
Figure 4. eQTL haplotype configurations that are predicted to increase pathogenic coding variant penetrance are enriched in individuals with cancer and autism spectrum disorder.
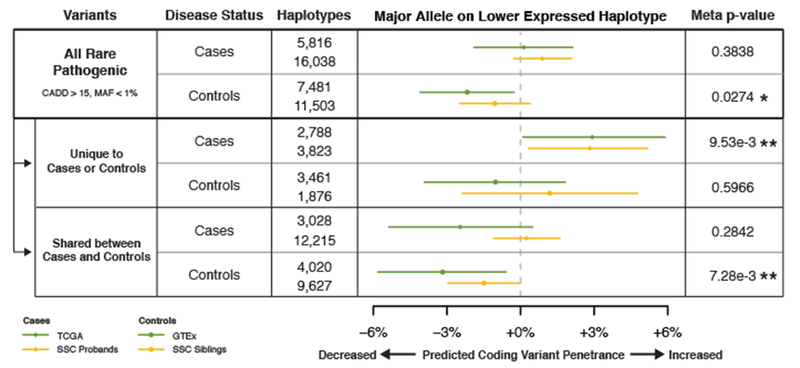
Analysis of eQTL and coding variant haplotype configurations in cases and controls for autism spectrum disorder (ASD) and cancer, using the top GTEx v6p eQTL per gene by p-value across all tissues. For ASD analysis, haplotype configurations generated from transmission phased genetic data of 2,304 SSC ASD affected probands (cases) and 1,712 of their unaffected siblings (controls) were used, and haplotypes were analyzed at ASD implicated genes. For cancer analysis, haplotype configurations generated from population and read-back phased germline whole genomes of 615 TCGA individuals (cases) and 620 whole genomes of v7 GTEx individuals (controls) were used, and haplotypes were analyzed at tumor suppressor genes. To enrich for putatively disease-causing variants, results were stratified based on if variants were restricted to cases or controls, or shared between both. Median estimates and 95% confidence intervals were generated using 100,000 bootstraps, and two-sided empirical p-values were generated from these confidence intervals and combined between cohorts using Fisher’s method to produce meta p-values (* p < 0.05, ** p < 0.01). See Methods – Gene Sets for description of gene sets used, Supplementary Figure 5 for description of eQTL coding variant haplotypes used for the analysis, Supplementary Figure 6 for results from benign variants and control genes, and Supplementary Table 3 for the full table of results, including individual cohort level p-values.