mutation found in pediatric glioblastoma. Science. 2013. May 17;340(6134):857–61. doi: 10.1126/science.1232245. PubMed PMID: ; PubMed Central PMCID: PMCPMC3951439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koch L Cancer genetics: Oncohistone pathology explained. Nat Rev Genet. 2016. July;17(7):375. doi: 10.1038/nrg.2016.71. PubMed PMID: ; eng. [DOI] [PubMed] [Google Scholar]
- 8.Zheng Y, Huang X, Kelleher NL. Epiproteomics: quantitative analysis of histone marks and codes by mass spectrometry. Curr Opin Chem Biol. 2016. August;33:142–50. doi: 10.1016/j.cbpa.2016.06.007. PubMed PMID: ; PubMed Central PMCID: PMCPMC5129744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Janssen KA, Sidoli S, Garcia BA. Recent Achievements in Characterizing the Histone Code and Approaches to Integrating Epigenomics and Systems Biology. Methods Enzymol. 2017;586:359–378. doi: 10.1016/bs.mie.2016.10.021. PubMed PMID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abmayr SM, Yao T, Parmely T, et al. Preparation of nuclear and cytoplas extracts from mammalian cells. Curr Protoc Mol Biol. 2006. August;Chapter 12 1. doi: 10.1002/0471142727.mb1201s75. PubMed PMID: [DOI] [PubMed] [Google Scholar]
- 11.Vermeulen M, Eberl HC, Matarese F, et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell. 2010. September 17;142(6):967–80. doi: 10.1016/j.cell.2010.08.020. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 12.Eberl HC, Spruijt CG, Kelstrup CD, et al. A map of general and specialized chromatin readers in mouse tissues generated by label-free interaction proteomics. Mol Cell. 2013. January 24;49(2):368–78. doi: 10.1016/j.molcel.2012.10.026. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 13.Bartke T, Vermeulen M, Xhemalce B, et al. Nucleosome-interacting proteins regulated by DNA and histone methylation. Cell. 2010. October 29;143(3):470–84. doi: 10.1016/j.cell.2010.10.012. PubMed PMID: ; PubMed Central PMCID: PMCPMC3640253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lambert JP, Mitchell L, Rudner A, et al. A novel proteomics approach for the discovery of chromatin-associated protein networks. Mol Cell Proteomics. 2009. April;8(4):870–82. doi: 10.1074/mcp.M800447-MCP200. PubMed PMID: ; PubMed Central PMCID: PMCPMC2667365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soldi M, Bonaldi T. The ChroP approach combines ChIP and mass spectrometry to dissect locus-specific proteomic landscapes of chromatin. J Vis Exp. 2014. April11(86). doi: 10.3791/51220. PubMed PMID: ; PubMed Central PMCID: PMCPMC4166860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mohammed H, Taylor C, Brown GD, et al. Rapid immunoprecipitation mass spectrometry of endogenous proteins (RIME) for analysis of chromatin complexes. Nat Protoc. 2016. February;11(2):316–26. doi: 10.1038/nprot.2016.020. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 17.Rafiee MR, Girardot C, Sigismondo G, et al. Expanding the Circuitry of Pluripotency by Selective Isolation of Chromatin-Associated Proteins. Mol Cell. 2016. November 03;64(3):624–635. doi: 10.1016/j.molcel.2016.09.019. PubMed PMID: ; PubMed Central PMCID: PMCPMC5101186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang CI, Alekseyenko AA, LeRoy G, et al. Chromatin proteins captured by ChIP-mass spectrometry are linked to dosage compensation in Drosophila. Nat Struct Mol Biol. 2013. February;20(2):202–9. doi: 10.1038/nsmb.2477. PubMed PMID: ; PubMed Central PMCID: PMCPMC3674866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zee BM, Alekseyenko AA, McElroy KA, et al. Streamlined discovery of cross- linked chromatin complexes and associated histone modifications by mass spectrometry. Proc Natl Acad Sci U S A. 2016. February 16;113(7):1784–9. doi: 10.1073/pnas.1522750113. PubMed PMID: ; PubMed Central PMCID: PMCPMC4763777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Byrum SD, Raman A, Taverna SD, et al. ChAP-MS: a method for identification of proteins and histone posttranslational modifications at a single genomic locus. Cell Rep. 2012. July 26;2(1):198–205. doi: 10.1016/j.celrep.2012.06.019. PubMed PMID: ; PubMed Central PMCID: PMCPMC3408609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waldrip ZJ, Byrum SD, Storey AJ, et al. A CRISPR-based approach for proteomic analysis of a single genomic locus. Epigenetics. 2014. September;9(9):1207–11. doi: 10.4161/epi.29919. PubMed PMID: ; PubMed Central PMCID: PMCPMC4169012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dejardin J, Kingston RE. Purification of proteins associated with specific genomic Loci. Cell. 2009. January 09;136(1):175–86. doi: 10.1016/j.cell.2008.11.045. PubMed PMID: ; PubMed Central PMCID: PMCPMC3395431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kliszczak AE, Rainey MD, Harhen B, et al. DNA mediated chromatin pull-down or the study of chromatin replication. Sci Rep. 2011; 1:95. doi: 10.1038/srep00095. PubMed PMID: ; PubMed Central PMCID: PMCPMC3216581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alabert C, Bukowski-Wills JC, Lee SB, et al. Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components. Nat Cell Biol. 2014. March;16(3):281–93. doi: 10.1038/ncb2918. PubMed PMID: ; PubMed Central PMCID: PMCPMC4283098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leitner A, Faini M, Stengel F, et al. Crosslinking and Mass Spectrometry: An Integrated Technology to Understand the Structure and Function of Molecular Machines. Trends Biochem Sci. 2016. January;41(1):20–32. doi: 10.1016/j.tibs.2015.10.008. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 26.Liu F, Rijkers DT, Post H, et al. Proteome-wide profiling of protein assen cross-linking mass spectrometry.Nat Methods. 2015. December;12(12):1179–84. doi: 10.1038/nmeth.3603. PubMed PMID: ; eng. [DOI] [PubMed] [Google Scholar]
- 27.Kramer K, Sachsenberg T, Beckmann BM, et al. Photo-cross-linking and high- resolution mass spectrometry for assignment of RNA-binding sites in RNA- binding proteins. Nat Methods. 2014. October;11(10):1064–70. doi: 10.1038/nmeth.3092. PubMed PMID: ; eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leitner A, Walzthoeni T, Aebersold R. Lysine-specific chemical cross-linking of protein complexes and identification of cross-linking sites using LC-MS/MS and the xQuest/xProphet software pipeline. Nat Protoc. 2014. January;9(1):120–37. doi: 10.1038/nprot.2013.168. PubMed PMID: ; eng. [DOI] [PubMed] [Google Scholar]
- 29.Li X, Li XD. Chemical proteomics approaches to examine novel histone posttranslational modifications. Curr Opin Chem Biol. 2015. February;24:80–90. doi: 10.1016/j.cbpa.2014.10.015. PubMed PMID: ; eng. [DOI] [PubMed] [Google Scholar]
- 30.**Nguyen VQ, Ranjan A, Stengel F, et al. Molecular architecture of the ATP- dependent chromatin-remodeling complex SWR1. Cell. 2013. September 12;154(6):1220–31. doi: 10.1016/j.cell.2013.08.018. PubMed PMID: ; PubMed Central PMCID: PMC3776929.eng.Since chromatin remodeling complexes are large multiprotein complexes they e difficult to analyze using x-ray crystallography. This paper couples cross-iking mass spectrometry and cryo-electron micrscopy to generate a structure of the SWR1 complex.
- 31.**Tosi A, Haas C, Herzog F, et al. Structure and subunit topology of the INO80 chromatin remodeler and its nucleosome complex. Cell. 2013. September 12;154(6):1207–19. dei: 10.1016/j.cell.2013.08.016. PubMed PMID: ;eng.Like the SWR1 paper above, this paper couples cress-linking mass spectrometry and cryo-electron micrscepy te generate a structure ef the Ino80 complex. It is likely that this combination of technologies will be used widely in the future to study chromatin remodeling complexes.
- 32.Ciferri C, Lander GC, Maiolica A, et al. Molecular architecture of human polycomb repressive complex 2. eLife. 2012. October 30;1:e00005. doi: 10.7554/eLife.00005. PubMed PMID: ; PubMed Central PMCID: PMC3482686. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim D, Setiaputra D, Jung T, et al. Molecular Architecture of Yeast Chromatin Assembly Factor 1. Sci Rep. 2016. May 25;6:26702. doi: 10.1038/srep26702. PubMed PMID: ; PubMed Central PMCID: PMC4879628. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sen P, Luo J, Hada A, et al. Loss of Snf5 Induces Formation of an Aberrant SWI/SNF Complex. Cell Rep. 2017. February 28;18(9):2135–2147. doi: 10.1016/j.celrep.2017.02.017. PubMed PMID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.*Kloetxys SL, Baymaz HI, Makowski M, et al. Towards elucidating the stability, dynamics and architecture of the nucleosome remodeling and deacetylase complex by using quantitative interaction proteomics. FEBS J. 2015. May;282(9):1774–85. doi: 10.1111/febs.12972. PubMed PMID: .In this study, the authors used quantitative proteomic and cross-linking mass spectrometry strategies to capture new insights into the NuRD complex.
- 36.Ong SE, Blagoev B, Kratchmarova I, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002. May;1(5):376–86. PubMed PMID: ; eng. [DOI] [PubMed] [Google Scholar]
- 37.Jakobsen L, Schroder JM, Larsen KM, et al. Centrosome isolation and analysis by mass spectrometry-based proteomics. Methods Enzymol. 2013;525:371–93. doi: 10.1016/B978-0-12-397944-5.00018-3. PubMed PMID: ; eng. [DOI] [PubMed] [Google Scholar]
- 38.Grolimund L, Aeby E, Hamelin R, et al. A quantitative telomeric chromatin isolation protocol identifies different telomeric states. Nat Commun. 2013;4:2848. doi: 10.1038/ncomms3848. PubMed PMID: ; eng. [DOI] [PubMed] [Google Scholar]
- 39.Kustatscher G, Wills KL, Furlan C, et al. Chromatin enrichment for proteomics. Nat Protoc. 2014. September;9(9):2090–9. doi: 10.1038/nprot.2014.142. PubMed PMID: ; PubMed Central PMCID: PMCPMC4300392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mews P, Zee BM, Liu S, et al. Histone methylation has dynamics distinct from those of histone acetylation in cell cycle reentry from quiescence. Mol Cell Biol. 2014. November;34(21):3968–80. doi: 10.1128/MCB.00763-14. PubMed PMID: ; PubMed Central PMCID: PMCPMC4386454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mews P, Berger SL. Exploring the Dynamic Relationship Between Cellular Metabolism and Chromatin Structure Using SILAC-Mass Spec and ChIPSequencing. Methods Enzymol. 2016;574:311–29. doi: 10.1016/bs.mie.2016.04.002. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 42.Cuomo A, Soldi M, Bonaldi T. SILAC-Based Quantitative Strategies for Accurate Histone Posttranslational Modification Profiling Across Multiple Biological Samples. Methods in molecular biology. 2017;1528:97–119. doi: 10.1007/978-1-4939-6630-1_7. PubMed PMID: ; eng. [DOI] [PubMed] [Google Scholar]
- 43.Hauer MH, Seeber A, Singh V, et al. Histone degradation in response to DNA damage enhances chromatin dynamics and recombination rates. Nat Struct Mol Biol. 2017. February;24(2):99–107. doi: 10.1038/nsmb.3347. PubMed PMID:. [DOI] [PubMed] [Google Scholar]
- 44.Zhang Y, Wen Z, Washburn MP, et al. Improving label-free quantitative proteomics strategies by distributing shared peptides and stabilizing variance. Anal Chem. 2015;87(9):4749–56. doi: 10.1021/ac504740p. PubMed PMID: ; eng. [DOI] [PubMed] [Google Scholar]
- 45.Sardiu ME, Cai Y, Jin J, et al. Probabilistic assembly of human protein interaction networks from label-free quantitative proteomics. Proc Natl Acad Sci U S A. 2008. February 05;105(5):1454–9. doi: 10.1073/pnas.0706983105. PubMed PMID: ; PubMed Central PMCID: PMCPMC2234165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Joshi P, Greco TM, Guise AJ, et al. The functional interactome landscape of the human histone deacetylase family. Mol Syst Biol. 2013;9:672. doi: 10.1038/msb.2013.26. PubMed PMID: ; PubMed Central PMCID: PMC3964310. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hauri S, Comoglio F, Seimiya M, et al. A High-Density Map for Navigating Human Polycomb Complexome. Cell Rep. 2016. October 04;17(2):583–595. doi: 10.1016/j.celrep.2016.08.096. PubMed PMID: ; eng. [DOI] [PubMed] [Google Scholar]
- 48.Kloet SL, Makowski MM, Baymaz HI, et al. The dynamic interactome and genomic targets of Polycomb complexes during stem-cell differentiation. Nat Struct Mol Biol. 2016. July;23(7):682–90. doi: 10.1038/nsmb.3248. PubMed PMID: ; PubMed Central PMCID: PMC4939079. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oliviero G, Brien GL, Waston A, et al. Dynamic Protein Interactions of the Polycomb Repressive Complex 2 during Differentiation of Pluripotent Cells. Mol Cell Proteomics. 2016. November;15(11):3450–3460. doi: 10.1074/mcp.M116.062240. PubMed PMID: ; PubMed Central PMCID: PMC5098042. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Waldron L, Steimle JD, Greco TM, et al. The Cardiac TBX5 Interactome Reveals a Chromatin Remodeling Network Essential for Cardiac Septation. Developmental cell. 2016. February 08;36(3):262–75. doi: 10.1016/j.devcel.2016.01.009. PubMed PMID: ; PubMed Central PMCID: PMC4920128. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smits AH, Jansen PW, Poser I, et al. Stoichiometry of chromatin-associated protein complexes revealed by label-free quantitative mass spectrometry-based proteomics. Nucleic Acids Res. 2013. January 07;41(1):e28. doi: 10.1093/nar/gks941. PubMed PMID: ; PubMed Central PMCID: PMCPMC3592467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gilmore JM, Sardiu ME, Groppe BD, et al. WDR76 Co-Localizes with Heterochromatin Related Proteins and Rapidly Responds to DNA Damage. PloS one. 2016; 11(6):e0155492. doi: 10.1371/journal.pone.0155492. PubMed PMID: ; PubMed Central PMCID: PMC4889050. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weems JC, Slaughter BD, Unruh JR, et al. Assembly of the Elongin A Ubiquitin Ligase Is Regulated by Genotoxic and Other Stresses. J Biol Chem. 2015. June 12;290(24):15030–41. doi: 10.1074/jbc.M114.632794. PubMed PMID: ; PubMed Central PMCID: PMCPMC4463447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saraf A, Cervantes S, Bunnik EM, et al. Dynamic and Combinatorial Landscape of Histone Modifications during the Intraerythrocytic Developmental Cycle of the Malaria Parasite. J Proteome Res. 2016. August 05;15(8):2787–801. doi: 10.1021/acs.jproteome.6b00366. PubMed PMID: ; eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Choi H, Kim S, Fermin D, et al. QPROT: Statistical method for testing expression using protein-level intensity data in label-free quantitative proteomics. J Proteomics. 2015. November 03;129:121–6. doi: 10.1016/j.jprot.2015.07.036. PubMed PMID: ; PubMed Central PMCID: PMCPMC4630079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pavelka N, Pelizzola M, Vizzardelli C, et al. A power law global error model for the identification of differentially expressed genes in microarray data. BMC Bioinformatics. 2004. December 17;5:203. doi: 10.1186/1471-2105-5-203. PubMed PMID: ; PubMed Central PMCID: PMCPMC545082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sowa ME, Bennett EJ, Gygi SP, et al. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009. July 23;138(2):389–403. doi: 10.1016/j.cell.2009.04.042. PubMed PMID: ; PubMed Central PMCID: PMCPMC2716422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Y, Wen Z, Washburn MP, et al. Refinements to label free proteome quantitation: how to deal with peptides shared by multiple proteins. Anal Chem. 2010. March 15;82(6):2272–81. doi: 10.1021/ac9023999. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 59.Choi H, Larsen B, Lin ZY, et al. SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat Methods. 2011. Jan;8(1):70–3. doi: 10.1038/nmeth.1541. PubMed PMID: ; PubMed Central PMCID: PMCPMC3064265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Choi H, Liu G, Mellacheruvu D, et al. Analyzing protein-protein interactions from affinity purification-mass spectrometry data with SAINT. Curr Protoc Bioinformatics. 2012. September;Chapter 8:Unit8 15. doi: 10.1002/0471250953.bi0815s39. PubMed PMID: ; PubMed Central PMCID: PMCPMC3446209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nesvizhskii AI. Computational and informatics strategies for identification of specific protein interaction partners in affinity purification mass spectrometry experiments. Proteomics. 2012. May;12(10):1639–55. doi: 10.1002/pmic.201100537. PubMed PMID: ; PubMed Central PMCID: PMCPMC3744239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Langley SR, Mayr M. Comparative analysis of statistical methods used for detecting differential expression in label-free mass spectrometry proteomics. J Proteomics. 2015. November 03;129:83–92. doi: 10.1016/j.jprot.2015.07.012. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 63.Ma’ayan A, MacArthur BD. New frontiers of network analysis in systems biology Dordrecht; New York: Springer; 2012. [Google Scholar]
- 64.Kaufman L, Rousseeuw PJ. Finding groups in data : an introduction to cluster analysis New York: Wiley; 1990. (Wiley series in probability and mathematical statistics Applied probability and statistics,). [Google Scholar]
- 65.Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015. January;43(Database issue):D447–52. doi: 10.1093/nar/gku1003. PubMed PMID: ; PubMed Central PMCID: PMC4383874. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang C, Gao S, Molascon AJ, et al. Bioinformatic and proteomic analysis of bulk histones reveals PTM crosstalk and chromatin features. J Proteome Res. 2014. July 03;13(7):3330–7. doi: 10.1021/pr5001829. PubMed PMID: ; PubMed Central PMCID: PMCPMC4096215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lum PY, Singh G, Lehman A, et al. Extracting insights from the shape of complex data using topology. Sci Rep. 2013;3:1236. doi: 10.1038/srep01236. PubMed PMID: ; PubMed Central PMCID: PMC3566620. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sardiu ME, Gilmore JM, Groppe BD, et al. Conserved abundance and topological features in chromatin-remodeling protein interaction networks. EMBO Rep. 2015. January;16(1):116–26. doi: 10.15252/embr.201439403. PubMed PMID: ; PubMed Central PMCID: PMCPMC4304735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sardiu ME, Gilmore JM, Groppe B, et al. Identification of Topological Network Modules in Perturbed Protein Interaction Networks. Sci Rep. 2017. March 08;7:43845. doi: 10.1038/srep43845. PubMed PMID: ; PubMed Central PMCID: PMCPMC5341041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gene Ontology C Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015. January;43(Database issue):D1049–56. doi: 10.1093/nar/gku1179. PubMed PMID: ; PubMed Central PMCID: PMCPMC4383973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gene Ontology C, Blake JA, Dolan M, et al. Gene Ontology annotations and resources. Nucleic Acids Res. 2013. January;41(Database issue):D530–5. doi: 10.1093/nar/gks1050. PubMed PMID: ; PubMed Central PMCID: PMCPMC3531070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huang da W, Sherman BT, Stephens R, et al. DAVID gene ID conversion tool. Bioinformation. 2008. July 30;2(10):428–30. PubMed PMID: ; PubMed Central PMCID: PMCPMC2561161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Warde-Farley D, Donaldson SL, Comes O, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010. July;38(Web Server issue):W214–20. doi: 10.1093/nar/gkq537. PubMed PMID: ; PubMed Central PMCID: PMCPMC2896186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Smoot ME, Ono K, Ruscheinski J, et al. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011. February 01;27(3):431–2. doi: 10.1093/bioinformatics/btq675. PubMed PMID: ; PubMed Central PMCID: PMC3031041. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Saito R, Smoot ME, Ono K, et al. A travel guide to Cytoscape plugins. Nat Methods. 2012. Nov;9(11):1069–76. doi: 10.1038/nmeth.2212. PubMed PMID: ; PubMed Central PMCID: PMC3649846. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tenga MJ, Lazar IM. Proteomic study reveals a functional network of cancer markers in the G1-Stage of the breast cancer cell cycle. BMC Cancer. 2014. September 24;14:710. doi: 10.1186/1471-2407-14-710. PubMed PMID: ; PubMed Central PMCID: PMCPMC4182858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee KK, Sardiu ME, Swanson SK, et al. Combinatorial depletion analysis to assemble the network architecture of the SAGA and ADA chromatin remodeling complexes. Mol Syst Biol. 2011. July 05;7:503. doi: 10.1038/msb.2011.40. PubMed PMID: ; PubMed Central PMCID: PMCPMC3159981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dutta A, Sardiu M, Gogol M, et al. Composition and Function of Mutant Swi/Snf Complexes. Cell Rep. 2017. February 28;18(9):2124–2134. doi: 10.1016/j.celrep.2017.01.058. PubMed PMID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.**Bantscheff M, Hopf C, Savitski MM, et al. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nature biotechnology. 2011. March;29(3):255–65. doi: 10.1038/nbt.1759. PubMed PMID: ;eng. This paper describes an impressive and systematic analysis of protein binding to 16 different immobilzed histone deacetylase inhibitors. The authors were able to determine kinetic parameters of protein binging and deconvolute protein complexes.
- 80.*Becher I, Dittmann A, Savitski MM, et al. Chemoproteomics reveals time- dependent binding of histone deacetylase inhibitors to endogenous repressor complexes. ACS chemical biology. 2014. August 15;9(8):1736–46. doi: 10.1021/cb500235n. PubMed PMID: ;eng. As a follow up to their previous work, the authors used their immoblized histone deacetylase strategy to study the time dependent binding of proteins and they found differneces in the binding of specific histone deacetylases and their associated proteins.
- 81.Sardiu ME, Smith KT, Groppe BD, et al. Suberoylanilide hydroxamic acid (SAHA)-induced dynamics of a human histone deacetylase protein interaction network. Mol Cell Proteomics. 2014. November;13(11):3114–25. doi: 10.1074/mcp.M113.037127. PubMed PMID: ; PubMed Central PMCID: PMCPMC4223495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Smith KT, Martin-Brown SA, Florens L, et al. Deacetylase inhibitors dissociate the histone-targeting ING2 subunit from the Sin3 complex. Chem Biol. 2010. January 29;17(1):65–74. doi: 10.1016/j.chembiol.2009.12.010. PubMed PMID: ; PubMed Central PMCID: PMC2819981. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013. October 17;502(7471):333–9. doi: 10.1038/nature12634. PubMed PMID: ; PubMed Central PMCID: PMC3927368. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kadoch C, Crabtree GR. Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Science advances. 2015. June;1(5):e1500447. doi: 10.1126/sciadv.1500447. PubMe PMID: ; PubMed Central PMCID: PMC4640607. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kustatscher G, Hegarat N, Wills KL, et al. Proteomics of a fuzzy organelle: interphase chromatin. EMBO J. 2014. March 18;33(6):648–64. doi: 10.1002/embj.201387614. PubMed PMID: ; PubMed Central PMCID: PMCPMC3983682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li M, Gray W, Zhang H, et al. Comparative shotgun proteomics using spectral count data and quasi-likelihood modeling. J Proteome Res. 2010. August 06;9(8):4295–305. doi: 10.1021/pr100527g. PubMed PMID: ; PubMed Central PMCID: PMCPMC2920032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kim SR, Nguyen TV, Seo NR, et al. Comparative proteomics: assessment of biological variability and dataset comparability. BMC Bioinformatics. 2015. April 17;16:121. doi: 10.1186/s12859-015-0561-9. PubMed PMID: ; PubMed Central PMCID: PMCPMC4704264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lakshminarasimhan M, Boanca G, Banks CA, et al. Proteomic and Genomic Analyses of the Rvb1 and Rvb2 Interaction Network upon Deletion of R2TP Complex Components. Mol Cell Proteomics. 2016. March;15(3):960–74. doi: 10.1074/mcp.M115.053165. PubMed PMID: ; PubMed Central PMCID: PMCPMC4813713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.**Herz HM, Morgan M, Gao X, et al. Histone H3 lysine-to-methionine mutants as a paradigm to study chromatin signaling. Science. 2014. August 29;345(6200):1065–70. doi: 10.1126/science.1255104. PubMed PMID: ; PubMed Central PMCID: PMC4508193.eng.In this study, an oncohistone present in highly aggressive pediatric gliomas was analyzed by quantitative proteomics and the differences in protein associations between the wild type and mutant histone provided novel insights into the mutation effects heterochromatic silencing.
- 90.Alekseyenko AA, Walsh EM, Zee BM, et al. Ectopic protein interactions within BRD4-chromatin complexes drive oncogenic megadomain formation in NUT midline carcinoma. Proc Natl Acad Sci U S A. 2017. May 23;114(21):E4184–E4192. doi: 10.1073/pnas.1702086114. PubMed PMID: ; PubMed Central PMCID: PMCPMC5448232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kurimchak AM, Shelton C, Duncan KE, et al. Resistance to BET Bromodomain Inhibitors Is Mediated by Kinome Reprogramming in Ovarian Cancer. Cell Rep. 2016. August 02;16(5):1273–86. doi: 10.1016/j.celrep.2016.06.091. PubMed PMID: ; PubMed Central PMCID: PMC4972668. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shu S, Lin CY, He HH, et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature. 2016. January 21;529(7586):413–7. doi: 10.1038/nature16508. PubMed PMID: ; PubMed Central PMCID: PMC4854653. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liang K, Volk AG, Haug JS, et al. Therapeutic Targeting of MLL Degradation Pathways in MLL-Rearranged Leukemia. Cell. 2017. January 12;168(1–2):59–72 e13. doi: 10.1016/j.cell.2016.12.011. PubMed PMID: ; PubMed Central PMCID: PMC5351781. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pettazzoni P, Pizzimenti S, Toaldo C, et al. Induction of cell cycle arrest and DNA damage by the HDAC inhibitor panobinostat (LBH589) and the lipid peroxidation end product 4-hydroxynonenal in prostate cancer cells. Free Radic Biol Med. 2011. January 15;50(2):313–22. doi: 10.1016/j.freeradbiomed.2010.11.011. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 95.**Mackmull MT, Iskar M, Parca L, et al. Histone Deacetylase Inhibitors (HDACi) Cause the Selective Depletion of Bromodomain Containing Proteins (BCPs). Mol Cell Proteomics. 2015. May;14(5):1350–60. doi: 10.1074/mcp.M114.042499. PubMed PMID: ; PubMed Central PMCID: PMC4424404.eng. In an interesting study suggesting a feedback loop between different chromatin remodeling complexes, the treatment of cells with histone deacetylase inhibitors resulted in downregualtion of bromodomain containing proteins, which themselves are the active targets of therapeutic intervention in cancer.
- 96.Wu Q, Cheng Z, Zhu J, et al. Suberoylanilide hydroxamic acid treatment reveals crosstalks among proteome, ubiquitylome and acetylome in non-small cell lung cancer A549 cell line. Sci Rep. 2015. March 31;5:9520. doi: 10.1038/srep09520. PubMed PMID: ; PubMed Central PMCID: PMCPMC4379480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Herz HM, Mohan M, Garrett AS, et al. Polycomb repressive complex 2- dependent and -independent functions of Jarid2 in transcriptional regulation in Drosophila. Mol Cell Biol. 2012. May;32(9):1683–93. doi: 10.1128/MCB.06503-11. PubMed PMID: ; PubMed Central PMCID: PMC3347239. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]