
Figure 3. Analytical workflows.
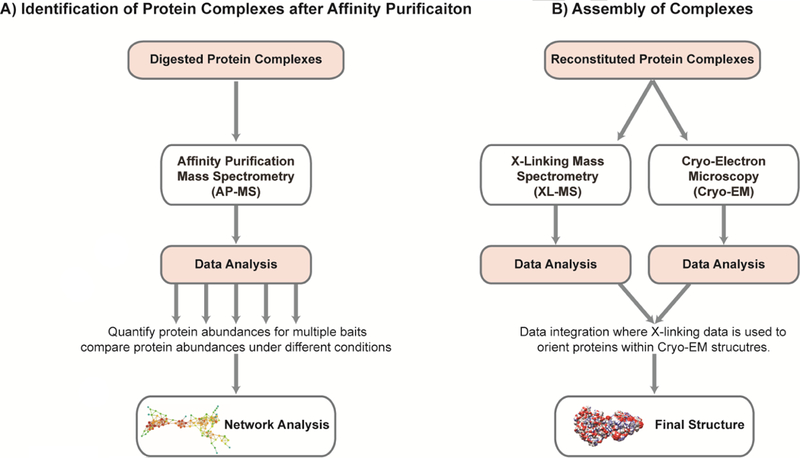
A brief overview of two workflows illustrating how quantitative proteomics is applied to the study of large protein complexes in chromatin biology. Both workflows progress in the same general manner, beginning with sample preparation, followed by mass spectrometry, and culminating in data analysis. However, each workflow utilizes different techniques. The first workflow, A) Identification of Protein Complexes, uses digested proteins and affinity purification mass spectrometry (AP-MS) to quantify and compare the protein abundances through the analysis of protein protein interactions (PPI) networks. The second workflow, B) Assembly of Protein Complexes, uses reconstituted protein complexes with both cryo- Electron Microscopy (cryo-EM) and cross-linking mass spectrometry (XL-MS) to determine the final structure of large protein complexes. Both methods are useful for determine the composition of large protein complexes in chromatin biology.