

Abstract
In the current study, we examined the role of Ezh2 as an epigenetic modifier for the enteric neural crest cell development through H3K27me3. Ezh2 conditional null mice were viable up to birth, but died within the first hour of life. In addition to craniofacial defects, Ezh2 conditional null mice displayed reduced number of ganglion cells in the enteric nervous system. RT-PCR and ChIP assays indicated aberrant up-regulation of Zic1, Pax3, and Sox10 and loss of H3K27me3 marks in the promoter regions of these genes in the myenteric plexus. Overall, these results suggest that Ezh2 is an important epigenetic modifier for the enteric neural crest cell development through repression of Zic1, Pax3, and Sox10.
Introduction
The neural crest cells (NCCs) are a multipotent cell population which have evolved among land vertebrates to develop a head, jaw, and sensory nervous system to adapt to predatory lifestyles [1]. The NCCs arise between the neural epithelium and epidermis after neurulation and migrate throughout the body to generate many different cell types. Depending on their rostro-caudal axis origin and cellular differentiation capabilities, NCCs can be divided into cranial, sacral, cardiac, trunk and vagal NCCs [2]. A number of different genes have been characterized to be important for the development of NCCs. Bone morphogenetic proteins (Bmps) and Wnt proteins induce NC formation. Pax3 and Zic1 specify the neural plate border [3, 4], and Sox10 sustains NCC multipotency [5]. Hox genes, which are critical for body segmentation and axial patterning [6, 7], also affect the development of NCCs [8]. Mutations in these genes can lead to neurocristopathies, which encompass a wide range of congenital diseases of neural crest cell origin. In some examples of neurocristopathies, such as Hirshsprung’s disease and Waardenburg syndrome, the enteric nervous system (ENS) is improperly developed, leading to intestinal aganglionosis in the distal portions of the colon [9]. Most of the ENS develops from vagal NCCs, and some contribution of sacral NCC have been seen in mice and chicks [10–12]. Although a number of genes have been characterized to be associated with the neurocristopathies, the penetrance for the mutant allele of these genes varies, suggesting a multigenic and potentially epigenetic involvement in manifestation of these disorders.
The polycomb group genes (PcGs) are epigenetic modifiers that play a significant role in regulating the expression of Hox genes and many other developmental transcription factors throughout embryonic development [13, 14]. The two major groups of PcGs include Polycomb repression complex 1 (PRC1) and Polycomb repression complex 2 (PRC2). These protein complexes are evolutionarily well conserved from flying insects to all mammals, including humans. These two complexes modify histones as an epigenetic signal: the ubiquitination on lysine 119 of histone 2B (H2BK119) by PRC1 and the methylation on lysine 27 of histone 3 (H3K27) by PRC2. In the case of PRC2, Enhancer of Zeste homolog 2 (Ezh2) is the histone methyltransferase, adding di- and tri-methyl groups to H3K27. The tri-methylation of H3K27 (H3K27me3) is usually associated with gene repression [15–17]. Complete deletion of Ezh2 leads to early embryonic lethality [18], and tissue specific deletions of Ezh2 display loss of cellular differentiation [19, 20]. In particular, deletion of Ezh2 in NCCs hinders the development of craniofacial bone structure in association with the de-repression of Hox genes in the first brachial arch (BA1) cells. In vitro culture of these BA1 cells demonstrated that differentiation factors such as GFAP and NF160 were properly expressed in Ezh2 conditional null cells [8]. However, these mutant NCCs have not been examined in later stages of embryonic development in vivo.
In the current study, we performed a series of conditional knockout experiments targeting Ezh2 to examine the effect of PRC2 in neural crest cell development in later stages of embryogenesis focusing on the ENS development. In addition to aberrant facial structures, conditional ablation of Ezh2 in NCCs led to improper development of ganglion cells in the ENS and derepression of Pax3, Zic1, and Sox10. Our results suggest a unique epigenetic role of Ezh2 in enteric neural crest cell development.
Results
Conditional knockout of Ezh2 in neural crest cells leads to perinatal lethality and abnormal facial structures
To characterize the function of Ezh2 during mouse NCC development, the mutant mice homozygous for the two loxP sites flanking exon 15 through 18 in 129P2/OlaHsd background were backcrossed with the Wnt1-Cre line in the 129/B6 mixed background, generating the Ezh2 homozygous mice in NCC (Ezh2flox/flox;Wnt1-CreT/+; hereafter referred to as ‘Ezh2 null’ mice). Exon deletion and subsequent reduced expression of Ezh2 were further confirmed through qRT-PCR (S1 Fig). Analysis of early and late embryos from this cross showed the expected Mendelian ratios, while an increased number of dead Ezh2 null mutants were observed among the pups found within the first day of birth (Fig 1A, 1B and 1C). The deletion of Ezh2 in NCC resulted in the lethality around the time of their birth (P0).
Fig 1. Various facial deformities and perinatal lethality in Ezh2 null mice.

A) Ezh2 null mice displays abnormal craniofacial structures, which are visible at E15.5 and continues through P0 (Ezh2 null, n = 30/30). B) H&E staining shows absence of frontal face and ‘pigeon’s chest’ (marked in asterisk) (Ezh2 null, n = 4/12). C) Kaplan-Meier survival curve demonstrates perinatal lethality in Ezh2 null mice within first 40 minutes after birth (n = 35, p = 0.0194, Fisher’s exact test). D) Absence of outer ear in Ezh2 null mice marked in white box. E) Absence of the outer ear in an H&E-stained transverse section of the head of a Ezh2 null P0 mouse, and higher magnification of an H&E-stained transverse section of the head revealing aberrant development of the middle ear in the Ezh2 null P0 mice. Wnt1-Cre-Red arrow: Developing outer ear (pinna and external ear canal), Black arrow: Developing middle ear (ossicles), Red asterisks: Developing inner ear (left: cochlea; right: cochlea + semicircular canals). Ezh2flox/flox;Wnt1-CreT/+- Red asterisks: Developing inner ear (cochlea + semicircular canals). There was no evidence of proper middle ear and outer ear development in any of the transverse sections of the head of the mutant mice. F) The dissected eyes of P0 pups have a 2/3 reduction in size in Ezh2 null mutants (n = 5/10) compared to the wild-type littermates. G) Circle: Shows primordium of oral cavity. Long arrow: Points to the space corresponding to the nasal chamber. Short arrow: points to a space that appears to correspond to the nasopharynx (junction of nasal cavity and pharynx). H) Insufficient closure of the eyelid can be seen in Ezh2 null P0 pups (marked arrowheads) (n = 30/30). H&E staining shows complete absence of cornea and conjunctiva. I) Toluidine blue staining of P0 pups showed staining only in the eyelid region of Ezh2 mutants (n = 4/4). J) Cleft lip (or bifid nose) visible in region of the upper lips of Ezh2 null mice. High magnification of an H&E-stained transverse section of the head of an Ezh2 mutant P0 mice. The arrow in red points to the nasal pits, and the surrounding cartilage plates are marked with (c) in red. The oral cavity in the center is rudimentary.
Microscopic analyses were performed to examine the facial features of the Ezh2 null pups (P0). The results are summarized as follows. First, all of the Ezh2 null pups displayed craniofacial defects (n = 30/30), which is consistent with previous observations from the similar mutant line (Ezh2flox/flox; Wnt1-Cre; Rosa26) at E17.5 stage [8] (Fig 1B and 1C). Mutant features in neonatal pups included absence of mandible (Fig 1G, S2, S3 and S6 Figs), absence of tongue (Fig 1G and S6 Fig), abnormal development of the middle ear (Fig 1E), and absence of nasofrontal plate with herniation of the brain (frontal lobe) and meninges through the extensive cranial bone defect (meningoencephalocele) (S2 Fig). Second, the Ezh2 null pups displayed absence of outer ear and microphthalmia (Fig 1D, 1F and 1B, and S2 Fig). Consistent with the fact that the pinna develops from NCCs [21], the pinnae of the outer ear were not visible in any of Ezh2 null mice (n = 30/30) (Fig 1D and 1E, and S6–S8 Figs). Some of the Ezh2 null mice displayed microphthalmia: the eye was approximately 2/3 the size of that in the wild-type littermates (n = 5/10) (Fig 1F). Third, a protruding chest (or ‘pigeon’s chest’-like defect) was observed in some Ezh2 null pups (n = 4/12) (Fig 1B asterisks). The Ezh2 null mice also exhibit defects in closure and merging of eyelids and upper lip (Fig 1H and 1J). All Ezh2 null mice (n = 30/30) had incomplete development of eyelids with immature mesenchyme covered by a thin layer of epithelium remaining in their place (Fig 1D and 1H). We performed toluidine blue staining of the mice to characterize the epithelial permeability barrier. As expected, staining was detected only at the site of the non-developed eyelids of the Ezh2 null mice (Fig 1I). The epithelial barrier was intact throughout the remaining face and body of the Ezh2 null neonatal pups, as the other areas of the body did not stain. Histologically, the Ezh2 null neonatal pups had no cornea and conjunctiva, resulting in a malformed anterior segment of the eyes (Fig 1H). Non-NCC lineage structures, however, such as the retina and the uvea, were similarly developed as those in the wild-type littermates (Fig 1H). The Ezh2 null mice also displayed incomplete fusion of the nasal placode (Fig 1J and S9 Fig). Gentle massaging of the stomach in Ezh2 null mutants after caesarian section released amniotic fluid from the mouth as it did in their wild-type littermates. This suggests that the oral cavity and the gastrointestinal tract are properly connected in the mutants. All mice, including the Ezh2 null mutants, also had well developed lungs, indicating adequate communication of the lower respiratory tract with the nasopharynx since fetal breathing of amniotic fluid is required for proper lung development. Overall, conditional deletion of Ezh2 in NCCs resulted in prenatal lethality and severe craniofacial defects.
Reduced number of ganglion cells in the enteric nervous system of Ezh2 null mice
The ganglion cells of the enteric nervous system (ENS) are also of NCC lineage. To examine proper development of ENS, series of AChE (acetylcholinesterase) histological staining were performed with the Ezh2 null mutants and the wild-type littermates [22]. The earliest visible difference in the morphology of these ganglion cells and nerve fibers through AChE staining was after E18.5, but was the most prominent in neonatal (P0) pups. Complete depletion of ganglion cells (aganglionosis) was visible in the distal colons of some Ezh2 null mice (n = 3 of 7 Ezh2 null mice) (Fig 2 –bottom panel) while milder reduction of ganglion cells (hypoganglionosis) were apparent in the other Ezh2 null mice (n = 4 of 7 Ezh2 null mice) (Fig 2 –middle panel). In the proximal colon, approximately 90% and 60% reduction of ganglion cells in comparison to its wild type littermate were displayed from the severe and mild cases of hypoganglionosis, respectively (p<0.0001) (Fig 2). In addition, the reduction of the enteric nervous system plexus was also observed in the small intestine and stomach of Ezh2 null mutants compared to those in the wild-type littermates (approximately 50% reduction, p = 0.0011) (S4 and S17 Figs). In sum, some of the Ezh2 null mice displayed aganglionosis in the distal colon and the hypoganglionosis was extended to the small intestine and the stomach.
Fig 2. Hypoganglionosis in Ezh2 null mice.

Whole-mount AChE staining in wild type and Ezh2 null E18.5 mice. AChE stained proximal and distal colon of wild-type littermate (top panel) and two Ezh2 mutants (bottom panel). The two Ezh2 null mice are representative of mild (middle panel, Ezh2 null n = 4/7, p<0.0001) hypogangliosis and severe (bottom panel, Ezh2 null n = 3/7, p<0.0001) aganglionosis cases. The bar graph is the quantification of the AChE staining in the proximal colon. (scale bar = 0.25 mm).
Expression level changes in the whole gut of Ezh2 null mice
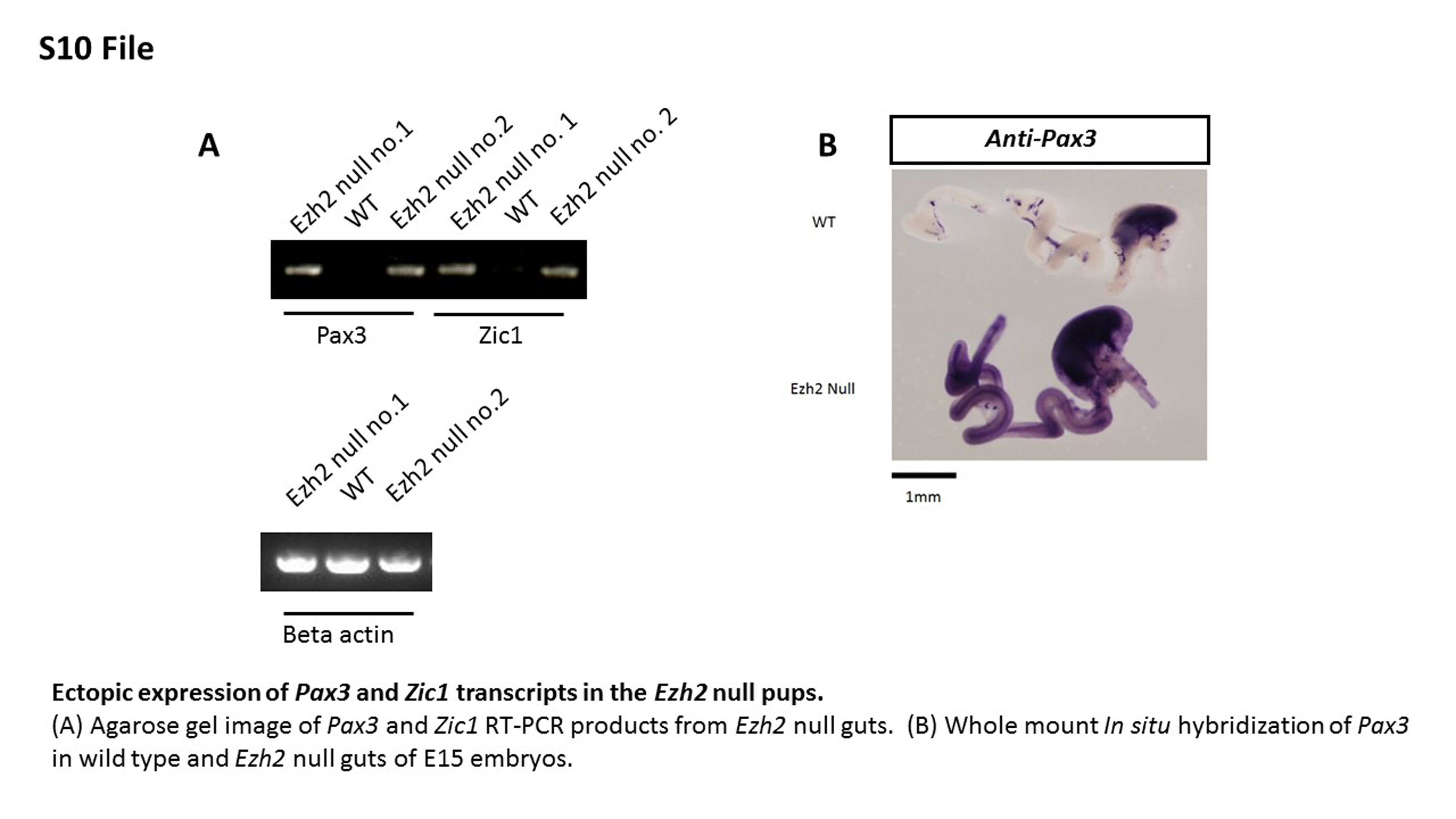
The effects of the Ezh2 deletion on the enteric nervous system were further analyzed at the molecular level in the following manner. First, we examined the expression levels of NCC developmental genes using the total RNA isolated from the guts of the Ezh2 null embryos and wild-type littermates at E18.5. Since previous studies observed over-expression of Hox genes in the BA1 cells of the Ezh2 null mutants, we also tested the expression levels of HoxA9, HoxA1, HoxA4, HoxA5, HoxB3, and HoxB5 (S5 Fig). According to the results, the majority of the tested NCC genes were down-regulated in the Ezh2 null embryos compared to their wild-type littermates (Fig 3). The most significant and consistent down-regulation was observed in Phox2b (n = 4, p = 0.0035) and Sox10 (n = 8, p = 0.0239). In contrast, two genes, Zic1 (n = 6, p = 0.003) and Pax3 (n = 6, p = 0.019), were 5 to 30 fold up-regulated in the Ezh2 null embryos compared to their wild-type littermates (Fig 3). A slight up-regulation of HoxA9 and HoxA5 was seen in some of the Ezh2 null mutants (n = 2), but the levels were not as significant as Pax3 or Zic1 (S5 Fig). Gel electrophoresis of the PCR product from whole gut cDNA confirmed the presence of Pax3 (226 bp) and Zic1 (235 bp) expression in Ezh2 null mutants and absence of expression in its wild-type littermates (S10 Fig). To examine the aberrant spatiotemporal gene expression of Pax3, series of whole mount RNA in situ hybridization were performed on the E15 gut. All wild-type embryo guts did not show staining with the anti-Pax3 probe (n = 6), while Ezh2 null embryo showed staining throughout the entire gut (n = 3) (S10 and S11 Figs).
Fig 3. NCC developmental gene expression in Ezh2 null mice guts.

NCC developmental gene expression levels were compared between wild-type (grey, Ezh2flox/+Wnt1-CreT/+, Ezh2flox/+ Wnt1-Cre+/+, Ezh2+/+ Wnt1-CreT/+, were all combined as wild type control), and Ezh2 mutant (red) gut tissues with qRT-PCR. The relative gene expression fold difference between Ezh2 null and wild type littermates are presented. Each bar graph represents a biological replicate. Among the eleven genes tested, four genes displayed significant expression changes in the Ezh2 null. Phox2b (n = 6, p = 0.0035) and Sox10 (n = 6, p = 0.00239) were down-regulated, and Zic1 (n = 6, p = 0.003) and Pax3 (n = 6, p = 0.0019) were up-regulated in the Ezh2 null mice.
Expression analysis and epigenetic changes in NCC genes of Ezh2 null mice in isolated myenteric plexus
To test if the changes in the expression of these NCC developmental genes are in the enteric nervous system, the myenteric plexus was isolated from P0 neonates in accordance with previously published protocols by Grundmann et al. [23] (Fig 4A). The expression of Pax3 (n = 3, p = 0.0010) and Zic1 (n = 3, p = 0.0071) was consistently derepressed in the myenteric plexus of the Ezh2 null mice, while no expression was observed in the wild-type littermate control. In contrast to the qRT-PCR data from the whole gut, in which the Sox10 was seen to be down-regulated in the Ezh2 null mice, a 20-fold up-regulation of Sox10 (n = 4, p = 0.0263) was detected in the Ezh2 null myenteric plexus cells. Overall, Pax3, Zic1 and Sox10 are aberrantly expressed in the isolated Ezh2 null myenteric plexus cells (Fig 4B).
Fig 4. NCC gene expression analysis and H3K27me3 ChIP assays in myenteric plexus.

A) Dissection and isolation of myenteric plexus from P0 wild-type and Ezh2 null mice for qRT-PCR and ChIP assays. B) qRT-PCR of Pax3, Zic1 and Sox10 in myenteric plexus. In the Ezh2 null myenteric plexus, Pax3 was upregulated 20–40 fold (n = 3, p = 0.0010), Sox10 was upregulated 5–30 fold (n = 3, p = 0.0263), and Zic1 was upregulated 40–100 fold (n = 3, p = 0.0071) compared to its wild type littermates. C) H3K27me3 ChIP-qPCR in Ezh2 mutant (in orange) and wild-type littermate (in grey) P0 gut tissues. The amount of precipitated DNA in promoter regions of NCC developmental genes and HoxA9 are presented as a relative value (%) to that of the input DNA normalized over a negative locus (beta actin) (y-axis). Student’s t-test (two-tailed) value shows significant difference in H3K27me3 enrichment between Ezh2 null (n = 6) and wild-type (n = 8) littermates (p<0.01).
Since the H3K27me3 histone modification mark is established through Ezh2 and this modification is associated with gene repression, ChIP assays with H3K27me3 antibodies (Upstate 07–449, Lot No. DAM1703508) were performed on the isolated myenteric plexus. These experiments utilized the chromatin isolated from the myenteric plexus of Ezh2 null and wild-type littermates at E18.5 (Fig 4A). According to the results, Pax3 and Zic1 showed the most significantly (p = 0.0011 and p = 0.0035, respectively) reduced levels of H3K27me3 enrichment in the Ezh2 null embryos (n = 6) compared to those from the wild-type littermates (n = 8) (Fig 4C). In sum, this series of analyses indicated up-regulation of Zic1, Pax3 and Sox10, and reduced levels of H3K27me3 modifications in the promoter regions of NCC developmental genes.
Reduced neuronal and glial cells and aberrant accumulation of SOX10 in Ezh2 null myenteric plexus
To examine the number of neurons and glial cells in the myenteric plexus, the longitudinal muscle attached to myenteric plexus (LMMPs) was isolated from the ileum and the proximal colon and were stained with a neuronal marker Hu [24] and a glial marker GFAP. The reduced number of neuron and glial cells were visible from E16 embryos (Fig 5), and this pattern of reduced neuronal and glial cells in the Ezh2 null mice continued in P0 neonates (Figs 6 and 7). On average, approximately 200 neuronal cells were counted per 300μm2 area in wild type P0 mice while approximately half of that amount of neurons in the same area was seen in the Ezh2 null myenteric plexus ranging from 39 to 138 neuronal cells (n = 8, p<0.001) (Fig 7). Immunohistochemistry of GFAP also showed much less amount of GFAP positive cells in the Ezh2 null mice compared to its littermate control in both E16 and P0 stage myenteric plexuses (n = 8, p<0.0001) (Figs 5 and 7).
Fig 5. Immunohistochemistry of Hu and GFAP in E16 Ezh2 null mice myenteric plexus.

The distribution of Hu expression (green) and GFAP expression (red) in wild type (top, n = 4) and Ezh2 null (bottom, n = 4) E16 myenteric plexus (scale bar = 100 μm).
Fig 6. Double immunofluorescence of Sox10 and Hu in myenteric plexus of P0 neonates.

Immunohistochemical analysis of Sox10 (red) in P0 neonates show non-overlapping expression of Sox10 (red) and Hu (green) in wild-type littermates (top, n = 4), while Ezh2 null mutants (n = 4) display aberrant overexpression of Sox10 proteins that partially overlap with Hu expression (bottom) (overlap of Sox10 and Hu marked with white arrow) (p<0.0001, scale bar = 100 μm).
Fig 7. Double immunofluorescence of Sox10 and GFAP in myenteric plexus of P0 neonates.

Co-expression analysis of Sox10 (red) and GFAP (green) in wild type (top panel, n = 4) and Ezh2 null (bottom panel, n = 4) myenteric plexus (p<0.001, scale bar = 100 μm).
As Sox10 is an early precursor marker for NCCs [25], and it is one of the genes which displayed contradicting results in the expression patterns of the whole gut compared to the isolated myenteric plexus (Figs 3 and 4), the expression patterns of Sox10 in the myenteric plexus of P0 stage neonates were examined with immunohistochemistry. During the enteric nervous system development, Sox10 is expressed all migrating progenitors of ENCCs, and as the cells differentiate into neuronal lineage, Sox10 is repressed in the neuronal cells and the expression of Sox10 remains only in the glial cells of the developed enteric nervous system [25, 26]. As expected, the SOX10 immunohistochemistry staining in Ezh2 wild type control mice also did not show much overlapping expression of SOX10 and Hu (Fig 6 –top panel), and almost complete overlapping expression of SOX10 and GFAP (Fig 7 –top panel). On the other hand, the Ezh2 null mice displayed aberrant overexpression of SOX10 proteins, some of which overlapped with Hu (Fig 6, bottom panel, white arrow) and some of which overlapped with GFAP (Fig 7 –bottom panel, white arrow). Other clusters of SOX10 overexpressing cells did not overlap with any of these markers (Figs 6 and 7). Overall, Ezh2 null mice displayed reduced number of neuronal cells and glial cells in the myenteric plexus of E16 embryos and P0 stage neonates, and overexpression of Sox10 proteins were detected in the myenteric plexus P0 stage neonates of Ezh2 null mice.
Discussion
In the current study, we characterized the function of Ezh2 as an epigenetic modifier in the development of cranial NCCs and ENCCs. First, we described the mutant phenotypes exhibited by the deletion of Ezh2 in NCCs. Then, we performed qRT-PCR, ChIP assays, and immunohistochemistry in the neonate myenteric plexus to test potential changes in the expression and epigenetic modification levels in NCC developmental genes in the Ezh2 null mice. The results suggest a role for Ezh2 as an epigenetic modifier for the development of ENCC in regulating the repression of Pax3, Zic1, and Sox10 during the development of the ENCCs.
The Ezh2 null mice were viable up to birth, but died shortly after (Fig 1). Consistent with the previous observation, these mice displayed multiple facial deformities, which include the absence of the jaw bones, underdevelopment of the middle ear, absence of Meckel’s cartilage, absence of nasofrontal plate with herniation of the brain and meninges (Fig 1) [8, 27]. Some of the newly described cranial NCC mutant phenotypes include absence of outer ear, incomplete development of the eyelids, absence of the cornea and conjunctiva, microphthalmia, and bifid nose (Fig 1). Ezh2 has been conditionally deleted in a large variety of murine cell types to examine its epigenetic properties during cellular differentiation. For example, conditional deletion of Ezh2 in skin cells (K14-Cre) led to reduced rate of epidermal differentiation [20], and deletion of Ezh2 in cortical progenitor cells (Emx1-Cre) led to improper development of neuronal cells in the brain [28]. Also, Ezh2 deletion in mesenchymal cells (Prrx1-Cre) led to many different skeletal abnormalities, which included domed head and premature fusion of the sutures in the skull [29, 30]. The function of Ezh2 in stem cells has been of much interest, yet its role in NCCs, which exhibit multipotent properties, has not been explored with other NCC related Cre lines, such as P0-Cre and Sox10-Cre mouse lines [31]. Currently, our studies and Schwarz et al. are the two publications, reporting the role of Ezh2 in cranial NCC development in murine models.
In addition to the cranial NCC defects, aberrant development of ENCCs were also often seen in Ezh2 null mice (Fig 2). The Ezh2 null mice displayed hypogangliosis in later stages of embryonic development (E18.5), characterized by hypoganglionosis in the distal colon and 30%-60% reduction in the stomach and small intestine enteric plexus in Ezh2 mutants (Fig 2, S4 and S17 Figs). Gene expression analysis in the E18.5 whole gut of Ezh2 null mice showed significant reduction of Sox10 and Phox2b expression and 5 to 30 fold up-regulation of Pax3 and Zic1 expression compared to its wild-type littermates (Fig 3). The spatial temporal expression of Pax3 was confirmed through whole mount in situ hybridization of E14.5 gut (S10 Fig) and the expression of the aberrant transcripts of Pax3, Zic1 and Sox10 were further confirmed in the isolated myenteric plexus cells (Fig 4). In the myenteric plexus, Sox10, which was down regulated in the whole gut seemed to also be surprisingly upregulated (Fig 4). The Immunohistochemistry of Ezh2 null myenteric plexus from E16 and P0 suggested reduced levels of both neuronal and glial cells and aberrant overexpression of Sox10 in P0 neonates (Figs 5, 6 and 7).
Two possible explanations can be suggested for the outcome of the hypoganglion mutant phenotype. First, the differentiation capacity of the ENCC precursor cells in Ezh2 null mice may be lost due to de-repression of Pax3 and Zic1. In Xenopus embryos, the over-expression of Pax3 and Zic1 was shown to induce the ectopic expression of neural crest differentiation downstream genes in the ventral region [32, 33]. During ENCC development, the neural plate border genes may need to be repressed through Ezh2, hence the loss of Ezh2 in these cells might lead to the aberrant expression of Pax3 and Zic1, resulting in competition for transcription factors or changes in chromatin structures of downstream gene enhancers. In the early NCC induction stage, Pax3 and Zic1 expression may have never been shut down due to the loss of H3K27me3 repression signals in the Ezh2 mutants. Aberrant expression of Pax3 in the later stages of NCC development has been reported to manifest cleft palate and abnormal osteogenesis in mice [34]. Zic1 has also been proposed to be repressed in migrating NCCs [35]. However, the derepression of Pax3 and Zic1 in the Ezh2 null mice may be limited to the ENCCs, since significant upregulation of these genes were not detected in the BA1 cells of Ezh2 null mice in previous studies [8]. Second, in addition to Pax3 and Zic1, Sox10 was also significantly up-regulated in the myenteric plexus cells. The conflicting results of Sox10 expression in the whole gut versus the myenteric plexus may be due to the overall reduced amount of NC specifier genes in the gut, but increased amount of undifferentiated cells in the myenteric plexus. Notably, aberrant expression of Sox10 has been linked to hypogangliosis in human patient studies and in mouse models [36–39]. The immunohistochemical staining of Sox10 in Ezh2 null mice seem to favor this scenario in that the clusters of Sox10 overexpressing cells overlap with neither neuronal nor glial cells (Figs 6 and 7) and seem to have adopted its own unique cell fate.
It is also possible that Hox genes are epigenetically altered in the ENS of the Ezh2 mutants, since Hox genes has been linked to megacolon phenotypes [40], and combined mutations in Ret and Hox genes have been previously linked to increased penetrance of Hirschsprung’s disease [41]. However, in the current studies, we have seen up-regulation of Pax3 and Zic1, but not HoxA9, which was one of the most significantly up-regulated genes in the BA1 cells of Ezh2 null mutants [8]. It is interesting that the upregulated genes between the cranial NCCs are not shared in the ENCCs. The transcriptional levels of other Hox genes, HoxA1, HoxA4, HoxA5, HoxB3, and HoxB5, which are involved in the enteric nervous system development [40, 42–45], were also tested. However, their gene expression levels were not significantly altered in the Ezh2 null mice (S5 Fig). Although ChIP assays suggest reduced H3K27me3 mark in HoxA9 promoter region, it is puzzling to see no changes in gene expression. A number of factors have been suggested to play a role in affecting the transcriptional machinery of PRC2, including how the PRC2 subunits such as Suz12, Eed, and Ezh1 are assembled, and how these complexes interacts with other cofactors such as Jarid2 and Aebp2 [46]. The removal of H3K27me3 alone have been demonstrated be insufficient to activate its downstream genes [47]. Future studies in comparing the Ezh2 null cranial NCCs to its ENCCs and identifying the types of transcription factors that may respond to the loss of H3K27me3 and recruit gene expression machinery to its downstream genes may help to characterize the mechanism in how Ezh2 affects NCC development. Also, systematic characterization of H3K27me1, H3K27me2, and H3K27me3 during ENCC development may help us to understand some discrepancies between the ChIP results and the expression data. The limitations of the H3K27me3 antibody [48] must also be considered, since it may not be able to distinguish H3K27me1, H3K27me2 and H3K27me3, in which their downstream gene expression outcomes can vary significantly [49].
Hirschsprung’s disease (HSCR) occurs 1 in every 5000 live birth. In the majority of the cases, the HSCR are isolated with no other congenital anomalies, while 10% of the cases are presented with other complications such as craniofacial or heart defects [50, 51]. Our mouse model presents aberrant development of the enteric neural crest cells which resembles the Hirschsprung’s disease phenotype. However, due to other complications, they are perinatally lethal (Fig 1). The Wnt1-Cre line have been a historically standard mouse line to study NCC development, and a number of epigenetic modifiers such as Dnmt [52] and Dicer [53] have also been conditionally deleted with the Wnt1-Cre mice (S1 Table). While the Wnt1-Cre line has provided much elucidation in understanding the biology of NCC development, this classical Wnt1-Cre mice have been shown to ectopically express Wnt1 during early stages of development [54]. This may explain the variable penetrance we were seeing in the gut of Ezh2 null mice (Fig 2). A recent new mouse line Wnt1-Cre2 have been presented as an improved mouse system to examine NCC development [54]. Careful investigation of the gastrointestinal tract with Ezh2 deletion of this mouse model would be desired in the near future. Nevertheless, this study is the first to suggest a potential epigenetic role of Ezh2 in enteric neural crest cell development.
NCCs played a significant role in vertebrate evolution, including adaptation to the environment through formation of the head, and advancement in sensory function as well as complex signaling between the gut and the brain [1, 55]. Further understanding the conserved transcriptional regulatory mechanisms involved in the differentiation of cranial NCCs compared to vagal and sacral NCCs may help us to understand the process by which these mutipotent cell lineages have evolved. Moreover, characterizing the epigenetic modifications involved in regulating the spatiotemporal expression of these transcription factors can add to another layer of deciphering the gene regulatory network responsible for NCC development. In this study, we show the importance of Ezh2’s role as an epigenetic modifier in cranial and non-cranial NCC development, and we propose the involvement of the H3K27me3 epigenetic mechanism in repressing some of the neural crest cell developmental genes which can manifest a Hirschsprung’s disease-like phenotype. The NCCs are a great cell system to decipher the function of epigenetic modifiers, since it can give rise to many different cell types in the body of vertebrates. We hope that these studies would shed light in the understanding of the functions of histone modifiers and their roles in manifesting various neurocristopathies.
Material and methods
Ethics statement
All the experiments related to mice were performed in accordance with the National Institutes of Health guidelines for care and use of animals, and also approved by the Louisiana State University Institutional Animal Care and Use Committee (IACUC), protocol #16–009.
Animals
The Ezh2 conditional knockout mice were derived as previously described [56]. Presence of a loxP site insertion was confirmed through polymerase chain reaction (PCR) amplification with the following primers: LoxF- CCCATTGAGAGTGCTGACTCA; LoxR- ACCTCGCTATGTGTAACCAGT, and F3.1- TCTTAGCACTTGCTTGTTCCCATTG. Expected fragment sizes were 100 bp without the loxP insertion and 120 bp with the loxP insertion (S1 Fig). Template DNA was acquired through mouse ear clips that had been digested in tail lysis buffer (0.1 M Tris-Cl, 5 mM EDTA, 0.2% SDS, 0.2 M NaCl, pH 8.0, 20 μg/ml Proteinase K). E15.5 and E18.5 embryos were harvested through timed mating between Ezh2flox/flox; Wnt1-CreT/+ double mutant backcrossed with Ezh2flox/flox. The survival curve was generated by combining four litters of cesarean sections (n = 40). Pups delivered through cesarean sections were massaged on the abdomen, and fluid was gently removed from the nasal and oral cavity using Kimwipes. The p value was derived using the Fisher exact statistical test. All mouse work was approved by the IACUC committee at Louisiana State University.
Histology
Embryos and neonatal mice were fixed for 24–28 hours in 10% formalin and subsequently moved into 70% ethanol for storage. Tissue were trimmed and routinely embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E).
Toluidine blue staining
Toluidine blue staining was performed as previously described [57]. In brief, unfixed neonates were incubated in 100% methanol for 5 minutes and then stained in 0.1% toluidine blue dye for 20 minutes.
AChE staining
Acetylcholinesterase (AChE) staining was performed according to a previously described protocol [22]. In brief, the dissected stomach and intestines were fixed in 4% paraformaldehyde for 2 hours at 4°C and moved to saturated sodium sulfate solution overnight at 4°C. Then, the tissues were incubated in the staining buffer (0.2 mM ethopropazine HCl, 4 mM acetylthiocholine iodide, 10 mM glycine, 2 mM cupric sulfate, and 65 mM sodium acetate pH 5.5) for 4–5 hours at room temperature. Lastly, acetylcholinesterase staining was completed by incubating the stomach and intestines in 1.25% sodium sulfide pH 6 for 1.5 minutes.
RT-PCR
The whole gut and the myenteric plexus (according to Grundmann et al. [23]) was isolated and snap frozen in liquid nitrogen. After adding Trizol (Invitrogen) into frozen tissues, the tissues were further minced into smaller fragments with sterile scissors in Trizol. Tissue fractions in Trizol were left on ice for 30 minutes and total RNA was isolated as described by manufacture’s protocol (Invitrogen). Isolated RNA was resuspended in nuclease-free water and cDNA was synthesized with MMLV reverse transcriptase (Invitrogen) according to manufacture’s protocol.
Chromatin immunoprecipitation assay
The P0 neonatal intestine was dissected and the myenteric plexus was isolated with Liberase TH (Roche) [23]. Tissues were homogenized in cold 1X PBS and fixed in 0.1% PFA v/v at 37°C for 20 minutes. After three consecutive washes with cold 1X PBS, the crosslinked tissues were placed in the lysis buffer (1% SDS, 10mM EDTA, 50mM Tris-Cl pH8.1) for sonication. Chromatin fraction was pre-cleared with protein A/G agarose beads (Cat. No. sc-2003, Santa Cruz) and pre-immune serum for 2 hours. After spinning down agarose beads, the remaining supernatant was used for immunoprecipitation with H3K27me3 antibody (Cat. No. 07–449, Upstate Biotech. Lot No. DAM1703508). The myenteric plexus isolated from two P0 neonates were combined per ChIP assay, and each ChIP assay was repeated at least three times (n = 8 wild-type, n = 6 Ezh2 null).
Whole mount RNA in situ hybridization
The E15 embryo guts were dissected and fixed in 4% PFA overnight in 4°C. The whole mount RNA in situ hybridization was performed as described previously [58] with anti-Pax3 probe hybridization at 68°C. The images were captured with Olympus DP70 camera attached to the Olympus SZX7 stereo microscope.
Immunohistochemical procedures
The longitudinal muscle attached to the myenteric plexus (LMMP) was isolated from embryonic day 16 (E16) and P0 guts and fixed for 30 minutes in room temperature in 4% paraformaldehyde. After twenty-minute treatment in 0.05% Triton-X in 1X PBS, the LMMPs were blocked for endogenous peroxidase activity by incubation in 3% hydrogen peroxide for one hour in room temperature. The LMMPs were pre-blocked in blocking solution (1% BSA, 0.3% Triton-X in 1X PBS) for 2 hours at room temperature, and incubated in blocking solution with primary antibodies with gentle rocking at room temperature overnight. The dilutions for the primary antibody are as follows: Sox10 antibody (Santa Cruz, cat. sc365692, 1:100), GFAP antibody (Santa Cruz, cat. 33673, 1:50), HuD antibody (Santa Cruz, cat. sc28299, 1:1000). The bound antibodies were detected with Alexa Flour 488 (Invitrogen, cat. A11001, 1:200) and Alexa Flour tyramide (Invitrogen, cat. B40942) according to its manufacture’s protocols. The cells were imaged on Leica DM2500 microscope and the images were captured with 18.2 Color Mosaic camera from Diagnostic Instruments, Incorporated.
Data analysis
The current data presented were obtained from at least three biological replicates from three different mice litters and at least two experimental replicates. The survival was assessed through the Kaplan-Meier curve, and the associated p value was derived from the Fisher exact test. The p values evaluating the penetrance of the mutant phenotypes were also derived through the Fisher exact test. The statistical significance of the real-time qRT-PCR values for expression analysis and ChIP assays was evaluated using the two-tailed Student’s t-test. Proper amplification of PCR products were validated through gel electrophoresis following qPCR reactions (S15 and S16 Figs). All the primer sequences are presented in the supporting information (S13 and S14 Figs). The neuronal cells were quantified by counting the number of neurons in 300μm2 area of at least eight images obtained from four mice of each wild type and mutant genotypes (n = 4 wild type, n = 4 Ezh2 null). The glial cells were counted in GFAP positive cluster in 300μm2 area of at least eight images from four mice of each wild type and mutant genotypes. The statistical significance was tested through two-tailed Student’s t-test. The density of the nerve fibers from the AChE staining were imaged in a set magnification and was quantified through skeletal reconstruction of the ganglion cells on image J and the number of intersections were counted and divided by the relative distance analyzed (S12 Fig).
Supporting information
This file contains qRT-PCR results of Ezh2 transcripts and agarose gel electrophoresis images of the Ezh2 genotyping.
(JPG)
{kind=link}
This file contains a set of images showing 2D and 3D microCT images of wild type and Ezh2 null pups.
(JPG)
{kind=link}
This file contains a set of images of wild type and Ezh2 null E15.5 embryos using micro CT.
(JPG)
{kind=link}
This file contains images of AChE stained small intestine in wild type and Ezh2 null pups.
(JPG)
{kind=link}
This file contains qRT-PCR results of Hox genes in the gastrointestinal tracts of Ezh2 null mice.
(JPG)
{kind=link}
This file contains a zoomed in view of H&E staining presented in Fig 1G.
(JPG)
{kind=link}
This file contains a zoomed in view of H&E staining presented in Fig 1E.
(JPG)
{kind=link}
This file contains a zoomed in view of H&E staining presented in Fig 1E.
(JPG)
{kind=link}
This file contains a zoomed in view of H&E staining presented in Fig 1J.
(JPG)
{kind=link}
This file contains agarose gel images of Pax3 and Zic1 RT-PCR products from Ezh2 null guts, and whole mount in situ hybridization of Pax3 in wild type and Ezh2 null guts of E15 embryos.
(JPG)
{kind=link}
This file contains an image of in situ hybridization with Pax3 sense probe.
(JPG)
{kind=link}
This file contains an image of an overview for quantifying the AChE stained images.
(JPG)
{kind=link}
This file contains the primer sequences for NCC qRT-PCR.
(DOCX)
This file contains the primer sequences for NCC ChIP assays.
(DOCX)
This file contains agarose gel electrophoresis image of all NCC cDNA primers used in this study.
(JPG)
{kind=link}
This file contains agarose gel electrophoresis images of all NCC ChIP assay primers used in this study.
(JPG)
{kind=link}
This file contains an image of wild type and Ezh2 null P0 stomachs.
(JPG)
{kind=link}
This file contains a table summarizing the results of epigenetic modifiers that were deleted with Wnt1-Cre. The phenotypes reported in this current study are marked in red.
(JPG)
{kind=link}
Acknowledgments
We thank the Kim lab members for their generous support and feedback in constructing the manuscript, and David Donze for sharing reagents and equipment needed in our studies.
Data Availability
All relevant data are within the paper and its Supporting information files.
Funding Statement
This work was supported by the National Institute of Health [J.K. R01-GM066225, R01-GM097074].
References
- 1.Gans C, Northcutt RG. Neural crest and the origin of the vertebrates: a new head. Science. 1983; 220: 268–274. 10.1126/science.220.4594.268 [DOI] [PubMed] [Google Scholar]
- 2.Bhatt S, Diaz R, Trainor PA. Signals and switches in mammalian neural crest cell differentiation. Cold Spring Harb Perspect Biol. 2013; 5: a008326 10.1101/cshperspect.a008326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Betancur P, Bronner-Fraser M, Sauka-Spengler T. Assembling neural crest regulatory circuits into a gene regulatory network. Annu. Rev. Cell Dev Biol. 2010; 26: 581–603. 10.1146/annurev.cellbio.042308.113245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Millet C, and Monsoro-Burq AH. Neural crest induction at the neural border in vertebrates. Dev Biol. 2012; 366: 22–33. 10.1016/j.ydbio.2012.01.013 [DOI] [PubMed] [Google Scholar]
- 5.Kim YJ, Lim H, Li Z, Oh Y, Kovlyagina I, Choi IY, et al. Generation of multipotent induced neural crest by direct re-programming of human postnatal fibroblasts with a single transcription factor. Cell Stem Cell. 2014; 15: 497–506. 10.1016/j.stem.2014.07.013 [DOI] [PubMed] [Google Scholar]
- 6.Maconochie M, Nonchev S, Morrison A, Krumlauf R. Paralogous Hox genes: function and regulation. Annu Rev Genet. 1996; 30: 529–556. 10.1146/annurev.genet.30.1.529 [DOI] [PubMed] [Google Scholar]
- 7.Holland PW, Garcia-Fernàndez J. Hox Genes and Chordate Evolution. Dev Biol. 1996; 173: 382–395. 10.1006/dbio.1996.0034 [DOI] [PubMed] [Google Scholar]
- 8.Schwarz D, Varum S, Zemke M, Schöler A, Baggiolini A, Draganova K, et al. Ezh2 is required for neural crest-derived cartilage and bone formation. Development. 2014; 141: 867–877. 10.1242/dev.094342 [DOI] [PubMed] [Google Scholar]
- 9.Moore SW, Johnson G. Acetylcholinesterase in Hirschsprung’s disease. Pediatr Surg Int. 2005; 4: 255–263. [DOI] [PubMed] [Google Scholar]
- 10.Serbedzija GN, Burgan S, Fraser S, Bronner-Fraser M. Vital dye labelling demonstrates a sacral neural crest contribution to the enteric nervous system of chick and mouse embryos. Development. 1991; 111: 857–866. [DOI] [PubMed] [Google Scholar]
- 11.Burns AJ, Champeval D, Le Douarin NM. Sacral neural crest cells colonise aganglionic hindgut in vivo but fail to compensate for lack of enteric ganglia. Dev Biol. 2000; 219: 30–43. 10.1006/dbio.1999.9592 [DOI] [PubMed] [Google Scholar]
- 12.Burns AJ, Douarin NM. The sacral neural crest contributes neurons and glia to the post-umbilical gut: spatiotemporal analysis of the development of the enteric nervous system. Development. 1998; 125: 4335–4347. [DOI] [PubMed] [Google Scholar]
- 13.Beuchle D, Struhl G, Müller J. Polycomb group proteins and heritable silencing of Drosophila Hox genes. Development. 2001; 128: 993–1004. [DOI] [PubMed] [Google Scholar]
- 14.Soshnikova N, Duboule D. Epigenetic temporal control of mouse Hox Genes in Vivo. Science. 2009; 324: 1320–1323. 10.1126/science.1171468 [DOI] [PubMed] [Google Scholar]
- 15.Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002; 298: 1039–1043. 10.1126/science.1076997 [DOI] [PubMed] [Google Scholar]
- 16.Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006; 441: 349–353. 10.1038/nature04733 [DOI] [PubMed] [Google Scholar]
- 17.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-resolution profiling of histone methylation in the human genome. Cell. 2007; 128: 823–837. [DOI] [PubMed] [Google Scholar]
- 18.O’Carroll D, Erhardt S, Pagani M, Barton SC, Surani MA, Jenuwein T. The Polycomb-Group gene Ezh2 is required for early mouse development. Mol Cell Biol. 2001; 21: 4330–4336. 10.1128/MCB.21.13.4330-4336.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang J, Taylor RJ, La Torre A, Wilken MS, Cox KE, Reh TA, et al. Ezh2 maintains retinal progenitor proliferation, transcriptional integrity, and the timing of late differentiation. Dev Biol. 2015; 403: 128–138. 10.1016/j.ydbio.2015.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ezhkova E, Pasolli HA, Parker JS, Stokes N, Su IH, Hannon G, et al. Ezh2 orchestrates gene expression for the stepwise differentiation of tissue-specific stem cells. Cell. 2009; 136: 1122–1135. 10.1016/j.cell.2008.12.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mallo M. Formation of the Outer and Middle Ear, Molecular Mechanisms. Curr Top Dev Biol. 2003; 57: 85–113. [DOI] [PubMed] [Google Scholar]
- 22.Enomoto H, Araki T, Jackman A, Heuckeroth RO, Snider WD, Johnson EM Jr., et al. GFR(alpha)1-deficient mice have deficits in the enteric nervous system and kidneys. Neuron. 1998; 21: 317–324. [DOI] [PubMed] [Google Scholar]
- 23.Grundmann D, Klotz M, Rabe H, Glanemann M, Schäfer KH. Isolation of high-purity myenteric plexus from adult human and mouse gastrointestinal tract. Sci. Report. 2016; 5: 9226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Furness JB, Robbins HL, Xiao J, Stebbing MJ, Nurgali K. Projections and chemistry of Dogiel type II neurons in the mouse colon. Cell Tissue Res. 2004; 317: 1–12. 10.1007/s00441-004-0895-5 [DOI] [PubMed] [Google Scholar]
- 25.Bondurand N, Sham MH. The role of SOX10 during enteric nervous system development. Dev Biol. 2013; 382: 330–343. 10.1016/j.ydbio.2013.04.024 [DOI] [PubMed] [Google Scholar]
- 26.Kim J, Lo L, Dormand E, Anderson DJ. SOX10 maintains multipotency and inhibits neuronal differentiation of neural crest stem cells. Neuron. 2003; 38: 17–31. [DOI] [PubMed] [Google Scholar]
- 27.Zemke M, Draganova K, Klug A, Schöler A, Zurkirchen L, Gay MH, et al. Loss of Ezh2 promotes a midbrain-to-forebrain identity switch by direct gene derepression and Wnt-dependent regulation. BMC Biol. 2015; 13: 103 10.1186/s12915-015-0210-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pereira JD, Sansom SN, Smith J, Dobenecher MW, Tarakhovsky A, Livesey FJ. Ezh2, the histone methyltransferase of PRC2, regulates the balance between self-renewal and differentiation in the cerebral cortex. Proc Natl Acad Sci USA. 2010; 107: 15957–15962. 10.1073/pnas.1002530107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dudakovic A, Camilleri ET, Xu F, Riester SM, McGee-Lawrence ME, Bradley EW, et al. Epigenetic control of skeletal development by the histone methyltransferase Ezh2. J Biol Chem. 2015; 290: 27604–27617. 10.1074/jbc.M115.672345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hemming S, Cakouros D, Codrington J, Vandyke K, Arthur A, Zannettino A et al. Ezh2 deletion in early mesenchyme compromises postnatal bone microarchitecture and structural integrity and accelerates remodeling. FASEB J. 2017; 31: 1011–1027. 10.1096/fj.201600748R [DOI] [PubMed] [Google Scholar]
- 31.Debbache J, Parfejevs V, Sommer L. Cre-driver lines used for genetic fate mapping of neural crest cells in the mouse: An overview. Genesis. 2018; 56: e23105 10.1002/dvg.23105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sato T, Sasai N, Sasai Y. Neural crest determination by co-activation of Pax3 and Zic1 genes in Xenopus ectoderm. Development. 2005; 132: 2355–2363. 10.1242/dev.01823 [DOI] [PubMed] [Google Scholar]
- 33.Hong CS, Saint-Jeannet JP. The activity of Pax3 and Zic1 regulates three distinct cell fates at the neural plate border. Mol Biol Cell. 2007; 18: 2192–2202. 10.1091/mbc.E06-11-1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu M, Li J, Engleka KA, Zhou B, Lu MM, Plotkin JB, et al. Persistent expression of Pax3 in the neural crest causes cleft palate and defective osteogenesis in mice. J Clin Invest. 2008; 118: 2076–2087. 10.1172/JCI33715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.The zic1 gene is expressed in chick somites but not in migratory neural crest. Sun Rhodes LS, Merzdorf CS. Gene Expr Patterns. 2006; 6: 539–545. 10.1016/j.modgep.2005.10.006 [DOI] [PubMed] [Google Scholar]
- 36.Sham MH, Lui VCH, Fu M, Tam PKH. Sox10 is abnormally expressed in aganglionic bowel of Hirschsprung’s disease infants. Gut. 2001; 49: 220–226. 10.1136/gut.49.2.220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Southard-Smith EM, Kos L, Pavan WJ. Sox10 mutation disrupts neural crest development in dom Hirschsprung mouse model. Nat Genet. 1998; 18: 60–64. 10.1038/ng0198-60 [DOI] [PubMed] [Google Scholar]
- 38.Herbarth B, Pingault V, Bondurand N, Kuhlbrodt K, Hermans-Borgmeyer I, Puliti A, et al. Mutations of the Sry-related Sox10 gene in Dominant megacolon, a mouse model for human Hirschsprung disease. Proc Natl Acad Sci USA. 1998; 95: 5161–5165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nagashimada M, Ohta H, Li C, Nakao K, Uesaka T, Brunet JF, et al. Autonomic neurocristopathy-associated mutations in PHOX2B dysregulate Sox10 expression. J Clin Invest. 2012; 122: 3145–3158. 10.1172/JCI63401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tennyson VM, Gershon MD, Sherman DL, Behringer RR, Raz R, Crotty DA, et al. Structural abnormalities associated with congenital megacolon in transgenic mice that overexpress the Hoxa-4 gene. Dev Dyn. 1993; 198: 28–53. 10.1002/aja.1001980105 [DOI] [PubMed] [Google Scholar]
- 41.Garcia-Barceló MM, Miao X, Lui VC, So MT, Ngan ES, Leon TY, et al. Correlation Between Genetic Variations in Hox Clusters and Hirschsprung’s Disease. Ann Hum Genet. 2007; 71: 526–536. 10.1111/j.1469-1809.2007.00347.x [DOI] [PubMed] [Google Scholar]
- 42.Aubin J, Chailler P, Ménard D, Jeannotte L. Loss of Hoxa5 gene function in mice perturbs intestinal maturation. American Journal of Physiology. 1999; 277. [DOI] [PubMed] [Google Scholar]
- 43.Chan KK, Chen YS, Yau TO, Fu M, Lui VC, Tam PK, et al. Hoxb3 vagal neural crest-specific enhancer element for controlling enteric nervous system development. Dev Dyn. 2005; 233: 473–483. 10.1002/dvdy.20347 [DOI] [PubMed] [Google Scholar]
- 44.Fu M, Lui VC, Sham MH, Cheung AN, Tam PK. HOXB5 expression is spatially and temporarily regulated in human embryonic gut during neural crest cell colonization and differentiation of enteric neuroblasts. Dev Dyn. 2003; 228: 1–10. 10.1002/dvdy.10350 [DOI] [PubMed] [Google Scholar]
- 45.Kam MKM, Cheung MCH, Zhu JJ, Cheng WWC, Sat EWY, Tam PKH, et al. Perturbation of Hoxb5 signaling in vagal and trunk neural crest cells causes apoptosis and neucristopathies in mice. Cell Death Differ. 2014; 21: 278–289. 10.1038/cdd.2013.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aranda S, Mas G, Di Croce L. Regulation of gene transcription by Polycomb proteins. Sci Adv. 2015; 1: e1500737 10.1126/sciadv.1500737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kashyap V, Gudas LJ, Brenet F, Funk P, Viale A, Scandura JM. Epigenomic reorganization of the clustered Hox genes in embryonic stem cells induced by retinoic acid. J Biol Chem. 2011; 286: 3250–3260. 10.1074/jbc.M110.157545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rothbart SB, Dickson BM, Raab JR, Grzybowski AT, Krajewski K.Guo AH, et al. An interactive database for the assessment of histone antibody specificity. Mol Cell. 2015; 59: 502–511. 10.1016/j.molcel.2015.06.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ferrari KJ, Scelfo A, Jammula S, Barozzi I, Stützer A, Fischle W, et al. Polycomb-dependent H3K27me1 & H3K27me2 regulate active transcription and enhancer fidelity. Mol Cell. 2014; 53: 49–62. 10.1016/j.molcel.2013.10.030 [DOI] [PubMed] [Google Scholar]
- 50.Amiel J, Lyonnet S. Hirschsprung disease, associated syndromes, and genetics: a review. J Med Genet. 2001; 38: 729–739. 10.1136/jmg.38.11.729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brooks AS, Breuning MH, Osinga J, vd Smagt JJ, Catman CE, Buys CH, et al. A consanguineous family with Hirschsprung disease, microcephaly, and mental retardation (Goldberg-Shprintzen syndrome). J Med Genet. 1999; 36: 485–489. [PMC free article] [PubMed] [Google Scholar]
- 52.Jacques-Fricke BT, Roffers-Agarwal J, Gammill LS. DNA methyltransferase 3b is dispensable for mouse neural crest development. PLoS One. 2012; 7: e47794 10.1371/journal.pone.0047794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang ZP, Chen JF, Regan JN, Maguire CT, Tang RH, Dong XR, et al. Loss of microRNAs in neural crest leads to cardiovascular syndromes resembling human congenital heart defects. Arterioscler Thromb Vasc Biol. 2010; 30: 2575–2586. 10.1161/ATVBAHA.110.213306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lewis AE, Vasudevan HN, O’Neill AK, Soriano P, Bush JO. The widely used Wnt-Cre transgene causes developmental phenotypes by ectopic activation of Wnt signaling. Dev Biol. 2013; 379: 229–234. 10.1016/j.ydbio.2013.04.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gershon MD. The enteric nervous system: a second brain. Hosp Pract. 1995; 34: 31–32. [DOI] [PubMed] [Google Scholar]
- 56.Su IH, Basavaraj A, Krutchinsky AN, Hobert O, Ullrich A, Chait BT, et al. Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat Immunol. 2003; 4: 124–131. 10.1038/ni876 [DOI] [PubMed] [Google Scholar]
- 57.Spazierer D, Fuchs P, Reipert S, Fischer I, Schmuth M, Lassmann H, et al. Epiplakin is dispensable for skin barrier function and for integrity of keratin network cytoarchitecture in simple and stratified epithelia. Mol Cell Biol. 2006; 26: 559–568. 10.1128/MCB.26.2.559-568.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zakin L, De Robertis EM. Inactivation of mouse twisted gastrulation reveals its role in promoting BMP4 activity during forebrain development. Development. 2004; 131: 413–424. 10.1242/dev.00946 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This file contains qRT-PCR results of Ezh2 transcripts and agarose gel electrophoresis images of the Ezh2 genotyping.
(JPG)
This file contains a set of images showing 2D and 3D microCT images of wild type and Ezh2 null pups.
(JPG)
This file contains a set of images of wild type and Ezh2 null E15.5 embryos using micro CT.
(JPG)
This file contains images of AChE stained small intestine in wild type and Ezh2 null pups.
(JPG)
This file contains qRT-PCR results of Hox genes in the gastrointestinal tracts of Ezh2 null mice.
(JPG)
This file contains a zoomed in view of H&E staining presented in Fig 1G.
(JPG)
This file contains a zoomed in view of H&E staining presented in Fig 1E.
(JPG)
This file contains a zoomed in view of H&E staining presented in Fig 1E.
(JPG)
This file contains a zoomed in view of H&E staining presented in Fig 1J.
(JPG)
This file contains agarose gel images of Pax3 and Zic1 RT-PCR products from Ezh2 null guts, and whole mount in situ hybridization of Pax3 in wild type and Ezh2 null guts of E15 embryos.
(JPG)
This file contains an image of in situ hybridization with Pax3 sense probe.
(JPG)
This file contains an image of an overview for quantifying the AChE stained images.
(JPG)
This file contains the primer sequences for NCC qRT-PCR.
(DOCX)
This file contains the primer sequences for NCC ChIP assays.
(DOCX)
This file contains agarose gel electrophoresis image of all NCC cDNA primers used in this study.
(JPG)
This file contains agarose gel electrophoresis images of all NCC ChIP assay primers used in this study.
(JPG)
This file contains an image of wild type and Ezh2 null P0 stomachs.
(JPG)
This file contains a table summarizing the results of epigenetic modifiers that were deleted with Wnt1-Cre. The phenotypes reported in this current study are marked in red.
(JPG)
Data Availability Statement
All relevant data are within the paper and its Supporting information files.